KV11.1, NaV1.5, and CaV1.2 Transporter Proteins as Antitarget for Drug Cardiotoxicity




Abstract
:1. Introduction
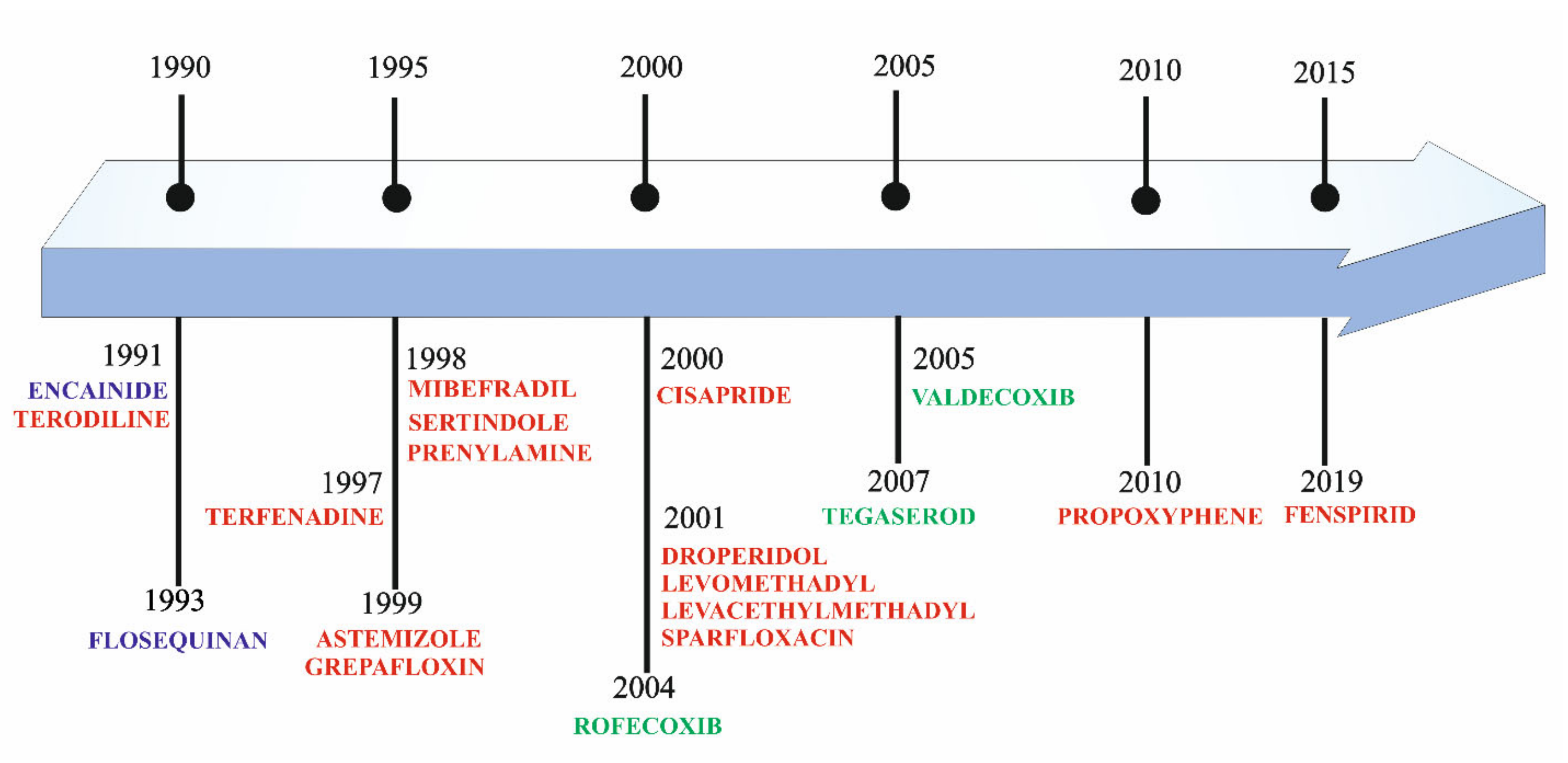
2. Historical Overview of Drug Cardiotoxicity
3. VGICs—Voltage-Gated Ion Channels
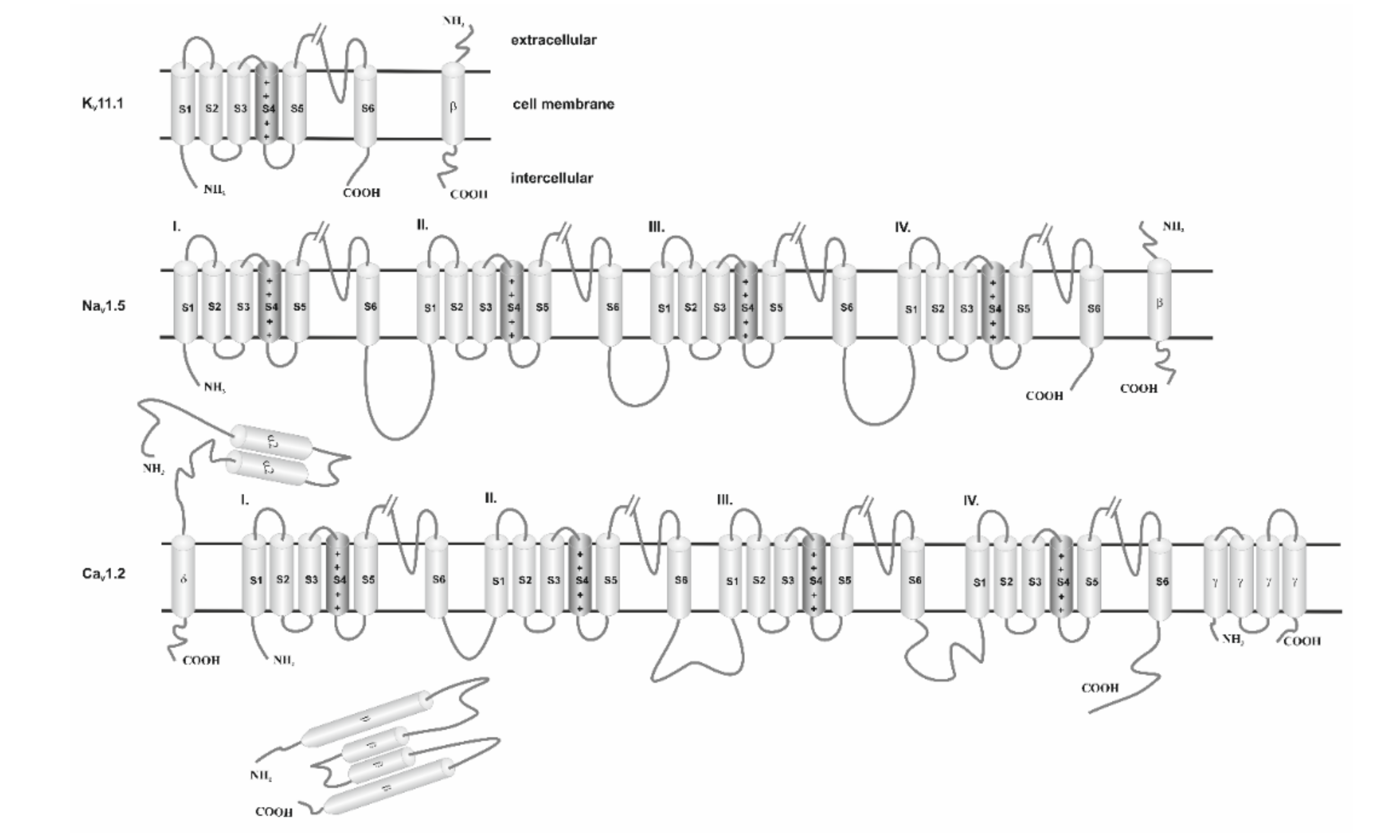
4. VGICs—Structure
4.1. Potassium Ion Channels
4.2. Calcium Ion Channels
4.3. Sodium Channels
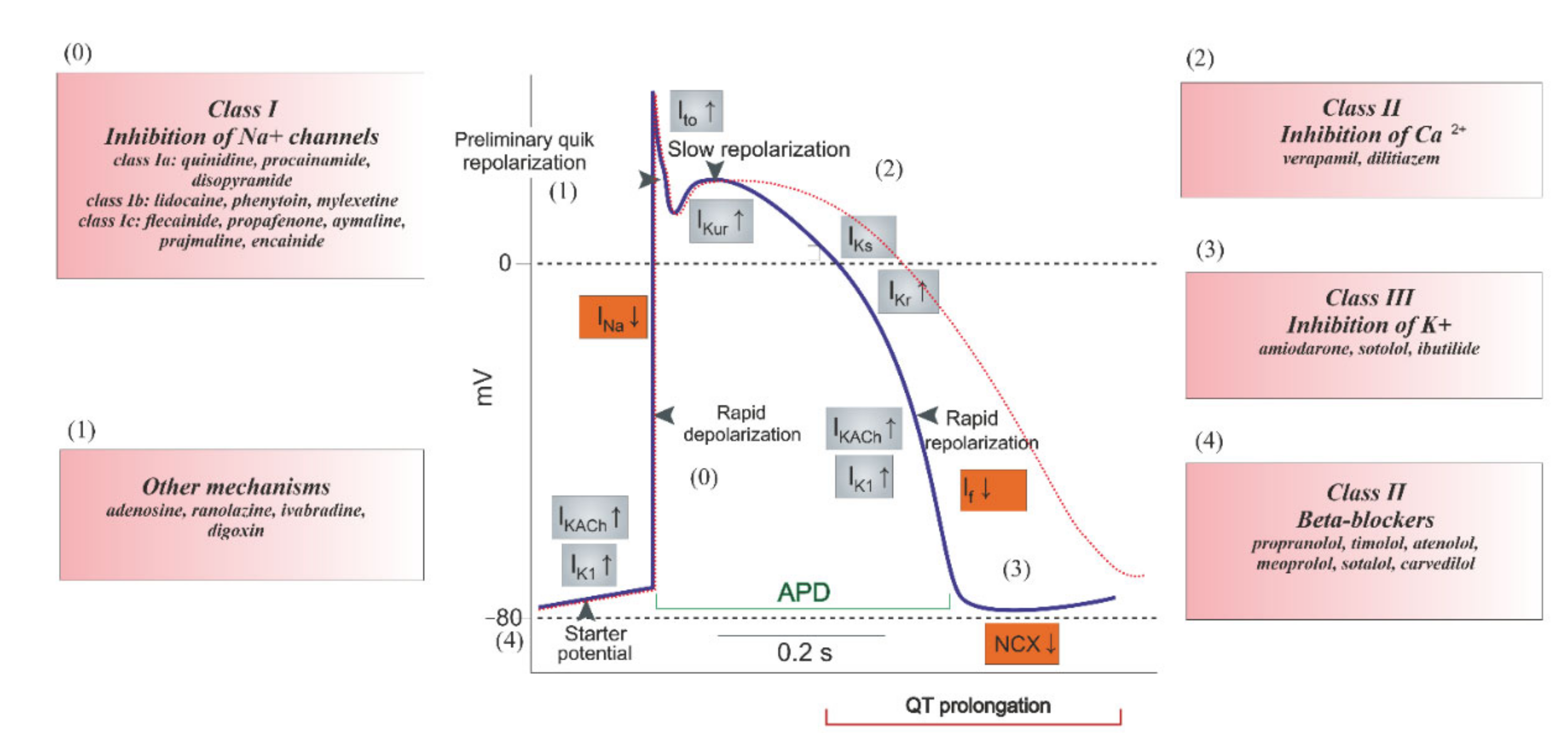
5. Mechanism of Ion Channel Inhibition
6. In Silico Methods for Testing the Risk of Cardiotoxicity
7. Conclusions and Perspective
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| VGICs | Voltage-Gated Ion Channels |
| VGKCs | Voltage-Gated Potassium Channels |
| VGNaCs | Voltage-Gated Sodium Channels |
| VGCaCs | Voltage-Gated Calcium Channels |
| hERG | Human Ether-à-go-go-Related Gene |
| TdP | Torsade the points |
| CiPA | Comprehensive In Vitro Proarrhythmia Assay |
| IC50 | Inhibition potency |
| Ki | Binding affinity |
References
- OECD Test No. 453: Combined Chronic Toxicity/Carcinogenicity Studies, OECD Guidelines for the Testing of Chemicals, Section 4. Available online: https://www.oecd-ilibrary.org/environment/test-no-453-combined-chronic-toxicity-carcinogenicity-studies_9789264071223-en (accessed on 29 October 2020).
- OECD, T.G.G. The Organisation for Economic Co-operation and Development; OEEC: Paris, France, 1960. [Google Scholar]
- Raghavan, M.; Fee, D.; Barkhaus, P.E. Generation and propagation of the action potential. In Neurology of Sexual and Bladder Disorders; Elsevier: Amsterdam, The Netherlands, 2019; Volume 160, pp. 3–22. [Google Scholar]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2015. [Google Scholar]
- Issa, Z.F.; Miller, J.M.; Zipes, D.P. Clinical Arrhythmology and Electrophysiology: A Companion to Braunwald’s Heart Disease. Clinical Arrhythmology and Electrophysiology: A Companion to Braunwald’s Heart Disease; Elsevier Health Sciences: Amsterdam, The Netherlands, 2019. [Google Scholar]
- Giegel, D.; Lewis, A.; Worland, P. Diversity versus Focus in Choosing Targets and Therapeutic Areas. In Comprehensive Medicinal Chemistry II.; Elsevier: Amsterdam, The Netherlands , 2007; pp. 753–770. [Google Scholar]
- Rubaiy, H.N. A Short Guide to Electrophysiology and Ion Channels. J. Pharm. Pharm. Sci. 2017, 20, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gingrich, K.J.; Yang, J. Molecular physiology. In Foundations of Anesthesia: Basic Sciences for Clinical Practice; Elsevier Health Sciences: Amsterdam, The Netherlands, 2006; 79p. [Google Scholar]
- Kirsch, G.E.; Kramer, J.; Bruening-Wright, A.; Obejero-Paz, C.; Brown, A.M. The Comprehensive In Vitro Proarrhythmia Assay (CiPA) Guide: A New Approach to Cardiac Risk Assessment; Charles River Laboratories International: Wilmington, MA, USA, 2016. [Google Scholar]
- Li, Z.; Ridder, B.J.; Han, X.; Wu, W.W.; Sheng, J.; Tran, P.N.; Wu, M.; Randolph, A.; Johnstone, R.H.; Mirams, G.R.; et al. Assessment of an In Silico Mechanistic Model for Proarrhythmia Risk Prediction Under the Ci PA Initiative. Clin. Pharmacol. Ther. 2019, 105, 466–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raschi, E.; Vasina, V.; Poluzzi, E.; de Ponti, F. The hERG K+ channel: Target and antitarget strategies in drug development. Pharmacol. Res. 2008, 57, 181–195. [Google Scholar] [CrossRef] [PubMed]
- Petkov, G.V. Ion channels. In Pharmacology; Elsevier: Amsterdam, The Netherlands, 2009; pp. 387–427. [Google Scholar]
- Cheung, S.; Parkinson, J.; Wählby-Hamrén, U.; Dota, C.D.; Kragh, A.M.; Bergenholm, L.; Vik, T.; Collins, T.; Arfvidsson, C.; Pollard, C.E.; et al. A tutorial on model informed approaches to cardiovascular safety with focus on cardiac repolarisation. J. Pharmacokinet. Pharmacodyn. 2018, 45, 365–381. [Google Scholar] [CrossRef] [PubMed]
- Sallam, K.; Li, Y.; Sager, P.T.; Houser, S.R.; Wu, J.C. Finding the Rhythm of Sudden Cardiac Death. Circ. Res. 2015, 116, 1989–2004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.-M.; Yu, M.-S.; Kazmi, S.R.; Oh, S.Y.; Rhee, K.-H.; Bae, M.-A.; Lee, B.H.; Shin, D.-S.; Oh, K.-S.; Ceong, H.; et al. Computational determination of hERG-related cardiotoxicity of drug candidates. BMC Bioinform. 2019, 20, 67–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woosley, R.D.; Romero, K.; Heise, C.W.; Gallo, T.; Tate, J.; Woosley, R.L. Summary of Torsades de Pointes (TdP) Reports Associated with Intravenous Drug Formulations Containing the Preservative Chlorobutanol. Drug Saf. 2019, 42, 907–913. [Google Scholar] [CrossRef]
- Guidance for Industry: E14 Clinical Evaluation of QT/QTc Interval Prolongation and Proarrhythmic Potential for Non-Antiarrhythmic Drugs; Food and Drug Administration: Rockville, MD, USA, 2005.
- ICH E14 Guideline: The Clinical Evaluation of QT/QTc Interval Prolongation and Proarrhythmic Potential for Non-Antiarrhythmic Drugs; International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use: Geneva, Switzerland, 2015.
- Li, Z.; Dutta, S.; Sheng, J.; Tran, P.N.; Wu, W.; Chang, K.; Mdluli, T.; Strauss, D.G.; Colatsky, T. Improving the In Silico Assessment of Proarrhythmia Risk by Combining hERG (Human Ether-à-go-go-Related Gene) Channel–Drug Binding Kinetics and Multichannel Pharmacology. Circ. Arrhythmia Electrophysiol. 2017, 10, e004628. [Google Scholar] [CrossRef]
- Hondeghem, L.; Carlsson, L.; Duker, G. Instability and Triangulation of the Action Potential Predict Serious Proarrhythmia, but Action Potential Duration Prolongation Is Antiarrhythmic. Circulation 2001, 103, 2004–2013. [Google Scholar] [CrossRef] [Green Version]
- Mirams, G.R.; Cui, Y.; Sher, A.; Fink, M.; Cooper, J.; Heath, B.M.; McMahon, N.C.; Gavaghan, D.J.; Noble, D. Simulation of multiple ion channel block provides improved early prediction of compounds’ clinical torsadogenic risk. Cardiovasc. Res. 2011, 91, 53–61. [Google Scholar] [CrossRef]
- Martin, R.L.; McDermott, J.S.; Salmen, H.J.; Palmatier, J.; Cox, B.F.; Gintant, G.A. The Utility of hERG and Repolarization Assays in Evaluating Delayed Cardiac Repolarization: Influence of Multi-Channel Block. J. Cardiovasc. Pharmacol. 2004, 43, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Blinova, K.; Stohlman, J.; Vicente, J.; Chan, D.; Johannesen, L.; Hortigon-Vinagre, M.P.; Zamora, V.; Smith, G.; Crumb, W.J.; Pang, L.; et al. Comprehensive Translational Assessment of Human-Induced Pluripotent Stem Cell Derived Cardiomyocytes for Evaluating Drug-Induced Arrhythmias. Toxicol. Sci. 2016, 155, 234–247. [Google Scholar] [CrossRef]
- Sager, P.T.; Gintant, G.; Turner, J.R.; Pettit, S.; Stockbridge, N. Rechanneling the cardiac proarrhythmia safety paradigm: A meeting report from the Cardiac Safety Research Consortium. Am. Hear. J. 2014, 167, 292–300. [Google Scholar] [CrossRef]
- Barker, B.S.; Young, G.T.; Soubrane, C.H.; Stephens, G.J.; Stevens, E.B.; Patel, M.K. Chapter 2—Ion Channels. In Conn’s Translational Neuroscience; Elsevier: Amsterdam, The Netherlands, 2017; pp. 11–43. [Google Scholar]
- Dunlop, J.; Bowlby, M.R.; Peri, R.; Vasilyev, D.; Arias, R. High-throughput electrophysiology: An emerging paradigm for ion-channel screening and physiology. Nat. Rev. Drug Discov. 2008, 7, 358–368. [Google Scholar] [CrossRef]
- Harmar, A.J.; Hills, R.A.; Rosser, E.M.; Jones, M.; Buneman, O.P.; Dunbar, D.R.; Greenhill, S.D.; Hale, V.A.; Sharman, J.L.; Bonner, T.I.; et al. IUPHAR-DB: The IUPHAR database of G protein-coupled receptors and ion channels. Nucleic Acids Res. 2008, 37, D680–D685. [Google Scholar] [CrossRef] [PubMed]
- Eranjan, R.; Logette, E.; Marani, M.; Herzog, M.; Tâche, V.; Scantamburlo, E.; Buchillier, V.; Markram, H. A Kinetic Map of the Homomeric Voltage-Gated Potassium Channel (Kv) Family. Front. Cell. Neurosci. 2019, 13, 13. [Google Scholar]
- Bennett, D.L.H.; Woods, C.G. Painful and painless channelopathies. Lancet Neurol. 2014, 13, 587–599. [Google Scholar] [CrossRef]
- Allen, N.M.; Weckhuysen, S.; Gorman, K.; King, M.D.; Lerche, H. Genetic potassium channel-associated epilepsies: Clinical review of the Kv family. Eur. J. Paediatr. Neurol. 2020, 24, 105–116. [Google Scholar] [CrossRef]
- Fernández-Ballester, G.; Fernández-Carvajal, A.; González-Ros, J.M.; Ferrer-Montiel, A. Ionic Channels as Targets for Drug Design: A Review on Computational Methods. Pharmaceutics 2011, 3, 932–953. [Google Scholar] [CrossRef] [Green Version]
- Amin, A.S.; Tan, H.L.; Wilde, A.A. Cardiac ion channels in health and disease. Hear. Rhythm. 2010, 7, 117–126. [Google Scholar] [CrossRef]
- Wulff, H.; Castle, N.A.; Pardo, L.A. Voltage-gated potassium channels as therapeutic targets. Nat. Rev. Drug Discov. 2009, 8, 982–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbott, G.W.; Sesti, F.; Splawski, I.; Buck, M.; Lehmann, M.H.; Timothy, K.W.; Keating, M.T.; Goldstein, S.A. MiRP1 Forms I Kr Potassium Channels with HERG and is Associated with Cardiac Arrhythmia. Cell 1999, 97, 175–187. [Google Scholar] [CrossRef] [Green Version]
- Isbrandt, D.; Friederich, P.; Solth, A.; Haverkamp, W.; Ebneth, A.; Borggrefe, M.; Funke, H.; Sauter, K.; Breithardt, G.; Pongs, O.; et al. Identification and functional characterization of a novel KCNE2 (MiRP1) mutation that alters HERG channel kinetics. J. Mol. Med. 2002, 80, 524–532. [Google Scholar] [CrossRef]
- Hu, B.; Zeng, W.-P.; Li, X.; Al-Sheikh, U.; Chen, S.-Y.; Ding, J. A conserved arginine/lysine-based motif promotes ER export of KCNE1 and KCNE2 to regulate KCNQ1 channel activity. Channels 2019, 13, 483–497. [Google Scholar] [CrossRef] [Green Version]
- Alexander, S.P.H.; Fabbro, D.; Kelly, E.; Mathie, A.; Peters, j.; Veale, E.L.; Armstrong, J.F.; Faccenda, E.; Harding, S.D.; Pawson, A.J.; et al. The Concise Guide to Pharmacology 2019/20: Enzymes. Br. J. Pharmacol. 2019, 176, S297–S396. [Google Scholar] [PubMed] [Green Version]
- Huang, H.; Pugsley, M.K.; Fermini, B.; Curtis, M.J.; Koerner, J.; Accardi, M.; Authier, S. Cardiac voltage-gated ion channels in safety pharmacology: Review of the landscape leading to the CiPA initiative. J. Pharmacol. Toxicol. Methods 2017, 87, 11–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; MacKinnon, R. Cryo-EM Structure of the Open Human Ether-à-go-go-Related K + Channel hERG. Cell 2017, 169, 422–430.e10. [Google Scholar] [CrossRef] [Green Version]
- Mobasheri, A.; Matta, C.; Uzieliene, I.; Budd, E.; Martín-Vasallo, P.; Bernotiene, E. The chondrocyte channelome: A narrative review. Jt. Bone Spine 2019, 86, 29–35. [Google Scholar] [CrossRef]
- Vicente, J.; Zusterzeel, R.; Johannesen, L.; Mason, J.; Sager, P.; Patel, V.; Matta, M.K.; Philip, S.; Liu, J.; Garnett, C.; et al. Mechanistic Model-Informed Proarrhythmic Risk Assessment of Drugs: Review of the “CiPA” Initiative and Design of a Prospective Clinical Validation Study. Clin. Pharmacol. Ther. 2017, 103, 54–66. [Google Scholar] [CrossRef]
- Orvos, P.; Kohajda, Z.; Szlovák, J.; Gazdag, P.; Árpádffy-Lovas, T.; Tóth, D.; Geramipour, A.; Tálosi, L.; Jost, N.; Varró, A.; et al. Evaluation of Possible Proarrhythmic Potency: Comparison of the Effect of Dofetilide, Cisapride, Sotalol, Terfenadine, and Verapamil on hERG and Native IKr Currents and on Cardiac Action Potential. Toxicol. Sci. 2018, 168, 365–380. [Google Scholar] [CrossRef]
- Ajayi, F.; Sun, H.; Perry, J. Adverse drug reactions: A review of relevant factors. J. Clin. Pharm. 2000, 40, 1093–1101. [Google Scholar]
- Li, M.; Ramos, L.G. Drug-Induced QT Prolongation and Torsades de Pointes. Pharm. Therap. 2017, 42, 473–477. [Google Scholar]
- Ringer, S. A further contribution regarding the influence of the different constituents of the blood on the contraction of the heart. J. Physiol. 1883, 4, 29–42. [Google Scholar] [CrossRef]
- Heilbrunn, L.V.; Wiercinski, F.J. The action of various cations on muscle protoplasm. J. Cell. Comp. Physiol. 1947, 29, 15–32. [Google Scholar] [CrossRef] [PubMed]
- Cosconati, S.; Marinelli, L.; Lavecchia, A.; Novellino, E. Characterizing the 1,4-Dihydropyridines Binding Interactions in the L-Type Ca2+ Channel: Model Construction and Docking Calculations. J. Med. Chem. 2007, 50, 1504–1513. [Google Scholar] [CrossRef] [PubMed]
- Zwanzger, P.; Eßer, D.; Nothdurfter, C.; Baghai, T.C.; Möller, H.-J.; Padberg, F.; Rupprecht, R. Effects of the GABA-reuptake Inhibitor Tiagabine on Panic and Anxiety in Patients with Panic Disorder. Pharmacopsychiatry 2009, 42, 266–269. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Rubaiy, H.N.; Chen, G.; Hallett, T.; Zaibi, N.; Zeng, B.; Saurabh, R.; Xu, S. T-type Ca2+ channel blocker mibefradil inhibits ORAI store-operated channels. J. Mol. Cell. Cardiol. 2020, 140, 26–27. [Google Scholar] [CrossRef]
- García, A.G.; García-De-Diego, A.M.; Gandia, L.; Borges, R.; García-Sancho, J. Calcium Signaling and Exocytosis in Adrenal Chromaffin Cells. Physiol. Rev. 2006, 86, 1093–1131. [Google Scholar] [CrossRef] [Green Version]
- Zamponi, G.W.; Striessnig, J.; Koschak, A.; Dolphin, A.C. The Physiology, Pathology, and Pharmacology of Voltage-Gated Calcium Channels and Their Future Therapeutic Potential. Pharmacol. Rev. 2015, 67, 821–870. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, F.; Flockerzi, V.; Kahl, S.; Wegener, J.W. L-Type CaV1.2 Calcium Channels: From In Vitro Findings to In Vivo Function. Physiol. Rev. 2014, 94, 303–326. [Google Scholar] [CrossRef] [Green Version]
- Balasubramanian, B.; Imredy, J.P.; Kim, D.; Penniman, J.; Lagrutta, A.; Salata, J.J. Optimization of Cav1.2 screening with an automated planar patch clamp platform. J. Pharmacol. Toxicol. Methods 2009, 59, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Striessnig, J.; Pinggera, A.; Kaur, G.; Bock, G.; Tuluc, P. L-type Ca2+ channels in heart and brain. Wiley Interdiscip. Rev. Membr. Transp. Signal. 2014, 3, 15–38. [Google Scholar] [CrossRef] [PubMed]
- Théophile, G. Discovery and Development of Calcium Channel Blockers. Front. Pharmacol. 2017, 8, 1–25. [Google Scholar]
- Reed, J.K.; Raftery, M.A. Properties of the tetrodotoxin binding component in plasma membranes isolated from Electrophorus electricus. Biochemistry 1976, 15, 944–953. [Google Scholar] [CrossRef]
- Chahine, M. Voltage-gated Sodium Channels: Structure, Function and Channelopathies; Springer: Cham, Switzerland, 2018; Volume 246. [Google Scholar]
- Wiffen, P.J.; Moore, R.A.; Aldington, D.; Cole, P.; Rice, A.S.; Lunn, M.P.; Hamunen, K.; Haanpää, M.; Kalso, E.; Derry, S. Antiepileptic drugs for neuropathic pain and fibromyalgia—An overview of Cochrane reviews. Cochrane Database Syst. Rev. 2013, 2013, 11. [Google Scholar] [CrossRef] [PubMed]
- de Marco, K.R.; Clancy, C.E. Cardiac Na Channels: Structure to Function. Curr. Top. Membr. 2016, 78, 287–311. [Google Scholar]
- Mantegazza, M.; Curia, G.; Biagini, G.; Ragsdale, D.S.; Avoli, M. Voltage-gated sodium channels as therapeutic targets in epilepsy and other neurological disorders. Lancet Neurol. 2010, 9, 413–424. [Google Scholar] [CrossRef]
- Bagnéris, C.; DeCaen, P.G.; Naylor, C.E.; Pryde, D.C.; Nobeli, I.; Clapham, D.E.; Wallace, B.A. Prokaryotic NavMs channel as a structural and functional model for eukaryotic sodium channel antagonism. Proc. Natl. Acad. Sci. USA 2014, 111, 8428–8433. [Google Scholar] [CrossRef] [Green Version]
- Yan, Z.; Zhou, Q.; Wang, L.; Wu, J.; Zhao, Y.; Huang, G.; Peng, W.; Shen, H.; Lei, J.; Yan, N. Structure of the Na v 1.4-β1 Complex from Electric Eel. Cell 2017, 170, 470–482. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Jin, X.; Huang, G.; Wu, K.; Lei, J.; Pan, X.; Yan, N. Structural basis for pore blockade of the human cardiac sodium channel Nav1.5 by tetrodotoxin and quinidine. bioRxiv 2019, 2019, 1–28. [Google Scholar]
- Huang, W.; Liu, M.; Yan, S.F.; Yan, N. Structure-based assessment of disease-related mutations in human voltage-gated sodium channels. Prot. Cell 2017, 8, 401–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loussouarn, G.; Sternberg, D.; Nicole, S.; Marionneau, C.; le Bouffant, F.; Toumaniantz, G.; Barc, J.; Malak, O.A.; Fressart, V.; Péreon, Y.; et al. Physiological and Pathophysiological Insights of Nav1.4 and Nav1.5 Comparison. Front. Pharmacol. 2016, 6, 1–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dib-Hajj, S.D.; Yang, Y.; Black, J.A.; Waxman, S.G. The NaV1.7 sodium channel: From molecule to man. Nat. Rev. Neurosci. 2012, 14, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Theile, J.W.; Cummins, T.R. Recent Developments Regarding Voltage-Gated Sodium Channel Blockers for the Treatment of Inherited and Acquired Neuropathic Pain Syndromes. Front. Pharmacol. 2011, 2, 54. [Google Scholar] [CrossRef] [Green Version]
- Du, Y.; Garden, D.P.; Wang, L.; Zhorov, B.S.; Dong, K. Identification of New Batrachotoxin-sensing Residues in Segment IIIS6 of the Sodium Channel. J. Biol. Chem. 2011, 286, 13151–13160. [Google Scholar] [CrossRef] [Green Version]
- Żelazny, P.; Uździcki, A.; Awgul, S.; Pawlik, A. Kanały jonowe jako punkty uchwytu leków stosowanych w terapii padaczki. Farm. Współczesna 2018, 11, 201–206. [Google Scholar]
- Lei, M.; Wu, L.; Terrar, D.A.; Huang, C.L.-H. Modernized Classification of Cardiac Antiarrhythmic Drugs. Circulation 2018, 138, 1879–1896. [Google Scholar] [CrossRef]
- Yellen, G. The moving parts of voltage-gated ion channels. Q. Rev. Biophys. 1998, 31, 239–295. [Google Scholar] [CrossRef]
- Chang, C.; Ray, A.; Swaan, P. In silico strategies for modeling membrane transporter function. Drug Discov. Today 2005, 10, 663–671. [Google Scholar] [CrossRef]
- Radchenko, E.; Rulev, Y.A.; Safanyaev, A.Y.; Palyulin, V.; Zefirov, N. Computer-aided estimation of the hERG-mediated cardiotoxicity risk of potential drug components. Dokl. Biochem. Biophys. 2017, 473, 128–131. [Google Scholar] [CrossRef]
- Recanatini, M.; Poluzzi, E.; Masetti, M.; Cavalli, A.; De Ponti, F. QT Prolongation Through hERG K+ Channel Blockade: Current Knowledge and Strategies for the Early Prediction During Drug Development. Med. Res. Rev. 2005, 36, 133–166. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.-M.; Temple, R.; Throckmorton, D.C.; Lesko, L.J. Drug Interaction Studies: Study Design, Data Analysis, and Implications for Dosing and Labeling. Clin. Pharmacol. Ther. 2007, 81, 298–304. [Google Scholar] [CrossRef]
- Zaręba, P.; Gryzło, B.; Malawska, K.; Sałat, K.; Höfner, G.C.; Nowaczyk, A.; Fijałowski, Ł.; Rapacz, A.; Podkowa, A.; Furgała, A.; et al. Novel GABA uptake inhibitors with enhanced inhibitory activity toward mGAT3/4 and their effect on pain threshold in mice. EJMCh 2020, 188, 111920. [Google Scholar] [CrossRef] [PubMed]
- Böck, M.C.; Höfner, G.; Wanner, K.T. N-Substituted Nipecotic Acids as (S)-SNAP-5114 Analogues with Modified Lipophilic Domains. ChemMedChem 2020, 15, 756–771. [Google Scholar] [CrossRef]
- Nowaczyk, A.; Fijałkowski, Ł.; Zaręba, P.; Sałat, K. Selective neuronal and astrocytic inhibition of human GABA transporter isoform 1 (hGAT1) inhibitors in the mechanism of epilepsy and pain-molecular docking and pharmacodynamics studies, part I. J. Mol. Grap. Model. 2018, 85, 171–181. [Google Scholar] [CrossRef]
- Bahnikova, M.; Matejoviâ, P.; Pasek, M.; Imurdová, M.; Imurda, J. Ajmaline-induced block of sodium current in rat ventricular myocytes. Scr. Med. 2002, 75, 169–177. [Google Scholar]
- Bébarová, M.; Matejovic, P.; Pásek, M.; Simurdová, M.; Simurda, J. Effect of ajmaline on action potential and ionic currents in rat ventricular myocytes. Gen. Physiol. Biophys. 2005, 24, 311–325. [Google Scholar] [PubMed]
- Kiesecker, C.; Zitron, E.; Bloehs, R.; Scholz, E.; Thomas, D.; Kreye, V.A.W.; Katus, H.A.; Schoels, W.; Karle, C.A.; Kiehn, J. Class Ia anti-arrhythmic drug ajmaline blocks HERG potassium channels: Mode of action. Naunyn Schmiedeberg’s Arch. Pharmacol. 2004, 370, 423–435. [Google Scholar] [CrossRef]
- Grima, M.; Schwartz, J.; Spach, M.; Velly, J. Anti-anginal arylalkylamines and sodium channels: [3H]-batrachotoxinin-A 20-alpha-benzoate and [3H]-tetracaine binding. Br. J. Pharmacol. 1986, 89, 641. [Google Scholar] [CrossRef]
- Lubic, S.P.; Nguyen, K.P.; Dave, B.; Giacomini, J.C. Antiarrhythmic agent amiodarone possesses calcium channel blocker properties. J. Cardiovasc. Pharmacol. 1994, 24, 707–714. [Google Scholar] [CrossRef]
- Leffler, A.; Reiprich, A.; Mohapatra, D.P.; Nau, C. Use-dependent block by lidocaine but not amitriptyline is more pronounced in tetrodotoxin (TTX)-Resistant Nav1. 8 than in TTX-sensitive Na+ channels. J. Pharmacol. Exp. Ther. 2007, 320, 354–364. [Google Scholar] [CrossRef] [Green Version]
- Zahradník, I.; Minarovič, I.; Zahradníková, A. Inhibition of the cardiac L-type calcium channel current by antidepressant drugs. J. Pharmacol. Exp. Ther. 2008, 324, 977–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, S.H.; Youm, J.B.; Lee, C.O.; Earm, Y.E.; Ho, W.K. Blockade of the HERG human cardiac K+ channel by the antidepressant drug amitriptyline. Br. J. Pharmacol. 2000, 129, 1474–1480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fossa, A.A.; de Pasquale, M.J.; Raunig, D.L.; Avery, M.J.; Leishman, D.J. The relationship of clinical QT prolongation to outcome in the conscious dog using a beat-to-beat QT-RR interval assessment. J. Pharmacol. Exp. Ther. 2002, 302, 828–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNeal, E.T.; Lewandowski, G.A.; Daly, J.W.; Creveling, C. [3H] Batrachotoxinin A 20. alpha.-benzoate binding to voltage-sensitive sodium channels: A rapid and quanitative assay for local anesthetic activity in a variety of drugs. J. Med. Chem. 1985, 28, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Testa, R.; Abbiati, G.; Ceserani, R.; Restelli, G.; Vanasia, A.; Barone, D.; Gobbi, M.; Mennini, T. Profile of in vitro binding affinities of neuroleptics at different rat brain receptors: Cluster analysis comparison with pharmacological and clinical profiles. Pharm. Res. 1989, 6, 571–577. [Google Scholar] [CrossRef]
- Sato, T.; Wu, B.; Kiyosue, T.; Arita, M. Effects of cibenzoline, a new class Ia antiarrhythmic drug, on various membrane ionic currents and action potentials of guinea-pig ventricular cells. Naunyn Schmiedeberg’s Arch. Pharmacol 1994, 350, 167–173. [Google Scholar] [CrossRef]
- Matsuoka, S.; Nawada, T.; Hisatome, I.; Miyamoto, J.; Hasegawa, J.; Kotake, H.; Mashiba, H. Comparison of Ca2+ channel inhibitory effects of cibenzoline with verapamil on guinea-pig heart. Gen. Pharmacol. Vasc. Syst. 1991, 22, 87–91. [Google Scholar] [CrossRef]
- Männikkö, R.; Overend, G.; Perrey, C.; Gavaghan, C.; Valentin, J.P.; Morten, J.; Armstrong, M.; Pollard, C. Pharmacological and electrophysiological characterization of nine, single nucleotide polymorphisms of the hERG-encoded potassium channel. Br. J. Pharmacol. 2010, 159, 102–114. [Google Scholar] [CrossRef] [Green Version]
- Mohammad, S.; Zhou, Z.; Gong, Q.; January, C.T. Blockage of the HERG human cardiac K+ channel by the gastrointestinal prokinetic agent cisapride. Am. J. Physiol. Content 1997, 273, H2534–H2538. [Google Scholar] [CrossRef]
- Ekins, S.; Crumb, W.J.; Sarazan, R.D.; Wikel, J.H.; Wrighton, S.A. “Three dimensional quantitative structure activity relationship for the inhibition of the hERG (human ether-a-gogo related gene) potassium channel,”. J. Pharmacol. Exp. Thera. 2002, 301, 427–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pauwels, P.J.; Laduron, P.M. TPP+ accumulation in rat brain synaptosomes as a probe for Na+ channels. Eur. J. Pharmacol. 1986, 132, 289–293. [Google Scholar] [CrossRef]
- Campiani, G.; Fiorini, I.; de Filippis, M.P.; Ciani, S.M.; Garofalo, A.; Nacci, V.; Giorgi, G.; Sega, A.; Botta, M.; Chiarini, A. Cardiovascular characterization of pyrrolo [2, 1-d][1, 5] benzothiazepine derivatives binding selectively to the peripheral-type benzodiazepine receptor (PBR): From dual PBR affinity and calcium antagonist activity to novel and selective calcium entry blockers. J. Med. Chem. 1996, 39, 2922–2938. [Google Scholar]
- Zhang, S.; Zhou, Z.; Gong, Q.; Makielski, J.C.; January, C.T. Mechanism of block and identification of the verapamil binding domain to HERG potassium channels. Circ. Res. 1999, 84, 989–998. [Google Scholar] [CrossRef] [Green Version]
- Johnson, R.E.; Silver, P.J.; Becker, R.; Birsner, N.C.; Bohnet, E.A.; Briggs, G.M.; Busacca, C.A.; Canniff, P.; Carabateas, P.M.; D’Ambra, T. 4, 5-Dihydro-3-(methanesulfonamidophenyl)-1-phenyl-1H-2, 4-benzodiazepines: A novel class III antiarrhythmic agents. J. Med. Chem. 1995, 38, 2551–2556. [Google Scholar] [CrossRef] [PubMed]
- Chadwick, C.C.; Ezrin, A.M.; O’Connor, B.; Volberg, W.A.; Smith, D.I.; Wedge, K.J.; Hill, R.J.; Briggs, G.M.; Pagani, E.D.; Silver, P.J. Identification of a specific radioligand for the cardiac rapidly activating delayed rectifier K+ channel. Circ. Res. 1993, 72, 707–714. [Google Scholar] [CrossRef] [Green Version]
- de Bruin, M.; Pettersson, M.; Meyboom, R.; Hoes, A.; Leufkens, H. Anti-HERG activity and the risk of drug-induced arrhythmias and sudden death. Eur. Heart J. 2005, 26, 590–597. [Google Scholar] [CrossRef]
- Redfern, W.; Carlsson, L.; Davis, A.; Lynch, W.; MacKenzie, I.; Palethorpe, S.; Siegl, P.; Strang, I.; Sullivan, A.; Wallis, R. Relationships between preclinical cardiac electrophysiology, clinical QT interval prolongation and torsade de pointes for a broad range of drugs: Evidence for a provisional safety margin in drug development. Cardiovasc. Res. 2003, 58, 32–45. [Google Scholar] [CrossRef]
- Schoemaker, H.; Claustre, Y.; Fage, D.; Rouquier, L.; Chergui, K.; Curet, O.; Oblin, A.; Gonon, F.; Carter, C.; Benavides, J. Neurochemical characteristics of amisulpride, an atypical dopamine D2/D3 receptor antagonist with both presynaptic and limbic selectivity. J. Pharmacol. Exp. Therap. 1997, 280, 83–97. [Google Scholar]
- Kirsch, G.E.; Trepakova, E.S.; Brimecombe, J.C.; Sidach, S.S.; Erickson, H.D.; Kochan, M.C.; Shyjka, L.M.; Lacerda, A.E.; Brown, A.M. Variability in the measurement of hERG potassium channel inhibition: Effects of temperature and stimulus pattern. J. Pharmacol. Toxicol. Methods 2004, 50, 93–101. [Google Scholar] [CrossRef]
- de Luca, A.; Talon, S.; De Bellis, M.; Desaphy, J.-F.; Franchini, C.; Lentini, G.; Catalano, A.; Corbo, F.; Tortorella, V.; Conte-Camerino, D. Inhibition of skeletal muscle sodium currents by mexiletine analogues: Specific hydrophobic interactions rather than lipophilia per se account for drug therapeutic profile. Naunyn Schmiedeberg’s Arch. Pharmacol. 2003, 367, 318–327. [Google Scholar] [CrossRef]
- Hu, X.; Qian, J. DDPH inhibited L-type calcium current and sodium current in single ventricular myocyte of guinea pig. Acta Pharmacol. Sin. 2001, 22, 415–419. [Google Scholar] [PubMed]
- Roche, O.; Trube, G.; Zuegge, J.; Pflimlin, P.; Alanine, A.; Schneider, G. A virtual screening method for prediction of the HERG potassium channel liability of compound libraries. ChemBioChem 2002, 3, 455–459. [Google Scholar] [CrossRef]
- Strege, P.R.; Bernard, C.E.; Ou, Y.; Gibbons, S.J.; Farrugia, G. Effect of mibefradil on sodium and calcium currents. Am. J. Physiol. Liver Physiol. 2005, 289, G249–G253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, I.; Wappl, E.; Herzog, A.; Mitterdorfer, J.; Glossmann, H.; Langer, T.; Striessnig, J. Conserved Ca2+-antagonist-binding properties and putative folding structure of a recombinant high-affinity dihydropyridine-binding domain. Biochem. J. 2000, 347, 829–836. [Google Scholar] [CrossRef]
- di Stilo, A.; Visentin, S.; Cena, C.; Gasco, A.M.; Ermondi, G.; Gasco, A. New 1, 4-dihydropyridines conjugated to furoxanyl moieties, endowed with both nitric oxide-like and calcium channel antagonist vasodilator activities. J. Med. Chem. 1998, 41, 5393–5401. [Google Scholar] [CrossRef]
- Zhabyeyev, P.; Missan, S.; Jones, S.E.; McDonald, T.F. Low-affinity block of cardiac K+ currents by nifedipine. Eur. J. Pharmacol. 2000, 401, 137–143. [Google Scholar] [CrossRef]
- Ehlert, F.J.; Roeske, W.R.; Itoga, E.; Yamamura, H.I. The binding of [3H] nitrendipine to receptors for calcium channel antagonists in the heart, cerebral cortex, and ileum of rats. Life Sci. 1982, 30, 2191–2202. [Google Scholar] [CrossRef]
- Pauwels, P.J.; Leysen, J.E.; Laduron, P.M. [3H] Batrachotoxinin A 20-α-benzoate binding to sodium channels in rat brain: Characterization and pharmacological significance. Eur. J. Pharmacol. 1986, 124, 291–298. [Google Scholar] [CrossRef]
- Reynolds, I.; Snowman, A.M.; Snyder, S.H. (-)-[3H] desmethoxyverapamil labels multiple calcium channel modulator receptors in brain and skeletal muscle membranes: Differentiation by temperature and dihydropyridines. J. Pharmacol. Exp. Ther. 1986, 237, 731–738. [Google Scholar]
- Paul, A.A.; Witchel, H.J.; Hancox, J.C. Inhibition of the current of heterologously expressed HERG potassium channels by flecainide and comparison with quinidine, propafenone and lignocaine. Br. J. Pharmacol. 2002, 136, 717–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanson, L.A.; Bass, A.S.; Gintant, G.; Mittelstadt, S.; Rampe, D.; Thomas, K. ILSI-HESI cardiovascular safety subcommittee initiative: Evaluation of three non-clinical models of QT prolongation. J. Pharmacol. Toxicol. Methods 2006, 54, 116–129. [Google Scholar] [CrossRef] [PubMed]
- Kongsamut, S.; Kang, J.; Chen, X.-L.; Roehr, J.; Rampe, D. A comparison of the receptor binding and HERG channel affinities for a series of antipsychotic drugs. Eur. J. Pharmacol. 2002, 450, 37–41. [Google Scholar] [CrossRef]
- Po, S.; Wang, D.; Yang, I.C.-H.; Johnson, J.; Nie, L.; Bennett, P. Modulation of HERG potassium channels by extracellular magnesium and quinidine. J. Cardiovasc. Pharmacol. 1999, 33, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, M.B.; Volders, P.G.; Stengl, M.; Späatjens, R.L.; Beekman, J.D.; Bischoff, U.; Kall, M.A.; Frederiksen, K.; Matz, J.; Vos, M.A. Electrophysiological safety of sertindole in dogs with normal and remodeled hearts. J. Pharmacol. Exp. Ther. 2003, 307, 776–784. [Google Scholar] [CrossRef]
- Kramer, J.; Obejero-Paz, C.A.; Myatt, G.; Kuryshev, Y.A.; Bruening-Wright, A.; Verducci, J.S.; Brown, A.M. MICE models: Superior to the HERG model in predicting Torsade de Pointes. Sci. Rep. 2013, 3, 2100. [Google Scholar] [CrossRef] [Green Version]
- Faivre, J.F.; Gout, B.; Bril, A. Tedisamil. Card. Drug Rev. 1995, 13, 33–50. [Google Scholar] [CrossRef]
- Jost, N.; Virág, L.; Hála, O.; Varro, A.; Thormahlen, D.; Papp, J.G. Effect of the antifibrillatory compound tedisamil (KC-8857) on transmembrane currents in mammalian ventricular myocytes. Curr. Med. Chem. 2004, 11, 3219–3228. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Name | hKV11.1/KCNH2 | hNaV1.5/SCN5A | hCaV1.2/CACNC1C |
|---|---|---|---|
| UniProtKB | Q12809 | Q14524 | Q13936 |
| pore-forming | 612–632: VTALYFTFSSLTSVGFGNVSP | 884–904: FFHAFLIIFRILCGEWIETMW | 694–715: QSLLTVFQILTGEDWNSVMYDG |
| ion selectivity sequence motif | GYG | DEKA | EEEE |
| cariac disease | Long Qt Syndrome; Short Qt Syndrome | Atrial Fibrillation, Familial, Brugada Syndrome; Cardiomyopathy, Dilated; Long Qt Syndrome; Progressive Familial Heart Block (Type Ia); Sick Sinus Syndrome, Autosomal Recessive; Sudden Infant Death Syndrome; Ventricular Fibrillation During Myocardial Infarction, Susceptibility to acquired arrhythmia | Long QT, Brugda Syndrome, Timothy Syndrome |
| sequence identity 1 | 88.66% | 66.70% | 70.3% |
| Drug | hNaV1.5 | hCaV1.2 | hKV11.1 |
|---|---|---|---|
| ajmaline | 5.09 [79] | 4.15 [80] | 5.98 [81] |
| amiodarone | 5.32 [82] | 5.57 [83] | 7.52 [21] |
| amitryptyline | 4.70 [84] | 4.94 [85] | 5.48 [86] |
| bepridil | 5.43 [21] | 6.68 [21] | 7.48 [87] |
| chlorpromazine | 5.37 [88] | n/a [88] | 5.83 [89] |
| cibenzoline | 5.11 [90] | 4.52 [91] | 4.65 [92] |
| cisapride | 4.83 [21] | n/a [21] | 8.19 [93] |
| desipramine | 5.82 [21] | 5.77 [21] | 5.86 [94] |
| diltiazem | 5.05 [95] | 6.35 [96] | 4.76 [97] |
| diphenhydramine | 4.39 [21] | 3.64 [21] | 5.28 [92] |
| dofetilide | 3.52 [98] | 4.22 [99] | 8.30 [100] |
| fluvoxamine | 4.40 [21] | 5.31 [21] | 5.51 [101] |
| haloperidol | 5.15 [102] | 5.77 [102] | 7.57 [103] |
| imipramine | 5.44 [88] | 5.08 [89] | 5.47 [101] |
| mexiletine | 4.37 [104] | 4.00 [105] | 4.30 [106] |
| mibefradil | 6.01 [107] | 6.81 [108] | 5.74 [92] |
| nifedipine | 4.43 [88] | 7.22 [109] | 3.56 [110] |
| nitredypine | 4.44 [88] | 9.46 [111] | 5.00 [101] |
| phenytoin | 4.31 [21] | 3.99 [21] | 4.00 [101] |
| pimozide | 7.27 [112] | 6.79 [113] | 7.70 [101] |
| prenylamine | 5.60 [21] | 5.91 [21] | 7.19 [21] |
| propafenone | 5.92 [21] | 5.74 [21] | 6.36 [114] |
| propranolol | 5.68 [21] | 4.74 [21] | 5.55 [115] |
| quetiapine | 4.77 [21] | 4.98 [21] | 5.24 [116] |
| quinidine | 4.78 [21] | 4.81 [21] | 6.52 [117] |
| risperidone | 3.99 [21] | 4.14 [21] | 6.82 [101] |
| sertindole | 5.64 [118] | 5.05 [118] | 7.85 [101] |
| sotalol | n/a [119] | n/a [119] | 7.07 [119] |
| tedisamil | 4.70 [120] | n/a [121] | 5.60 [101] |
| terfenadine | 6.01 [21] | 6.43 [21] | 8.05 [115] |
| thioridazine | 5.74 [21] | 5.89 [21] | 7.48 [101] |
| verapamile | 4.38 [21] | 7.00 [21] | 6.84 [97] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kowalska, M.; Nowaczyk, J.; Nowaczyk, A. KV11.1, NaV1.5, and CaV1.2 Transporter Proteins as Antitarget for Drug Cardiotoxicity. Int. J. Mol. Sci. 2020, 21, 8099. https://doi.org/10.3390/ijms21218099
Kowalska M, Nowaczyk J, Nowaczyk A. KV11.1, NaV1.5, and CaV1.2 Transporter Proteins as Antitarget for Drug Cardiotoxicity. International Journal of Molecular Sciences. 2020; 21(21):8099. https://doi.org/10.3390/ijms21218099
Chicago/Turabian StyleKowalska, Magdalena, Jacek Nowaczyk, and Alicja Nowaczyk. 2020. "KV11.1, NaV1.5, and CaV1.2 Transporter Proteins as Antitarget for Drug Cardiotoxicity" International Journal of Molecular Sciences 21, no. 21: 8099. https://doi.org/10.3390/ijms21218099