Agonist Binding and G Protein Coupling in Histamine H2 Receptor: A Molecular Dynamics Study


Abstract
:1. Introduction
2. Results and Discussion
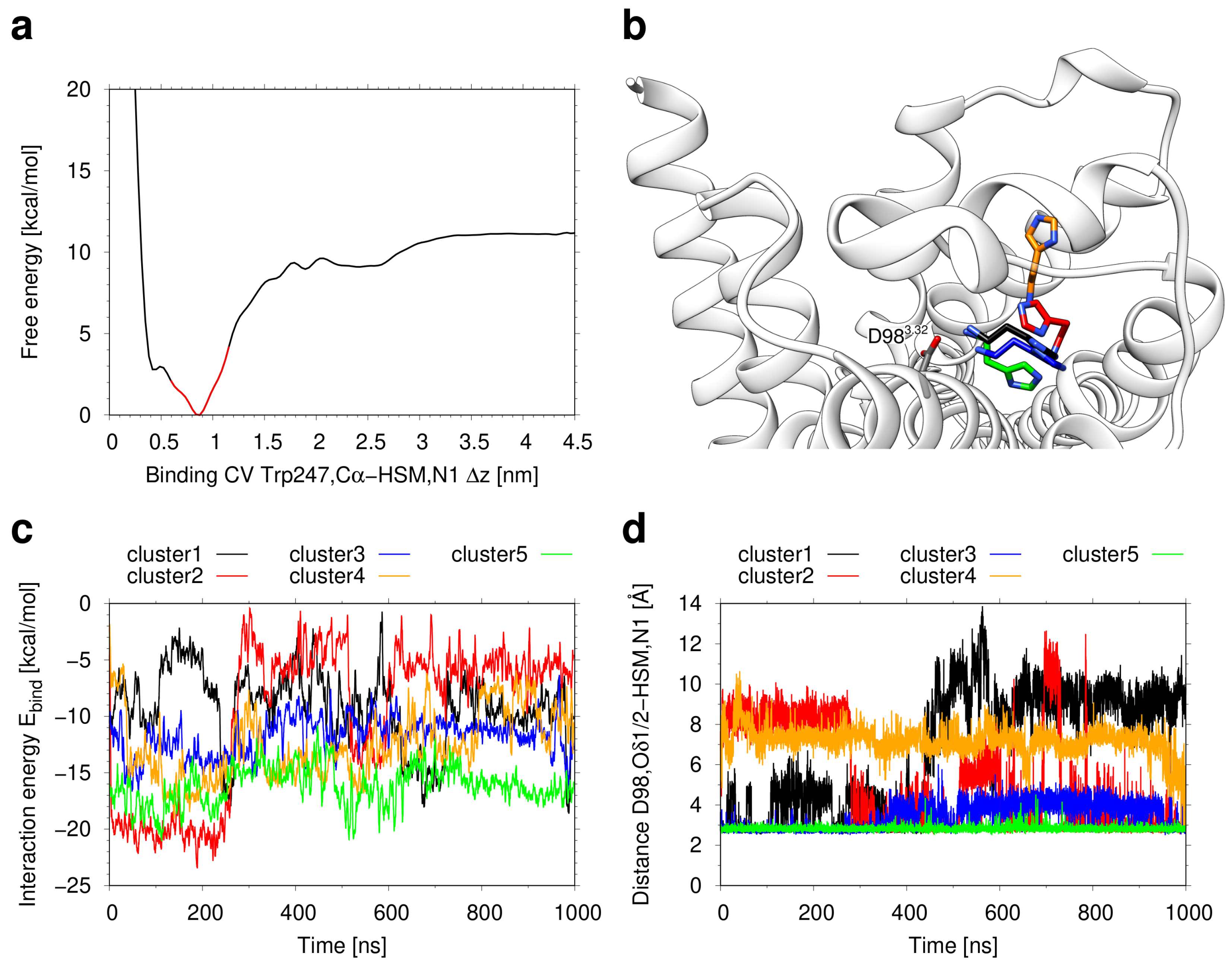
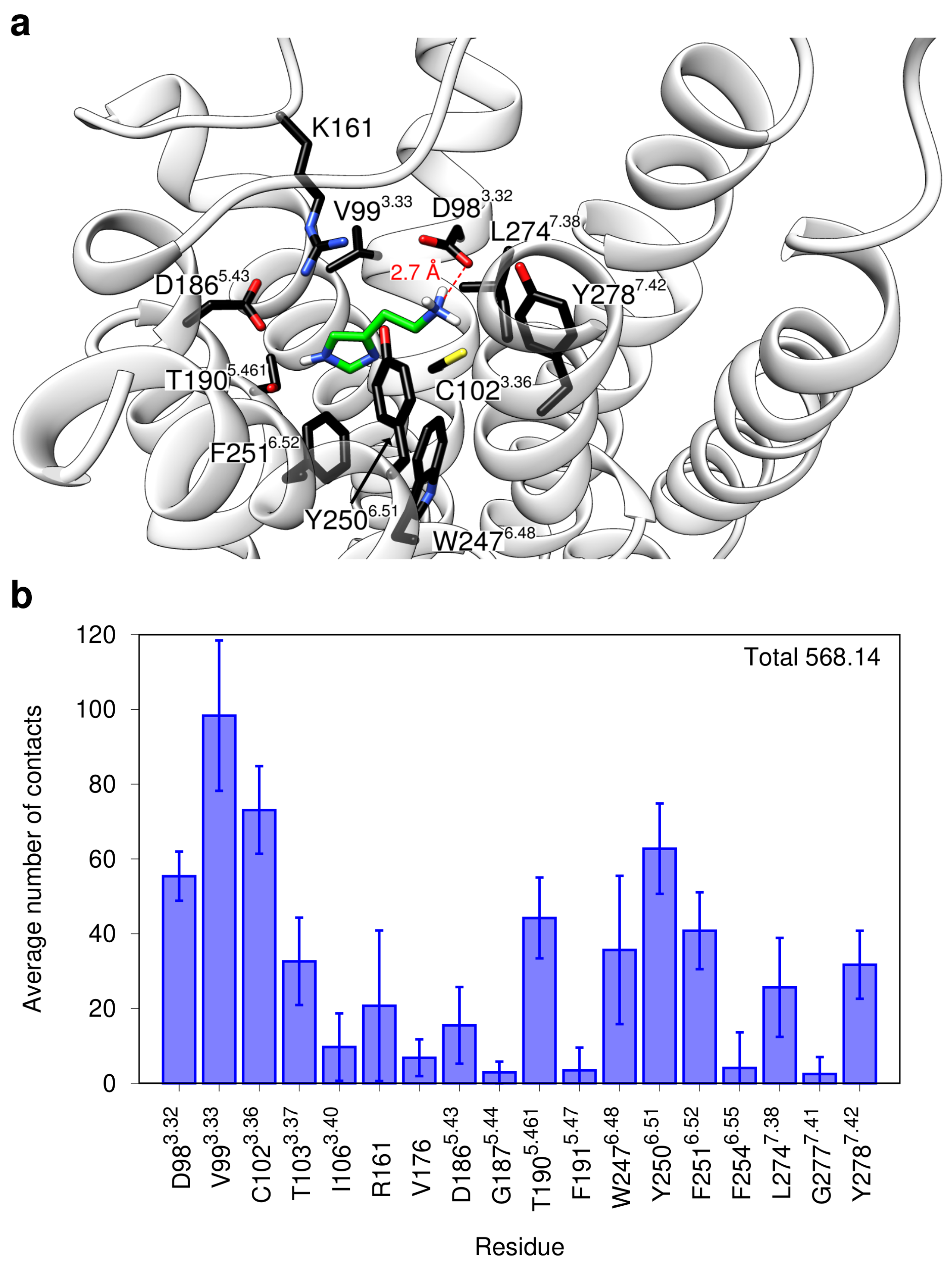
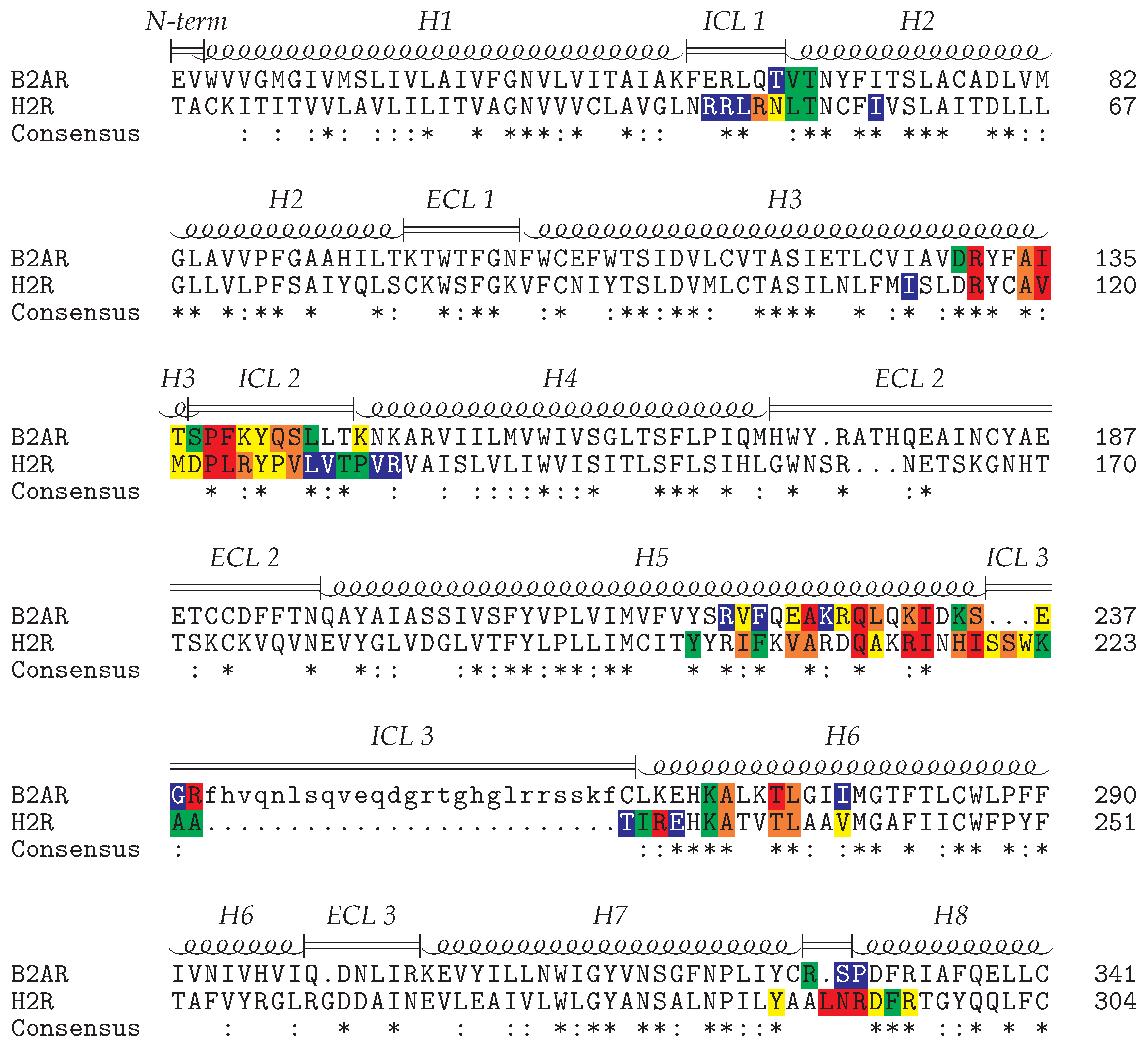
2.1. Modeling of the H2R in the Active State
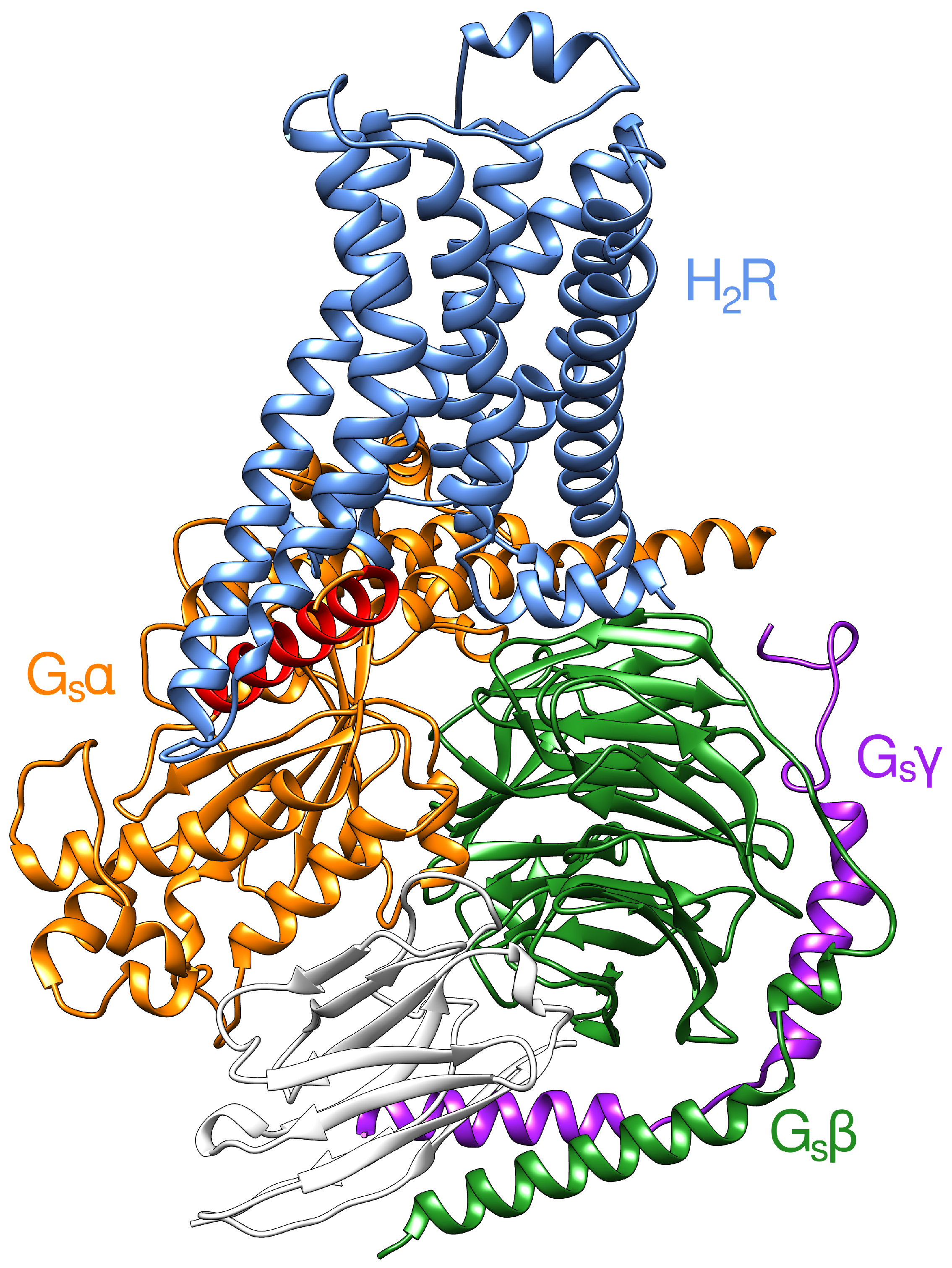
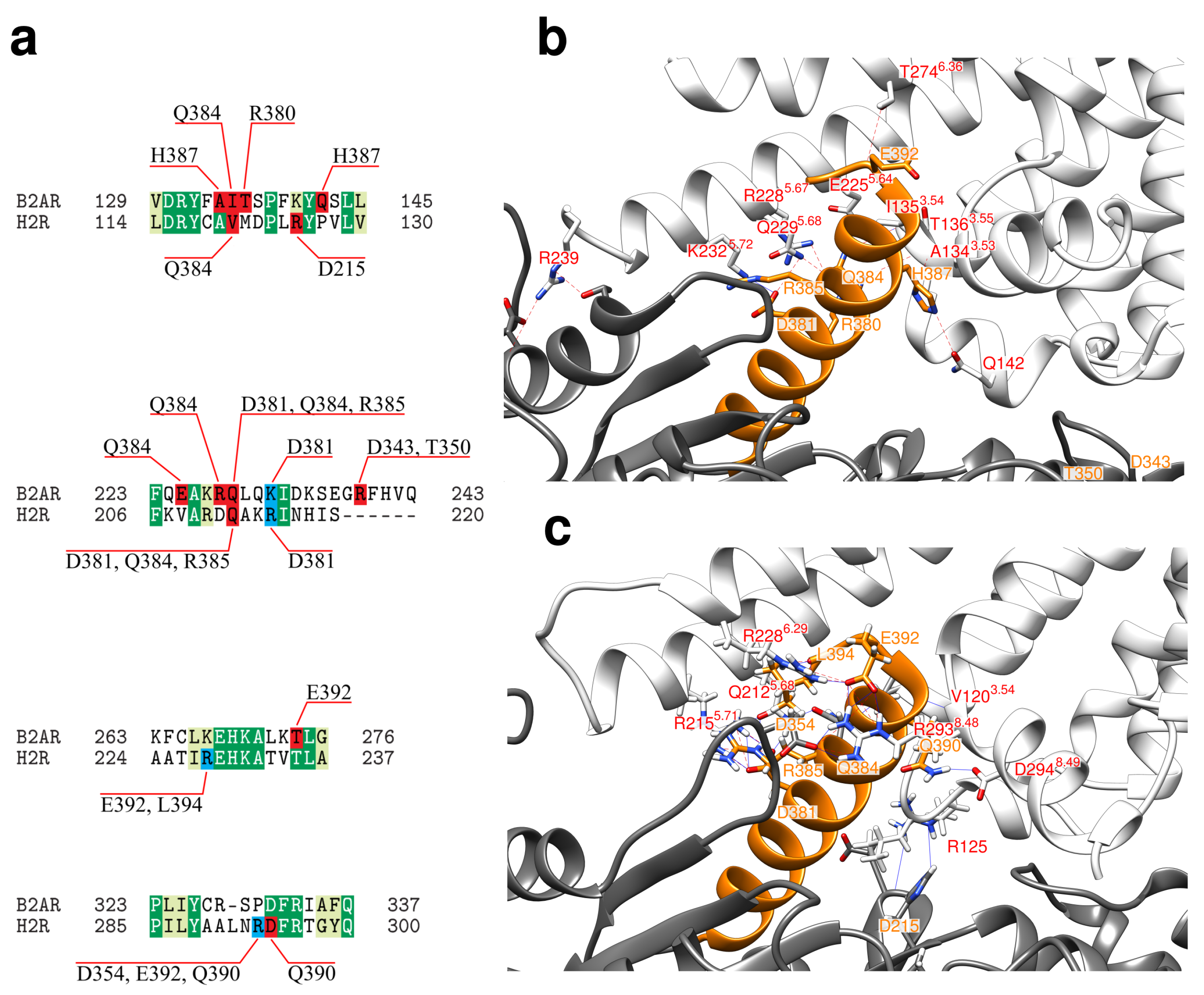
2.2. Interface between the Receptor and the Gs Protein
- (i)
- (ii)
- As recently demonstrated for 2AR, transient interactions observed between the GPCR and GDP-bound Gs protein may represent an intermediate on the way to the formation of the final complex and may contribute to coupling specificity [53].
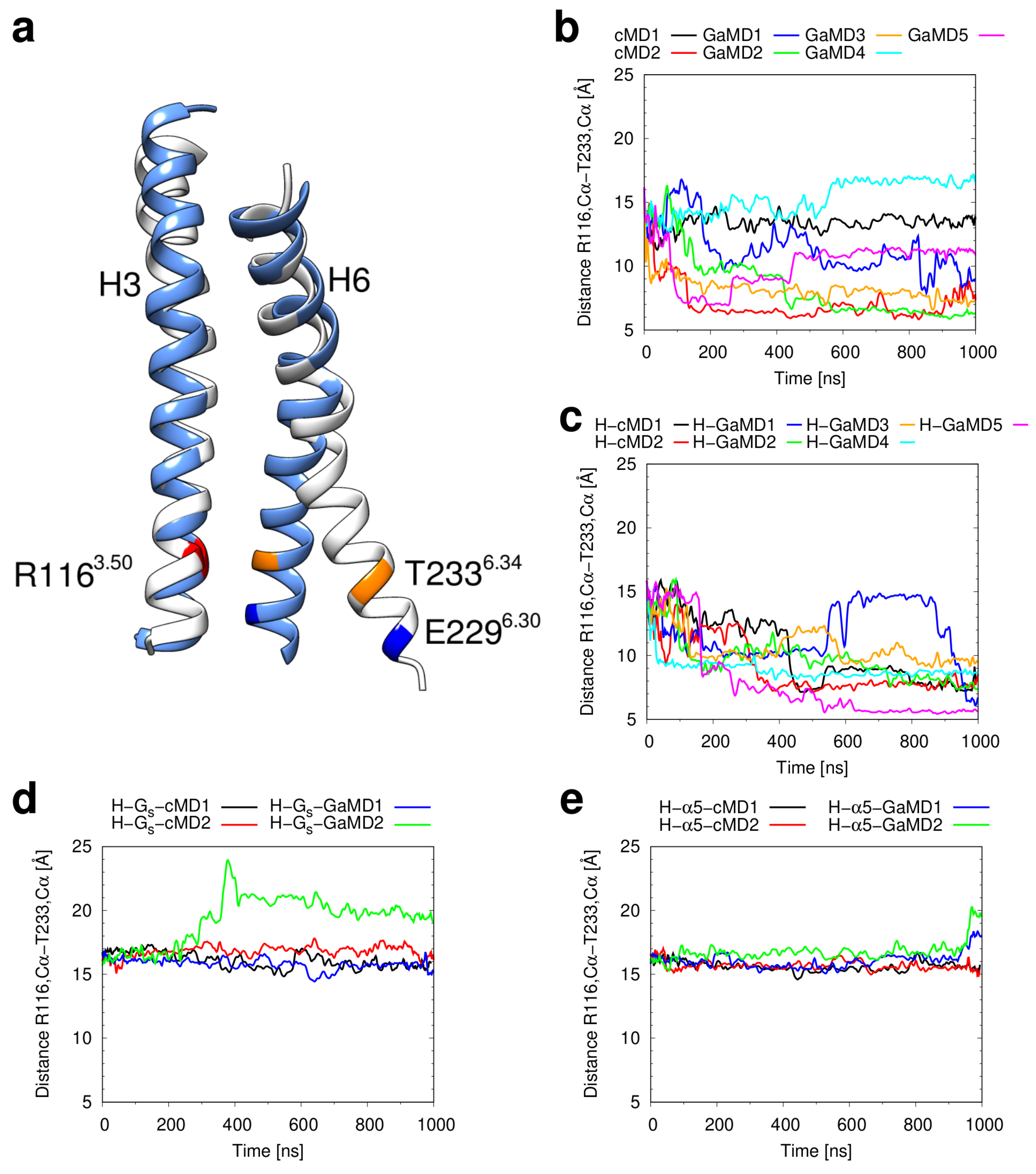
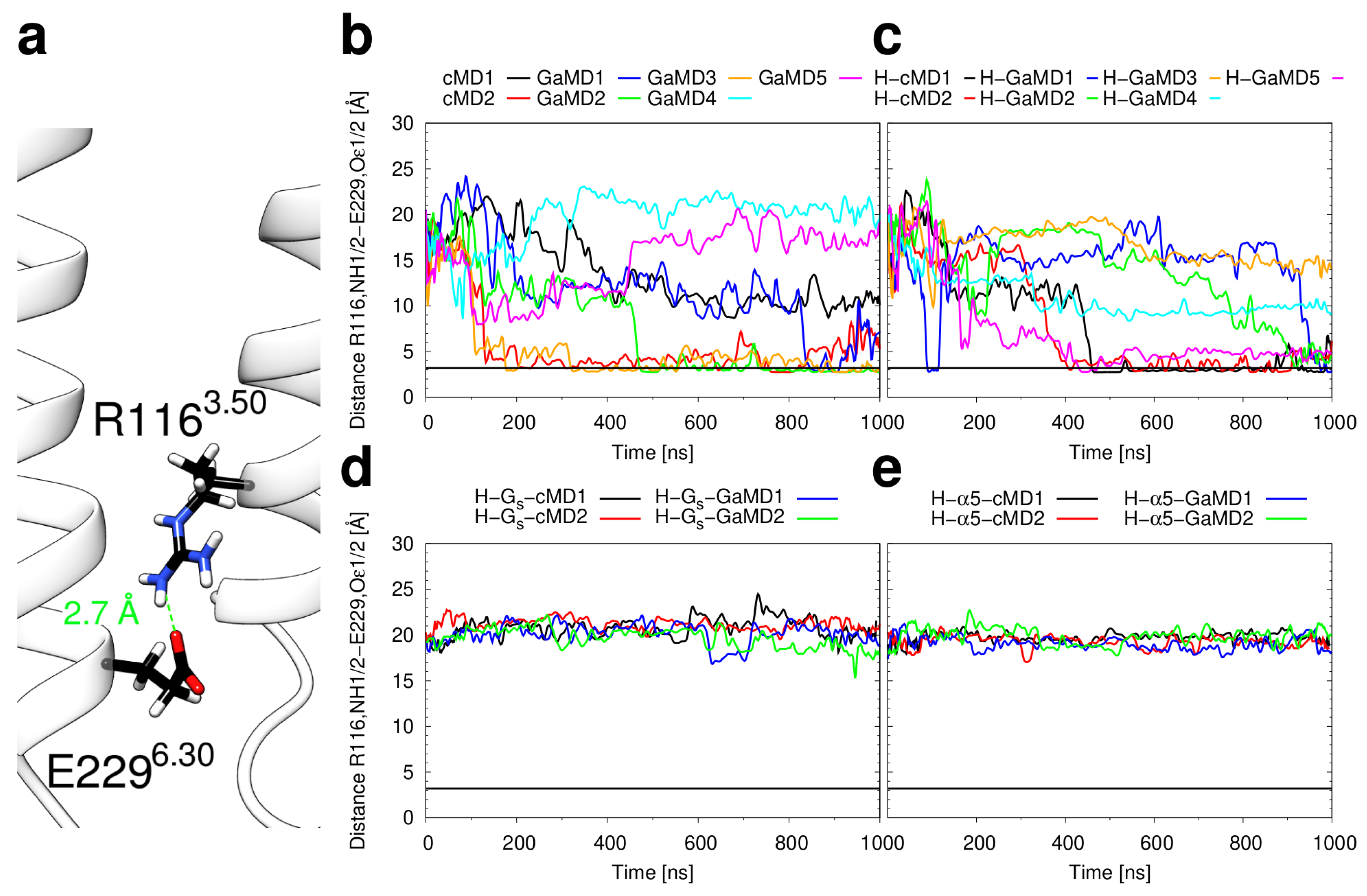
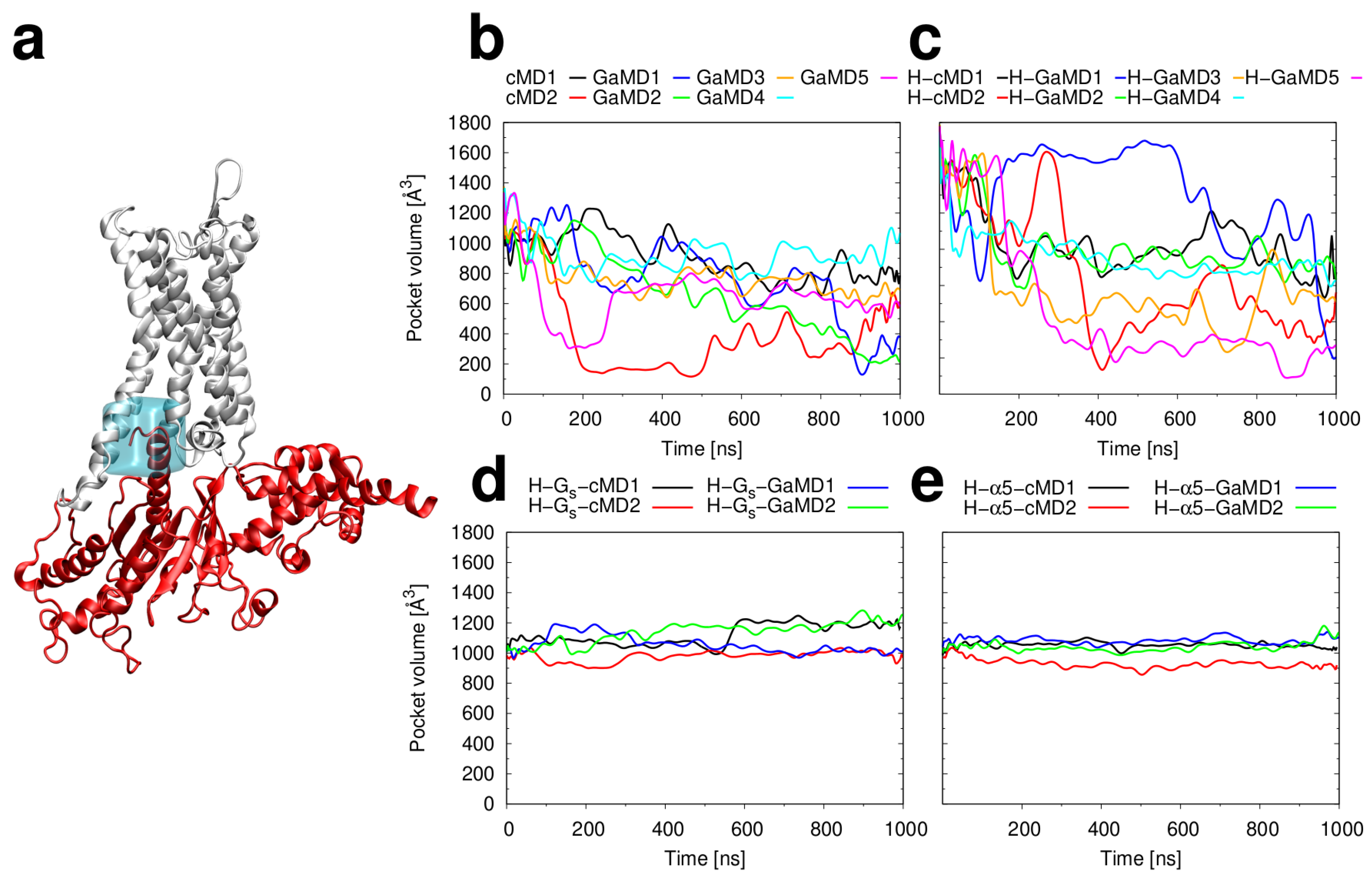
2.3. Conformational Stability of the Active H2R
3. Materials and Methods
3.1. Molecular Modeling of the H2R-Gs Complex
3.2. Overview of the Molecular Dynamics Simulations
3.3. Investigation of H2R Conformational Stability
3.4. Structural Analysis
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Parsons, M.E.; Ganellin, C.R. Histamine and its receptors. Br. J. Pharmacol. 2006, 147, S127–S135. [Google Scholar] [CrossRef] [Green Version]
- Seifert, R.; Strasser, A.; Schneider, E.H.; Neumann, D.; Dove, S.; Buschauer, A. Molecular and cellular analysis of human histamine receptor subtypes. Trends Pharmacol. Sci. 2013, 34, 33–58. [Google Scholar] [CrossRef] [Green Version]
- Rosenbaum, D.M.; Rasmussen, S.G.; Kobilka, B.K. The structure and function of G-protein-coupled receptors. Nature 2009, 459, 356–363. [Google Scholar] [CrossRef] [Green Version]
- Panula, P.; Chazot, P.L.; Cowart, M.; Gutzmer, R.; Leurs, R.; Liu, W.L.S.; Stark, H.; Thurmond, R.L.; Haas, H.L. International Union of Basic and Clinical Pharmacology. XCVIII. Histamine Receptors. Pharmacol. Rev. 2015, 67, 601–655. [Google Scholar] [CrossRef] [Green Version]
- Seifert, R.; Wenzel-Seifert, K. Constitutive activity of G-protein-coupled receptors: Cause of disease and common property of wild-type receptors. Naunyn-Schmiedeberg Arch. Pharmacol. 2002, 366, 381–416. [Google Scholar] [CrossRef]
- Thurmond, R.L.; Gelfand, E.W.; Dunford, P.J. The role of histamine H 1 and H 4 receptors in allergic inflammation: The search for new antihistamines. Nat. Rev. Drug Discov. 2008, 7, 41–53. [Google Scholar] [CrossRef]
- Del Valle, J.; Gantz, I. Novel insights into histamine H2receptor biology. Am. J. Physiol. Gastrointest. Liver Physiol. 1997, 273, G987–G996. [Google Scholar] [CrossRef]
- Brimblecombe, R.; Duncan, W.; Durant, G.; Emmett, J.; Ganellin, C.; Leslie, G.; Parsons, M. Characterization and development of cimetidine as a histamine H2-receptor antagonist. Gastroenterology 1978, 74, 339–347. [Google Scholar] [CrossRef]
- Shamburek, R.; Schubert, M. Control of gastric acid secretion. Histamine H2-receptor antagonists and H+ K (+)-ATPase inhibitors. Gastroenterol. Clin. N. Am. 1992, 21, 527–550. [Google Scholar]
- Matsuda, N.; Jesmin, S.; Takahashi, Y.; Hatta, E.; Kobayashi, M.; Matsuyama, K.; Kawakami, N.; Sakuma, I.; Gando, S.; Fukui, H.; et al. Histamine H1 and H2 receptor gene and protein levels are differentially expressed in the hearts of rodents and humans. J. Pharmacol. Exp. Ther. 2004, 309, 786–795. [Google Scholar] [CrossRef]
- Leary, P.J.; Tedford, R.J.; Bluemke, D.A.; Bristow, M.R.; Heckbert, S.R.; Kawut, S.M.; Krieger, E.V.; Lima, J.A.; Masri, C.S.; Ralph, D.D.; et al. Histamine H2 receptor antagonists, left ventricular morphology, and heart failure risk: The MESA study. J. Am. Coll. Cardiol. 2016, 67, 1544–1552. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Shen, L.; Li, X.; Luo, T.; Wei, X.; Zhang, J.; Cao, S.; Huang, X.; Fukushima, Y.; Bin, J.; et al. Disruption of histamine H2 receptor slows heart failure progression through reducing myocardial apoptosis and fibrosis. Clin. Sci. 2014, 127, 435–448. [Google Scholar] [CrossRef] [PubMed]
- Latorraca, N.R.; Venkatakrishnan, A.; Dror, R.O. GPCR dynamics: Structures in motion. Chem. Rev. 2017, 117, 139–155. [Google Scholar] [CrossRef] [PubMed]
- Weis, W.I.; Kobilka, B.K. The molecular basis of G protein–coupled receptor activation. Annu. Rev. Biochem. 2018, 87, 897–919. [Google Scholar] [CrossRef]
- Laio, A.; Parrinello, M. Escaping free-energy minima. Proc. Natl. Acad. Sci. USA 2002, 99, 12562–12566. [Google Scholar] [CrossRef] [Green Version]
- Laio, A.; Gervasio, F.L. Metadynamics: A method to simulate rare events and reconstruct the free energy in biophysics, chemistry and material science. Rep. Prog. Phys. 2008, 71, 126601. [Google Scholar] [CrossRef]
- Barducci, A.; Bonomi, M.; Parrinello, M. Metadynamics. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 826–843. [Google Scholar] [CrossRef]
- Shang, Y.; Yeatman, H.R.; Provasi, D.; Alt, A.; Christopoulos, A.; Canals, M.; Filizola, M. Proposed mode of binding and action of positive allosteric modulators at opioid receptors. ACS Chem. Biol. 2016, 11, 1220–1229. [Google Scholar] [CrossRef]
- Provasi, D.; Bortolato, A.; Filizola, M. Exploring molecular mechanisms of ligand recognition by opioid receptors with metadynamics. Biochemistry 2009, 48, 10020–10029. [Google Scholar] [CrossRef] [Green Version]
- Saleh, N.; Saladino, G.; Gervasio, F.; Haensele, E.; Banting, L.; Whitley, D.; Sopkova-de Oliveira Santos, J.; Bureau, R.; Clark, T. A Three-Site Mechanism for Agonist/Antagonist Selective Binding to Vasopressin Receptors. Angew. Chem. 2016, 128, 8140–8144. [Google Scholar] [CrossRef]
- Milanos, L.; Saleh, N.; Kling, R.C.; Kaindl, J.; Tschammer, N.; Clark, T. Identification of two distinct sites for antagonist and biased agonist binding to the human chemokine receptor CXCR3. Angew. Chem. 2016, 128, 15503–15507. [Google Scholar] [CrossRef]
- Saleh, N.; Hucke, O.; Kramer, G.; Schmidt, E.; Montel, F.; Lipinski, R.; Ferger, B.; Clark, T.; Hildebrand, P.W.; Tautermann, C.S. Multiple binding sites contribute to the mechanism of mixed agonistic and positive allosteric modulators of the cannabinoid CB1 receptor. Angew. Chem. 2018, 130, 2610–2615. [Google Scholar] [CrossRef]
- Miao, Y.; Feher, V.A.; McCammon, J.A. Gaussian accelerated molecular dynamics: Unconstrained enhanced sampling and free energy calculation. J. Chem. Theory Comput. 2015, 11, 3584–3595. [Google Scholar] [CrossRef] [PubMed]
- Abrams, C.; Bussi, G. Enhanced sampling in molecular dynamics using metadynamics, replica-exchange, and temperature-acceleration. Entropy 2014, 16, 163–199. [Google Scholar] [CrossRef] [Green Version]
- Brown, B.P.; Zhang, Y.K.; Westover, D.; Yan, Y.; Qiao, H.; Huang, V.; Du, Z.; Smith, J.A.; Ross, J.S.; Miller, V.A.; et al. On-target resistance to the mutant-selective EGFR inhibitor osimertinib can develop in an allele-specific manner dependent on the original EGFR-activating mutation. Clin. Cancer Res. 2019, 25, 3341–3351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, Y.; McCammon, J.A. Gaussian accelerated molecular dynamics: Theory, implementation, and applications. In Annual Reports in Computational Chemistry; Elsevier: Amsterdam, The Netherlands, 2017; Volume 13, pp. 231–278. [Google Scholar]
- Pang, Y.T.; Miao, Y.; Wang, Y.; McCammon, J.A. Gaussian accelerated molecular dynamics in NAMD. J. Chem. Theory Comput. 2017, 13, 9–19. [Google Scholar] [CrossRef]
- Peng, Y.; Cao, S.; Kiselar, J.; Xiao, X.; Du, Z.; Hsieh, A.; Ko, S.; Chen, Y.; Agrawal, P.; Zheng, W.; et al. A metastable contact and structural disorder in the estrogen receptor transactivation domain. Structure 2019, 27, 229–240. [Google Scholar] [CrossRef] [Green Version]
- Salawu, E.O. The Impairment of TorsinA’s Binding to and Interactions With Its Activator: An Atomistic Molecular Dynamics Study of Primary Dystonia. Front. Mol. Biosci. 2018, 5, 64. [Google Scholar] [CrossRef] [Green Version]
- Chuang, C.H.; Chiou, S.j.; Cheng, T.L.; Wang, Y.T. A molecular dynamics simulation study decodes the Zika virus NS5 methyltransferase bound to SAH and RNA analogue. Sci. Rep. 2018, 8, 1–9. [Google Scholar]
- Liao, J.M.; Wang, Y.T. In silico studies of conformational dynamics of Mu opioid receptor performed using gaussian accelerated molecular dynamics. J. Biomol. Struct. Dyn. 2019, 37, 166–177. [Google Scholar] [CrossRef]
- Miao, Y.; McCammon, J.A. Mechanism of the G-protein mimetic nanobody binding to a muscarinic G-protein-coupled receptor. Proc. Natl. Acad. Sci. USA 2018, 115, 3036–3041. [Google Scholar] [CrossRef] [Green Version]
- Miao, Y.; McCammon, J.A. Graded activation and free energy landscapes of a muscarinic G-protein–coupled receptor. Proc. Natl. Acad. Sci. USA 2016, 113, 12162–12167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawnikar, S.; Miao, Y. Pathway and mechanism of drug binding to chemokine receptors revealed by accelerated molecular simulations. Future Med. Chem. 2020, 12, 1213–1225. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.T.; Chan, Y.H. Understanding the molecular basis of agonist/antagonist mechanism of human mu opioid receptor through gaussian accelerated molecular dynamics method. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattarai, A.; Wang, J.; Miao, Y. G-protein-coupled receptor–membrane interactions depend on the receptor activation state. J. Comput. Chem. 2020, 41, 460–471. [Google Scholar] [CrossRef]
- Palermo, G.; Miao, Y.; Walker, R.C.; Jinek, M.; McCammon, J.A. CRISPR-Cas9 conformational activation as elucidated from enhanced molecular simulations. Proc. Natl. Acad. Sci. USA 2017, 114, 7260–7265. [Google Scholar] [CrossRef] [Green Version]
- Park, J.B.; Kim, Y.H.; Yoo, Y.; Kim, J.; Jun, S.H.; Cho, J.W.; El Qaidi, S.; Walpole, S.; Monaco, S.; García-García, A.A.; et al. Structural basis for arginine glycosylation of host substrates by bacterial effector proteins. Nat. Commun. 2018, 9, 1–15. [Google Scholar] [CrossRef]
- Ricci, C.G.; Chen, J.S.; Miao, Y.; Jinek, M.; Doudna, J.A.; McCammon, J.A.; Palermo, G. Deciphering off-target effects in CRISPR-Cas9 through accelerated molecular dynamics. ACS Cent. Sci. 2019, 5, 651–662. [Google Scholar] [CrossRef] [Green Version]
- Sibener, L.V.; Fernandes, R.A.; Kolawole, E.M.; Carbone, C.B.; Liu, F.; McAffee, D.; Birnbaum, M.E.; Yang, X.; Su, L.F.; Yu, W.; et al. Isolation of a structural mechanism for uncoupling T cell receptor signaling from peptide-MHC binding. Cell 2018, 174, 672–687. [Google Scholar] [CrossRef]
- Söldner, C.A.; Horn, A.H.; Sticht, H. A Metadynamics-Based Protocol for the Determination of GPCR-Ligand Binding Modes. Int. J. Mol. Sci. 2019, 20, 1970. [Google Scholar] [CrossRef] [Green Version]
- Hill, S.; Ganellin, C.; Timmerman, H.; Schwartz, J.; Shankley, N.; Young, J.; Schunack, W.; Levi, R.; Haas, H. International Union of Pharmacology. XIII. Classification of histamine receptors. Pharmacol. Rev. 1997, 49, 253–278. [Google Scholar] [PubMed]
- Rasmussen, S.G.; DeVree, B.T.; Zou, Y.; Kruse, A.C.; Chung, K.Y.; Kobilka, T.S.; Thian, F.S.; Chae, P.S.; Pardon, E.; Calinski, D.; et al. Crystal structure of the β-2-adrenergic receptor–Gs protein complex. Nature 2011, 477, 549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saleh, N.; Ibrahim, P.; Saladino, G.; Gervasio, F.L.; Clark, T. An efficient metadynamics-based protocol to model the binding affinity and the transition state ensemble of G-protein-coupled receptor ligands. J. Chem. Inf. Model. 2017, 57, 1210–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kooistra, A.; Kuhne, S.; De Esch, I.; Leurs, R.; De Graaf, C. A structural chemogenomics analysis of aminergic GPCRs: Lessons for histamine receptor ligand design. Br. J. Pharmacol. 2013, 170, 101–126. [Google Scholar] [CrossRef] [Green Version]
- Gantz, I.; DelValle, J.; Wang, L.; Tashiro, T.; Munzert, G.; Guo, Y.; Konda, Y.; Yamada, T. Molecular basis for the interaction of histamine with the histamine H2 receptor. J. Biol. Chem. 1992, 267, 20840–20843. [Google Scholar]
- Sinha, N.; Smith-Gill, S.J. Electrostatics in protein binding and function. Curr. Protein Pept. Sci. 2002, 3, 601–614. [Google Scholar] [CrossRef]
- Inoue, A.; Raimondi, F.; Kadji, F.M.N.; Singh, G.; Kishi, T.; Uwamizu, A.; Ono, Y.; Shinjo, Y.; Ishida, S.; Arang, N.; et al. Illuminating G-protein-coupling selectivity of GPCRs. Cell 2019, 177, 1933–1947. [Google Scholar] [CrossRef]
- Venkatakrishnan, A.; Deupi, X.; Lebon, G.; Tate, C.G.; Schertler, G.F.; Babu, M.M. Molecular signatures of G-protein-coupled receptors. Nature 2013, 494, 185–194. [Google Scholar] [CrossRef]
- Kang, Y.; Kuybeda, O.; de Waal, P.W.; Mukherjee, S.; Van Eps, N.; Dutka, P.; Zhou, X.E.; Bartesaghi, A.; Erramilli, S.; Morizumi, T.; et al. Cryo-EM structure of human rhodopsin bound to an inhibitory G protein. Nature 2018, 558, 553–558. [Google Scholar] [CrossRef]
- Koehl, A.; Hu, H.; Maeda, S.; Zhang, Y.; Qu, Q.; Paggi, J.M.; Latorraca, N.R.; Hilger, D.; Dawson, R.; Matile, H.; et al. Structure of the μ-opioid receptor–G i protein complex. Nature 2018, 558, 547–552. [Google Scholar] [CrossRef]
- Rose, A.S.; Elgeti, M.; Zachariae, U.; Grubmüller, H.; Hofmann, K.P.; Scheerer, P.; Hildebrand, P.W. Position of transmembrane helix 6 determines receptor G protein coupling specificity. J. Am. Chem. Soc. 2014, 136, 11244–11247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Xu, X.; Hilger, D.; Aschauer, P.; Tiemann, J.K.; Du, Y.; Liu, H.; Hirata, K.; Sun, X.; Guixà-González, R.; et al. Structural insights into the process of GPCR-G protein complex formation. Cell 2019, 177, 1243–1251. [Google Scholar] [CrossRef] [PubMed]
- Filipek, S. Molecular switches in GPCRs. Curr. Opin. Struct. Biol. 2019, 55, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Staus, D.P.; Strachan, R.T.; Manglik, A.; Pani, B.; Kahsai, A.W.; Kim, T.H.; Wingler, L.M.; Ahn, S.; Chatterjee, A.; Masoudi, A.; et al. Allosteric nanobodies reveal the dynamic range and diverse mechanisms of G-protein-coupled receptor activation. Nature 2016, 535, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Dror, R.O.; Arlow, D.H.; Borhani, D.W.; Jensen, M.; Piana, S.; Shaw, D.E. Identification of two distinct inactive conformations of the β2-adrenergic receptor reconciles structural and biochemical observations. Proc. Natl. Acad. Sci. USA 2009, 106, 4689–4694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romo, T.D.; Grossfield, A.; Pitman, M.C. Concerted interconversion between ionic lock substates of the β2 adrenergic receptor revealed by microsecond timescale molecular dynamics. Biophys. J. 2010, 98, 76–84. [Google Scholar] [CrossRef] [Green Version]
- Dror, R.O.; Arlow, D.H.; Maragakis, P.; Mildorf, T.J.; Pan, A.C.; Xu, H.; Borhani, D.W.; Shaw, D.E. Activation mechanism of the β2-adrenergic receptor. Proc. Natl. Acad. Sci. USA 2011, 108, 18684–18689. [Google Scholar] [CrossRef] [Green Version]
- Frei, J.N.; Broadhurst, R.W.; Bostock, M.J.; Solt, A.; Jones, A.J.; Gabriel, F.; Tandale, A.; Shrestha, B.; Nietlispach, D. Conformational plasticity of ligand-bound and ternary GPCR complexes studied by 19 F NMR of the β 1-adrenergic receptor. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Murphree, L.J.; Marshall, M.A.; Rieger, J.M.; MacDonald, T.L.; Linden, J. Human A2A adenosine receptors: High-affinity agonist binding to receptor-G protein complexes containing Gβ4. Mol. Pharmacol. 2002, 61, 455–462. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, B.; Nehmé, R.; Warne, T.; Leslie, A.G.; Tate, C.G. Structure of the adenosine A 2A receptor bound to an engineered G protein. Nature 2016, 536, 104–107. [Google Scholar] [CrossRef]
- Leurs, R.; Smit, M.J.; Menge, W.M.; Timmerman, H. Pharmacological characterization of the human histamine H2 receptor stably expressed in Chinese hamster ovary cells. Br. J. Pharmacol. 1994, 112, 847–854. [Google Scholar] [CrossRef] [Green Version]
- Isogai, S.; Deupi, X.; Opitz, C.; Heydenreich, F.M.; Tsai, C.J.; Brueckner, F.; Schertler, G.F.; Veprintsev, D.B.; Grzesiek, S. Backbone NMR reveals allosteric signal transduction networks in the β 1-adrenergic receptor. Nature 2016, 530, 237–241. [Google Scholar] [CrossRef]
- DeVree, B.T.; Mahoney, J.P.; Vélez-Ruiz, G.A.; Rasmussen, S.G.; Kuszak, A.J.; Edwald, E.; Fung, J.J.; Manglik, A.; Masureel, M.; Du, Y.; et al. Allosteric coupling from G protein to the agonist-binding pocket in GPCRs. Nature 2016, 535, 182–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Šali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Söldner, C.A.; Horn, A.H.; Sticht, H. Binding of histamine to the H1 receptor—A molecular dynamics study. J. Mol. Model. 2018, 24, 346. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Case, D.; Ben-Shalom, I.; Brozell, S.; Cerutti, D.; Cheatham, T., III; Cruzeiro, V.; Darden, T.; Duke, R.; Ghoreishi, D.; Gilson, M.; et al. AMBER 18; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Bonomi, M.; Branduardi, D.; Bussi, G.; Camilloni, C.; Provasi, D.; Raiteri, P.; Donadio, D.; Marinelli, F.; Pietrucci, F.; Broglia, R.A.; et al. PLUMED: A portable plugin for free-energy calculations with molecular dynamics. Comput. Phys. Commun. 2009, 180, 1961–1972. [Google Scholar] [CrossRef] [Green Version]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Ponder, J.W.; Case, D.A. Force fields for protein simulations. In Advances in Protein Chemistry; Elsevier: Amsterdam, The Netherlands, 2003; Volume 66, pp. 27–85. [Google Scholar]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Berendsen, H.J.; Postma, J.v.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Accounts Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Donini, O.; Reyes, C.M.; Kollman, P.A. Biomolecular simulations: Recent developments in force fields, simulations of enzyme catalysis, protein-ligand, protein-protein, and protein-nucleic acid noncovalent interactions. Annu. Rev. Biophys. Biomol. Struct. 2001, 30, 211–243. [Google Scholar] [CrossRef] [Green Version]
- Durrant, J.D.; Votapka, L.; Sørensen, J.; Amaro, R.E. POVME 2.0: An enhanced tool for determining pocket shape and volume characteristics. J. Chem. Theory Comput. 2014, 10, 5047–5056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Jonsson, A.L.; Beuming, T.; Shelley, J.C.; Voth, G.A. Ligand-dependent activation and deactivation of the human adenosine A2A receptor. J. Am. Chem. Soc. 2013, 135, 8749–8759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Williams, T.; Kelley, C. Gnuplot 4.6: An Interactive Plotting Program. Available online: http://gnuplot.sourceforge.net/ (accessed on 1 January 2013).
- Beitz, E. TeXshade: Shading and labeling of multiple sequence alignments using LaTeX2e. Bioinformatics 2000, 16, 135–139. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Binding Energy [kcal/mol] |
|---|---|
| H-cMD1 | –13.13 ± 0.02 |
| H-cMD2 | –13.09 ± 0.04 |
| H-Gs-cMD1 | –14.36 ± 0.03 |
| H-Gs-cMD2 | –13.87 ± 0.04 |
| H-5-cMD1 | –13.41 ± 0.03 |
| H-5-cMD2 | –11.76 ± 0.04 |
| Simulation Name | Runs × Time | H2R | HSM | Gs | #Atoms | #Water | #DOPC | Water Box Dimensions |
|---|---|---|---|---|---|---|---|---|
| Deduction of histamine (HSM) binding mode | ||||||||
| Multiple walker metadynamics | 32 × 48 ns | ✓ | ✓ | ✓ | 355,534 | 82,961 | 638 | 14.5 Å × 14.5 Å × 16.5 Å |
| Refinement cMD cluster{1–5} | 5 × 1 s | ✓ | ✓ | ✓ | 355,534 | 82,961 | 638 | 14.5 Å × 14.5 Å × 16.5 Å |
| Validation of stability and switchability | ||||||||
| cMD{1–2} GaMD{1–5} | 7 × 1 s | ✓ | × | × | 125,620 | 27,519 | 278 | 9.6 Å × 9.7 Å × 13.0 Å |
| H-cMD{1–2} H-GaMD{1–5} | 7 × 1 s | ✓ | ✓ | × | 125,638 | 27,519 | 278 | 9.6 Å × 9.7 Å × 13.0 Å |
| H-Gs-cMD{1–2} H-Gs-GaMD{1–2} | 4 × 1 s | ✓ | ✓ | ✓ | 355,534 | 82,961 | 638 | 14.5 Å × 14.5 Å × 16.5 Å |
| H-5-cMD{1–2} H-5-GaMD{1–2} | 4 × 1 s | ✓ | ✓ | ⋋ | 342,067 | 110,592 | 638 | 14.5 Å × 14.5 Å × 16.5 Å |
| Simulation Name | Total E Boost [kcal/mol] | Dihedral Energy Boost [kcal/mol] |
|---|---|---|
| GaMD1 | 7.19 ± 3.11 | 6.61 ± 2.67 |
| GaMD2 | 7.17 ± 3.06 | 6.56 ± 2.67 |
| GaMD3 | 7.47 ± 3.16 | 6.72 ± 2.70 |
| GaMD4 | 7.70 ± 3.19 | 6.63 ± 2.69 |
| GaMD5 | 7.64 ± 3.16 | 6.05 ± 2.55 |
| H-GaMD1 | 8.52 ± 3.48 | 6.24 ± 2.59 |
| H-GaMD2 | 7.59 ± 3.18 | 5.94 ± 2.55 |
| H-GaMD3 | 7.45 ± 3.14 | 6.26 ± 2.61 |
| H-GaMD4 | 7.56 ± 3.22 | 6.16 ± 2.57 |
| H-GaMD5 | 7.27 ± 3.10 | 6.15 ± 2.57 |
| H-Gs-GaMD1 | 34.72 ± 6.85 | 6.68 ± 2.76 |
| H-Gs-GaMD2 | 7.89 ± 3.56 | 7.51 ± 2.94 |
| H-5-GaMD1 | 10.21 ± 4.39 | 6.68 ± 2.73 |
| H-5-GaMD2 | 7.81 ± 4.00 | 6.87 ± 2.79 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Conrad, M.; Söldner, C.A.; Miao, Y.; Sticht, H. Agonist Binding and G Protein Coupling in Histamine H2 Receptor: A Molecular Dynamics Study. Int. J. Mol. Sci. 2020, 21, 6693. https://doi.org/10.3390/ijms21186693
Conrad M, Söldner CA, Miao Y, Sticht H. Agonist Binding and G Protein Coupling in Histamine H2 Receptor: A Molecular Dynamics Study. International Journal of Molecular Sciences. 2020; 21(18):6693. https://doi.org/10.3390/ijms21186693
Chicago/Turabian StyleConrad, Marcus, Christian A. Söldner, Yinglong Miao, and Heinrich Sticht. 2020. "Agonist Binding and G Protein Coupling in Histamine H2 Receptor: A Molecular Dynamics Study" International Journal of Molecular Sciences 21, no. 18: 6693. https://doi.org/10.3390/ijms21186693