Umbilical Cord-Derived Mesenchymal Stem Cells Are Able to Use bFGF Treatment and Represent a Superb Tool for Immunosuppressive Clinical Applications

Abstract
:1. Introduction
2. Results
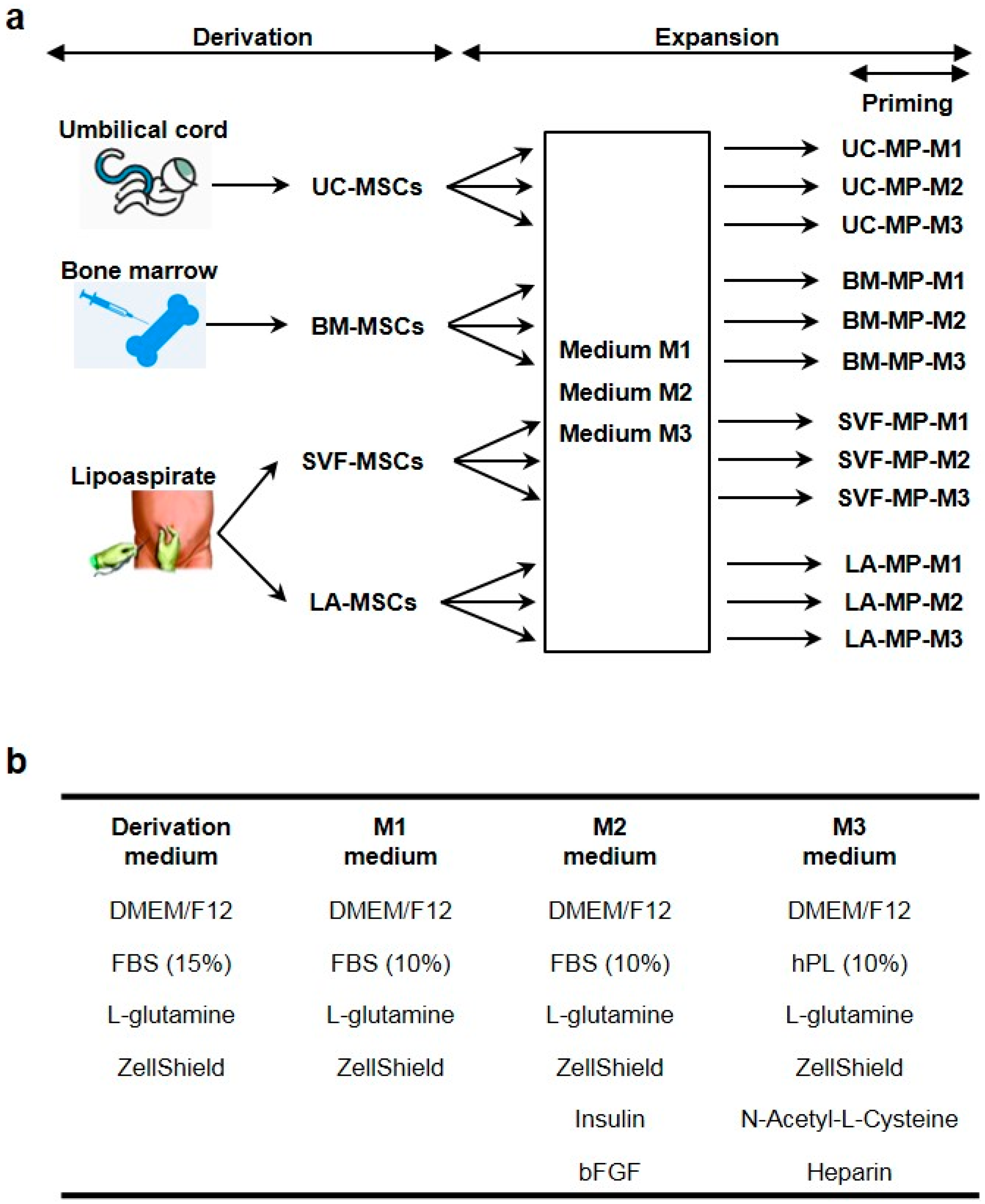
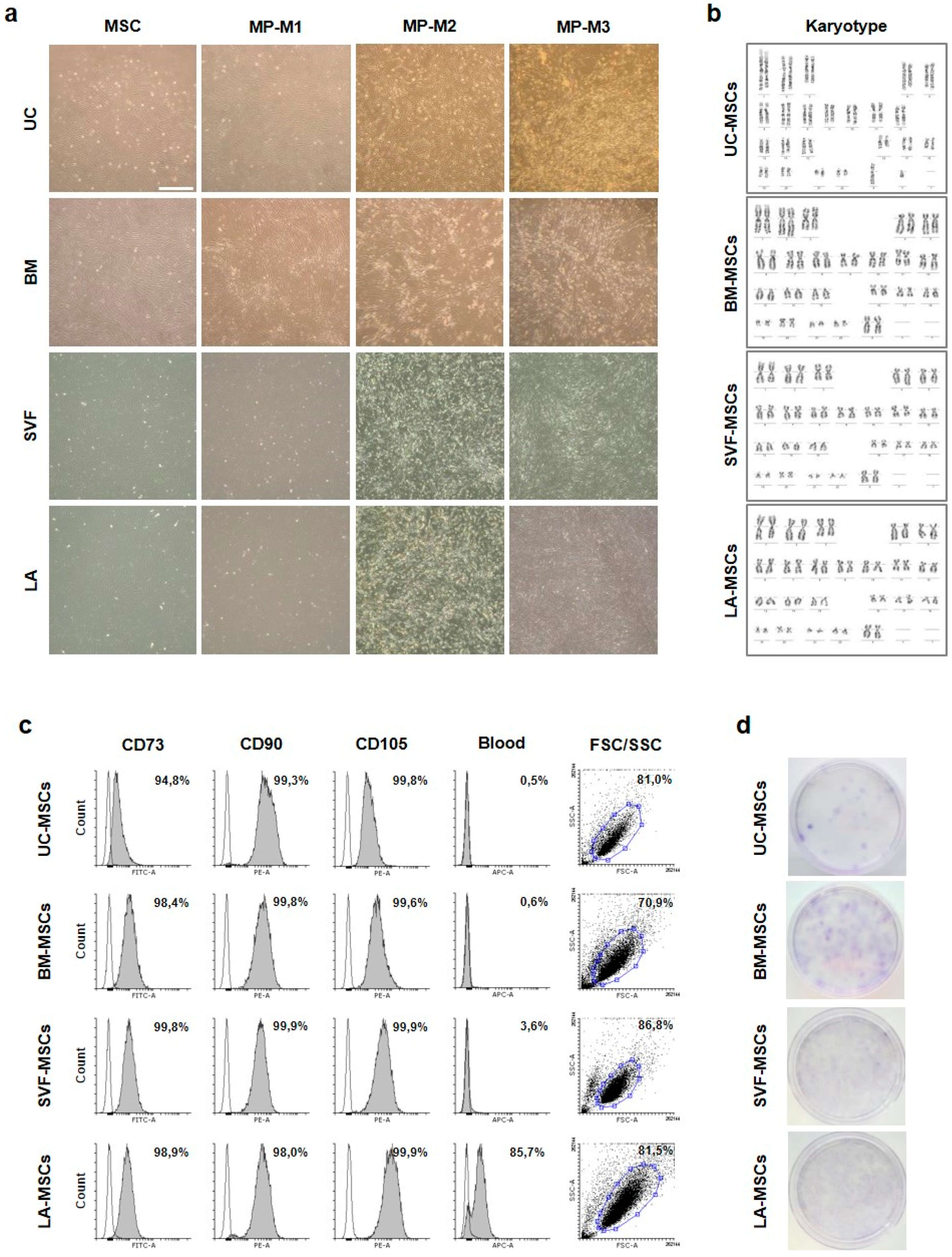
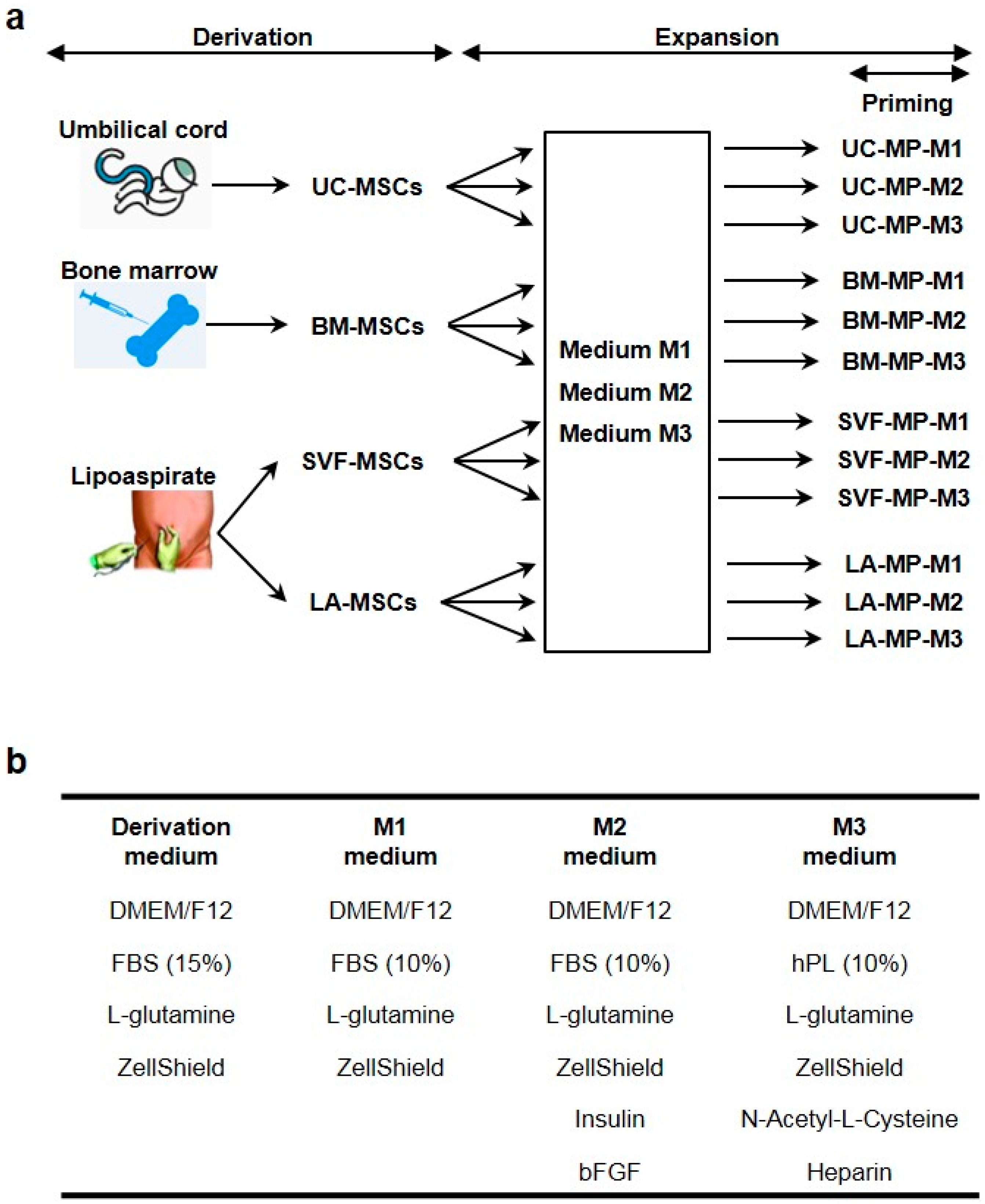
2.1. Derivation and Characterization of Tissue-Specific MSCs
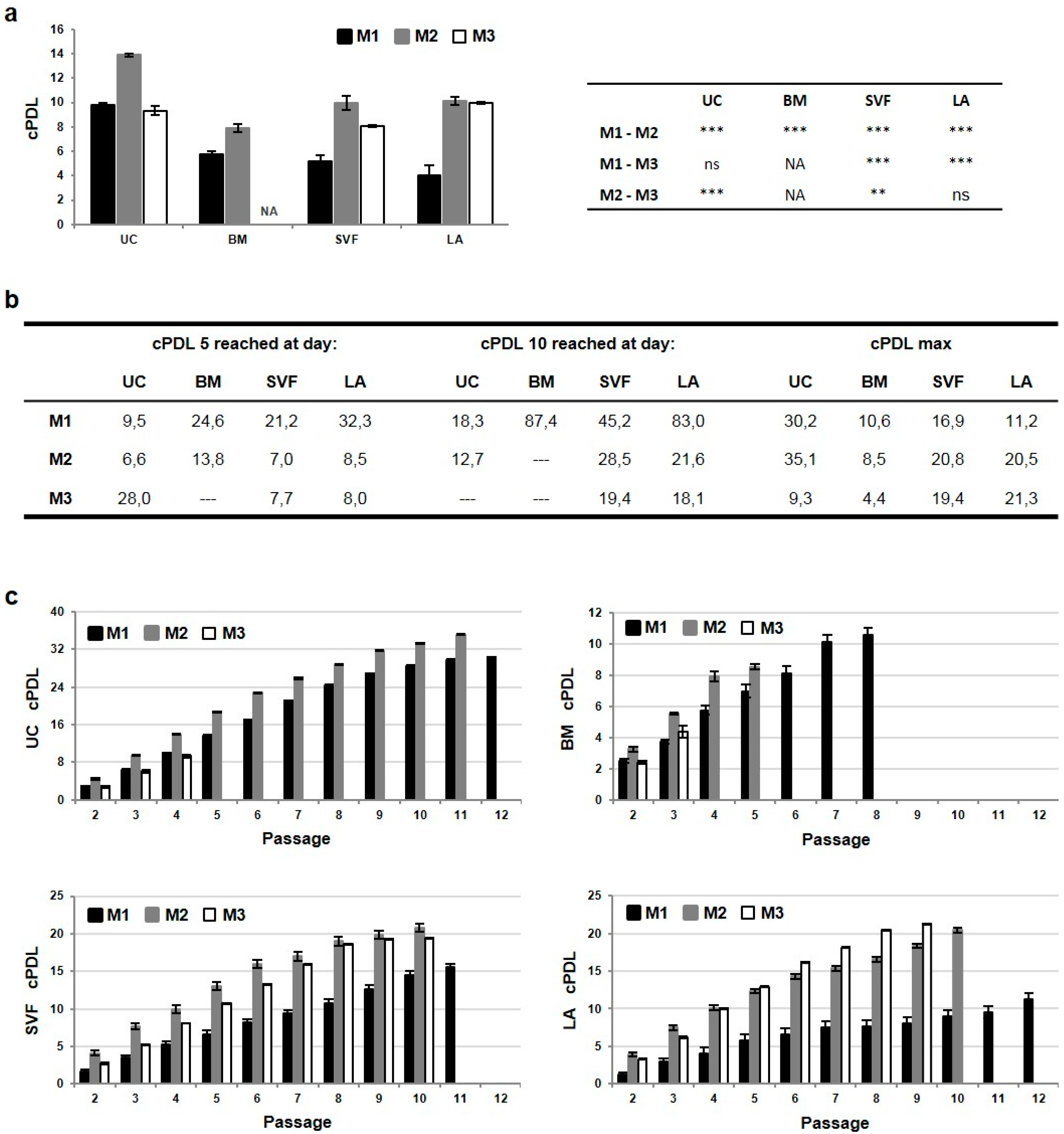
2.2. The Highest Proliferative Capacity of UC-MSCs Is Further Enhanced in M2 Media
2.3. Priming of MSCs Expanded in M2 Media Interferes with Cell Proliferation, CFU-F Ability and CD90 Expression
2.4. Priming of MSC Induces Expression of Immunosuppressive Surface Molecules
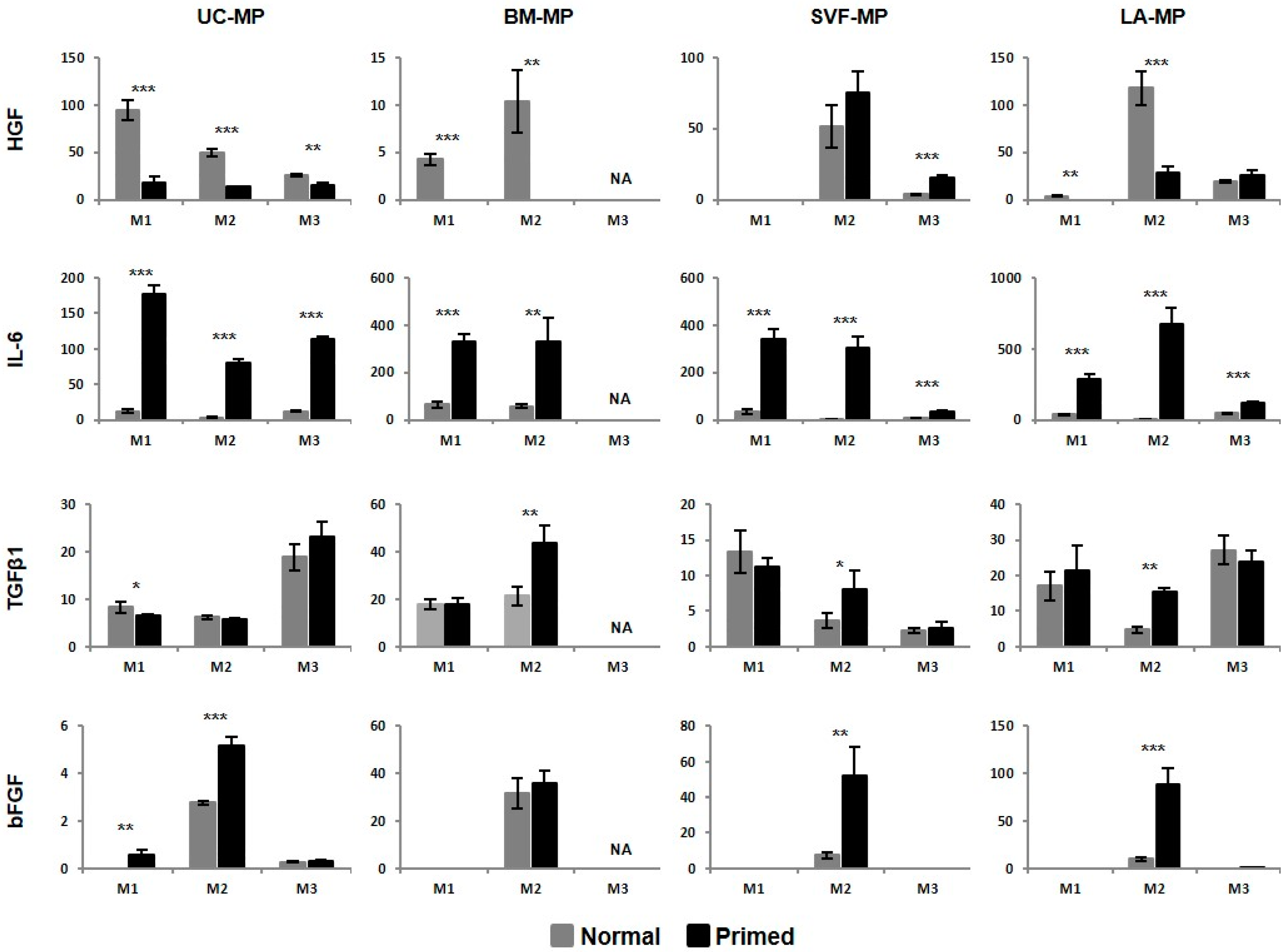
2.5. MSCs Respond to Priming by Increased IDO Activity and IL-6 Production
2.6. MSCs from all Source Tissues Are Able to Suppress T-Cell Proliferation
3. Discussion
4. Materials and Methods
4.1. Umbilical Cord, Bone Marrow and Lipoaspirate Samples
4.2. Derivation of MSCs
4.3. Expansion and Priming of Tissue-Specific MSCs
4.4. Characterization of MSCs
4.5. Immunophenotyping by Flow Cytometry
4.6. Trilineage Differentiation
4.7. Colorimetric Assay of Indoleamine 2,3-Dioxygenase Activity
4.8. Enzyme-Linked Immunosorbent Assay
4.9. Mixed Lymphocyte Reaction
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AT | Adipose tissue |
| ATMP | Advanced therapy medicinal product |
| bFGF | Basic fibroblast growth factor |
| cPDL | Cumulative population doubling level |
| BM | Bone marrow |
| CFSE | Carboxyfluorescein succinimidyl ester |
| CFU-F | Colony-forming unit–fibroblastic |
| DMEM | Dulbecco’s modified Eagle’s medium |
| FBS | Fetal bovine serum |
| hPL | Human platelet lysate |
| IDO | Indoleamine 2,3-dioxygenase |
| IFN-γ | Interferon gamma |
| IL-6 | Intrleukin-6 |
| ISCT | International Society for Cellular Therapy |
| LA | Adipose fraction of lipoaspirate |
| MLR | Mixed lymphocyte reaction |
| MP | Medicinal product |
| MSC | Mesenchymal stem cell |
| PBMC | Peripheral blood mononuclear cell |
| PBS | Phosphate buffered saline |
| PDT | Population doubling time |
| RBC | Red blood cell |
| rMFI | Relative mean fluorescence intensity |
| SD | Standard deviation |
| SVF | Stromal vascular fraction of lipoaspirate |
| TGF-β | Transforming growth factor beta |
| TNF-α | Tumor necrosis factor alpha |
| UC | Umbilical cord |
References
- Horwitz, E.M.; Le Blanc, K.; Dominici, M.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.C.; Deans, R.J.; Krause, D.S.; Keating, A.; Therapy, I.S.f.C. Clarification of the nomenclature for msc: The international society for cellular therapy position statement. Cytotherapy 2005, 7, 393–395. [Google Scholar] [CrossRef] [PubMed]
- Pittenger, M.F.; Mackay, A.M.; Beck, S.C.; Jaiswal, R.K.; Douglas, R.; Mosca, J.D.; Moorman, M.A.; Simonetti, D.W.; Craig, S.; Marshak, D.R. Multilineage potential of adult human mesenchymal stem cells. Science 1999, 284, 143–147. [Google Scholar] [CrossRef] [Green Version]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The international society for cellular therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Friedenstein, A.J.; Petrakova, K.V.; Kurolesova, A.I.; Frolova, G.P. Heterotopic of bone marrow. Analysis of precursor cells for osteogenic and hematopoietic tissues. Transplantation 1968, 6, 230–247. [Google Scholar] [CrossRef]
- Chu, D.T.; Nguyen Thi Phuong, T.; Tien, N.L.B.; Tran, D.K.; Minh, L.B.; Thanh, V.V.; Gia Anh, P.; Pham, V.H.; Thi Nga, V. Adipose tissue stem cells for therapy: An update on the progress of isolation, culture, storage, and clinical application. J. Clin. Med. 2019, 8, 917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arutyunyan, I.; Elchaninov, A.; Makarov, A.; Fatkhudinov, T. Umbilical cord as prospective source for mesenchymal stem cell-based therapy. Stem Cells Int. 2016, 2016, 6901286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.S.; Park, Y.B.; Ha, C.W.; Kim, J.A.; Heo, J.C.; Han, W.J.; Oh, S.Y.; Choi, S.J. Different characteristics of mesenchymal stem cells isolated from different layers of full term placenta. PLoS ONE 2017, 12, e0172642. [Google Scholar] [CrossRef] [Green Version]
- Ledesma-Martínez, E.; Mendoza-Núñez, V.M.; Santiago-Osorio, E. Mesenchymal stem cells derived from dental pulp: A review. Stem Cells Int. 2016, 2016, 4709572. [Google Scholar] [CrossRef] [Green Version]
- Al-Nbaheen, M.; Vishnubalaji, R.; Ali, D.; Bouslimi, A.; Al-Jassir, F.; Megges, M.; Prigione, A.; Adjaye, J.; Kassem, M.; Aldahmash, A. Human stromal (mesenchymal) stem cells from bone marrow, adipose tissue and skin exhibit differences in molecular phenotype and differentiation potential. Stem Cell Rev. Rep. 2013, 9, 32–43. [Google Scholar] [CrossRef] [Green Version]
- Sangeetha, K.N.; Vennila, R.; Secunda, R.; Sakthivel, S.; Pathak, S.; Jeswanth, S.; Surendran, R. Functional variations between mesenchymal stem cells of different tissue origins: A comparative gene expression profiling. Biotechnol. Lett. 2020, 42, 1287–1304. [Google Scholar] [CrossRef]
- Chen, J.Y.; Mou, X.Z.; Du, X.C.; Xiang, C. Comparative analysis of biological characteristics of adult mesenchymal stem cells with different tissue origins. Asian Pac. J. Trop. Med. 2015, 8, 739–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozlowska, U.; Krawczenko, A.; Futoma, K.; Jurek, T.; Rorat, M.; Patrzalek, D.; Klimczak, A. Similarities and differences between mesenchymal stem/progenitor cells derived from various human tissues. World J. Stem Cells 2019, 11, 347–374. [Google Scholar] [CrossRef] [PubMed]
- Stenderup, K.; Justesen, J.; Clausen, C.; Kassem, M. Aging is associated with decreased maximal life span and accelerated senescence of bone marrow stromal cells. Bone 2003, 33, 919–926. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S.M.; Glowacki, J. Age-related decline in the osteogenic potential of human bone marrow cells cultured in three-dimensional collagen sponges. J. Cell Biochem. 2001, 82, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Nishida, S.; Endo, N.; Yamagiwa, H.; Tanizawa, T.; Takahashi, H.E. Number of osteoprogenitor cells in human bone marrow markedly decreases after skeletal maturation. J. Bone Miner. Metab. 1999, 17, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Kern, S.; Eichler, H.; Stoeve, J.; Klüter, H.; Bieback, K. Comparative analysis of mesenchymal stem cells from bone marrow, umbilical cord blood, or adipose tissue. Stem Cells 2006, 24, 1294–1301. [Google Scholar] [CrossRef] [PubMed]
- Zuk, P.A.; Zhu, M.; Ashjian, P.; De Ugarte, D.A.; Huang, J.I.; Mizuno, H.; Alfonso, Z.C.; Fraser, J.K.; Benhaim, P.; Hedrick, M.H. Human adipose tissue is a source of multipotent stem cells. Mol. Biol. Cell 2002, 13, 4279–4295. [Google Scholar] [CrossRef]
- Saccardi, R.; Mancardi, G.L.; Solari, A.; Bosi, A.; Bruzzi, P.; Di Bartolomeo, P.; Donelli, A.; Filippi, M.; Guerrasio, A.; Gualandi, F.; et al. Autologous hsct for severe progressive multiple sclerosis in a multicenter trial: Impact on disease activity and quality of life. Blood 2005, 105, 2601–2607. [Google Scholar] [CrossRef] [Green Version]
- Di Nicola, M.; Carlo-Stella, C.; Magni, M.; Milanesi, M.; Longoni, P.D.; Matteucci, P.; Grisanti, S.; Gianni, A.M. Human bone marrow stromal cells suppress t-lymphocyte proliferation induced by cellular or nonspecific mitogenic stimuli. Blood 2002, 99, 3838–3843. [Google Scholar] [CrossRef]
- Tse, W.T.; Pendleton, J.D.; Beyer, W.M.; Egalka, M.C.; Guinan, E.C. Suppression of allogeneic t-cell proliferation by human marrow stromal cells: Implications in transplantation. Transplantation 2003, 75, 389–397. [Google Scholar] [CrossRef]
- Gao, X.; Song, L.; Shen, K.; Wang, H.; Qian, M.; Niu, W.; Qin, X. Bone marrow mesenchymal stem cells promote the repair of islets from diabetic mice through paracrine actions. Mol. Cell Endocrinol. 2014, 388, 41–50. [Google Scholar] [CrossRef]
- Del Fattore, A.; Luciano, R.; Pascucci, L.; Goffredo, B.M.; Giorda, E.; Scapaticci, M.; Fierabracci, A.; Muraca, M. Immunoregulatory effects of mesenchymal stem cell-derived extracellular vesicles on t lymphocytes. Cell Transplant. 2015, 24, 2615–2627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, G.; Zhang, L.; Zhao, X.; Xu, G.; Zhang, Y.; Roberts, A.I.; Zhao, R.C.; Shi, Y. Mesenchymal stem cell-mediated immunosuppression occurs via concerted action of chemokines and nitric oxide. Cell Stem Cell 2008, 2, 141–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ti, D.; Hao, H.; Fu, X.; Han, W. Mesenchymal stem cells-derived exosomal micrornas contribute to wound inflammation. Sci. China Life Sci. 2016, 59, 1305–1312. [Google Scholar] [CrossRef] [PubMed]
- Negi, N.; Griffin, M.D. Effects of mesenchymal stromal cells on regulatory t cells: Current understanding and clinical relevance. Stem Cells 2020, 38, 596–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krampera, M.; Galipeau, J.; Shi, Y.; Tarte, K.; Sensebe, L.; MSC Committee of the International Society for Cellular Therapy (ISCT). Immunological characterization of multipotent mesenchymal stromal cells--the international society for cellular therapy (isct) working proposal. Cytotherapy 2013, 15, 1054–1061. [Google Scholar] [CrossRef] [Green Version]
- Chinnadurai, R.; Copland, I.B.; Patel, S.R.; Galipeau, J. Ido-independent suppression of t cell effector function by ifn-γ-licensed human mesenchymal stromal cells. J. Immunol. 2014, 192, 1491–1501. [Google Scholar] [CrossRef]
- Chinnadurai, R.; Copland, I.B.; Garcia, M.A.; Petersen, C.T.; Lewis, C.N.; Waller, E.K.; Kirk, A.D.; Galipeau, J. Cryopreserved mesenchymal stromal cells are susceptible to t-cell mediated apoptosis which is partly rescued by ifnγ licensing. Stem Cells 2016, 34, 2429–2442. [Google Scholar] [CrossRef] [Green Version]
- Chinnadurai, R.; Rajan, D.; Ng, S.; McCullough, K.; Arafat, D.; Waller, E.K.; Anderson, L.J.; Gibson, G.; Galipeau, J. Immune dysfunctionality of replicative senescent mesenchymal stromal cells is corrected by ifnγ priming. Blood Adv. 2017, 1, 628–643. [Google Scholar] [CrossRef]
- Rovira Gonzalez, Y.I.; Lynch, P.J.; Thompson, E.E.; Stultz, B.G.; Hursh, D.A. In vitro cytokine licensing induces persistent permissive chromatin at the indoleamine 2,3-dioxygenase promoter. Cytotherapy 2016, 18, 1114–1128. [Google Scholar] [CrossRef] [Green Version]
- Pourgholaminejad, A.; Aghdami, N.; Baharvand, H.; Moazzeni, S.M. The effect of pro-inflammatory cytokines on immunophenotype, differentiation capacity and immunomodulatory functions of human mesenchymal stem cells. Cytokine 2016, 85, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Gorin, C.; Rochefort, G.Y.; Bascetin, R.; Ying, H.; Lesieur, J.; Sadoine, J.; Beckouche, N.; Berndt, S.; Novais, A.; Lesage, M.; et al. Priming dental pulp stem cells with fibroblast growth factor-2 increases angiogenesis of implanted tissue-engineered constructs through hepatocyte growth factor and vascular endothelial growth factor secretion. Stem Cells Transl. Med. 2016, 5, 392–404. [Google Scholar] [CrossRef] [PubMed]
- Sivanathan, K.N.; Rojas-Canales, D.M.; Hope, C.M.; Krishnan, R.; Carroll, R.P.; Gronthos, S.; Grey, S.T.; Coates, P.T. Interleukin-17a-induced human mesenchymal stem cells are superior modulators of immunological function. Stem Cells 2015, 33, 2850–2863. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.; Lee, S.; Ju, H.; Kim, Y.; Heo, J.; Lee, H.Y.; Choi, K.C.; Son, J.; Oh, Y.M.; Kim, I.G.; et al. Valproic acid enforces the priming effect of sphingosine-1 phosphate on human mesenchymal stem cells. Int. J. Mol. Med. 2017, 40, 739–747. [Google Scholar] [CrossRef]
- Fujisawa, K.; Takami, T.; Okada, S.; Hara, K.; Matsumoto, T.; Yamamoto, N.; Yamasaki, T.; Sakaida, I. Analysis of metabolomic changes in mesenchymal stem cells on treatment with desferrioxamine as a hypoxia mimetic compared with hypoxic conditions. Stem Cells 2018, 36, 1226–1236. [Google Scholar] [CrossRef] [Green Version]
- Takeda, K.; Ning, F.; Domenico, J.; Okamoto, M.; Ashino, S.; Kim, S.H.; Jeong, Y.Y.; Shiraishi, Y.; Terada, N.; Sutherland, E.R.; et al. Activation of p70s6 kinase-1 in mesenchymal stem cells is essential to lung tissue repair. Stem Cells Transl. Med. 2018, 7, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Popa, M.A.; Mihai, M.C.; Constantin, A.; Şuică, V.; Ţucureanu, C.; Costache, R.; Antohe, F.; Dubey, R.K.; Simionescu, M. Dihydrotestosterone induces pro-angiogenic factors and assists homing of msc into the cardiac tissue. J. Mol. Endocrinol. 2018, 60, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Yoon, Y.M.; Lee, S.H. Hypoxic preconditioning promotes the bioactivities of mesenchymal stem cells via the hif-1α-grp78-akt axis. Int. J. Mol. Sci. 2017, 18, 1320. [Google Scholar] [CrossRef]
- Lee, S.G.; Joe, Y.A. Autophagy mediates enhancement of proangiogenic activity by hypoxia in mesenchymal stromal/stem cells. Biochem. Biophys. Res. Commun. 2018, 501, 941–947. [Google Scholar] [CrossRef]
- Li, B.; Li, C.; Zhu, M.; Zhang, Y.; Du, J.; Xu, Y.; Liu, B.; Gao, F.; Liu, H.; Cai, J.; et al. Hypoxia-induced mesenchymal stromal cells exhibit an enhanced therapeutic effect on radiation-induced lung injury in mice due to an increased proliferation potential and enhanced antioxidant ability. Cell Physiol. Biochem. 2017, 44, 1295–1310. [Google Scholar] [CrossRef]
- Sun, X.; Su, W.; Ma, X.; Zhang, H.; Sun, Z.; Li, X. Comparison of the osteogenic capability of rat bone mesenchymal stem cells on collagen, collagen/hydroxyapatite, hydroxyapatite and biphasic calcium phosphate. Regen. Biomater. 2018, 5, 93–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Shi, J.; Zhang, M.; Chen, Y.; Wang, X.; Zhang, L.; Tian, Z.; Yan, Y.; Li, Q.; Zhong, W.; et al. Mesenchymal stem cell-laden anti-inflammatory hydrogel enhances diabetic wound healing. Sci. Rep. 2015, 5, 18104. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Ali, F.; Mohsin, S.; Akhtar, S.; Mehmood, A.; Choudhery, M.S.; Khan, S.N.; Riazuddin, S. Preconditioning diabetic mesenchymal stem cells with myogenic medium increases their ability to repair diabetic heart. Stem Cell Res. Ther. 2013, 4, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jossen, V.; van den Bos, C.; Eibl, R.; Eibl, D. Manufacturing human mesenchymal stem cells at clinical scale: Process and regulatory challenges. Appl. Microbiol. Biotechnol. 2018, 102, 3981–3994. [Google Scholar] [CrossRef] [Green Version]
- Tozetti, P.A.; Caruso, S.R.; Mizukami, A.; Fernandes, T.R.; da Silva, F.B.; Traina, F.; Covas, D.T.; Orellana, M.D.; Swiech, K. Expansion strategies for human mesenchymal stromal cells culture under xeno-free conditions. Biotechnol. Prog. 2017, 33, 1358–1367. [Google Scholar] [CrossRef]
- Mizukami, A.; Fernandes-Platzgummer, A.; Carmelo, J.G.; Swiech, K.; Covas, D.T.; Cabral, J.M.; da Silva, C.L. Stirred tank bioreactor culture combined with serum-/xenogeneic-free culture medium enables an efficient expansion of umbilical cord-derived mesenchymal stem/stromal cells. Biotechnol. J. 2016, 11, 1048–1059. [Google Scholar] [CrossRef]
- Cunha, B.; Aguiar, T.; Carvalho, S.B.; Silva, M.M.; Gomes, R.A.; Carrondo, M.J.T.; Gomes-Alves, P.; Peixoto, C.; Serra, M.; Alves, P.M. Bioprocess integration for human mesenchymal stem cells: From up to downstream processing scale-up to cell proteome characterization. J. Biotechnol. 2017, 248, 87–98. [Google Scholar] [CrossRef]
- Timmins, N.E.; Kiel, M.; Günther, M.; Heazlewood, C.; Doran, M.R.; Brooke, G.; Atkinson, K. Closed system isolation and scalable expansion of human placental mesenchymal stem cells. Biotechnol. Bioeng. 2012, 109, 1817–1826. [Google Scholar] [CrossRef]
- Mizukami, A.; Orellana, M.D.; Caruso, S.R.; de Lima Prata, K.; Covas, D.T.; Swiech, K. Efficient expansion of mesenchymal stromal cells in a disposable fixed bed culture system. Biotechnol. Prog. 2013, 29, 568–572. [Google Scholar] [CrossRef]
- Haack-Sørensen, M.; Follin, B.; Juhl, M.; Brorsen, S.K.; Søndergaard, R.H.; Kastrup, J.; Ekblond, A. Culture expansion of adipose derived stromal cells. A closed automated quantum cell expansion system compared with manual flask-based culture. J. Transl. Med. 2016, 14, 319. [Google Scholar] [CrossRef] [Green Version]
- Lambrechts, T.; Papantoniou, I.; Rice, B.; Schrooten, J.; Luyten, F.P.; Aerts, J.M. Large-scale progenitor cell expansion for multiple donors in a monitored hollow fibre bioreactor. Cytotherapy 2016, 18, 1219–1233. [Google Scholar] [CrossRef] [PubMed]
- Nold, P.; Brendel, C.; Neubauer, A.; Bein, G.; Hackstein, H. Good manufacturing practice-compliant animal-free expansion of human bone marrow derived mesenchymal stroma cells in a closed hollow-fiber-based bioreactor. Biochem. Biophys. Res. Commun. 2013, 430, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Rojewski, M.T.; Fekete, N.; Baila, S.; Nguyen, K.; Fürst, D.; Antwiler, D.; Dausend, J.; Kreja, L.; Ignatius, A.; Sensebé, L.; et al. Gmp-compliant isolation and expansion of bone marrow-derived mscs in the closed, automated device quantum cell expansion system. Cell Transplant. 2013, 22, 1981–2000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanley, P.J.; Mei, Z.; Durett, A.G.; Cabreira-Hansen, M.a.G.; Cabreira-Harrison, M.a.G.; Klis, M.; Li, W.; Zhao, Y.; Yang, B.; Parsha, K.; et al. Efficient manufacturing of therapeutic mesenchymal stromal cells with the use of the quantum cell expansion system. Cytotherapy 2014, 16, 1048–1058. [Google Scholar] [CrossRef] [Green Version]
- Guess, A.J.; Daneault, B.; Wang, R.; Bradbury, H.; La Perle, K.M.D.; Fitch, J.; Hedrick, S.L.; Hamelberg, E.; Astbury, C.; White, P.; et al. Safety profile of good manufacturing practice manufactured interferon γ-primed mesenchymal stem/stromal cells for clinical trials. Stem Cells Transl. Med. 2017, 6, 1868–1879. [Google Scholar] [CrossRef]
- Li, W.J.; Tuli, R.; Okafor, C.; Derfoul, A.; Danielson, K.G.; Hall, D.J.; Tuan, R.S. A three-dimensional nanofibrous scaffold for cartilage tissue engineering using human mesenchymal stem cells. Biomaterials 2005, 26, 599–609. [Google Scholar] [CrossRef]
- Apel, A.; Groth, A.; Schlesinger, S.; Bruns, H.; Schemmer, P.; Büchler, M.W.; Herr, I. Suitability of human mesenchymal stem cells for gene therapy depends on the expansion medium. Exp. Cell Res. 2009, 315, 498–507. [Google Scholar] [CrossRef]
- Sotiropoulou, P.A.; Perez, S.A.; Salagianni, M.; Baxevanis, C.N.; Papamichail, M. Characterization of the optimal culture conditions for clinical scale production of human mesenchymal stem cells. Stem Cells 2006, 24, 462–471. [Google Scholar] [CrossRef] [Green Version]
- Bahsoun, S.; Coopman, K.; Akam, E.C. The impact of cryopreservation on bone marrow-derived mesenchymal stem cells: A systematic review. J. Transl. Med. 2019, 17, 397. [Google Scholar] [CrossRef]
- Lin, Y.; Hogan, W.J. Clinical application of mesenchymal stem cells in the treatment and prevention of graft-versus-host disease. Adv. Hematol. 2011, 2011, 427863. [Google Scholar] [CrossRef] [Green Version]
- Subbanna, P.K. Mesenchymal stem cells for treating gvhd: In-vivo fate and optimal dose. Med. Hypotheses 2007, 69, 469–470. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.T.; Xie, Y.; Li, D.S.; Zhu, Y.Y.; Zhang, X.L.; Feng, Y.L.; Chen, Y.P.; Xu, L.J.; Liao, P.F.; Wang, G. Comparison of biological characteristics of mesenchymal stem cells derived from the human umbilical cord and decidua parietalis. Mol. Med. Rep. 2019, 20, 633–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beeravolu, N.; Khan, I.; McKee, C.; Dinda, S.; Thibodeau, B.; Wilson, G.; Perez-Cruet, M.; Bahado-Singh, R.; Chaudhry, G.R. Isolation and comparative analysis of potential stem/progenitor cells from different regions of human umbilical cord. Stem Cell Res. 2016, 16, 696–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramasamy, R.; Tong, C.K.; Yip, W.K.; Vellasamy, S.; Tan, B.C.; Seow, H.F. Basic fibroblast growth factor modulates cell cycle of human umbilical cord-derived mesenchymal stem cells. Cell Prolif. 2012, 45, 132–139. [Google Scholar] [CrossRef]
- Nekanti, U.; Mohanty, L.; Venugopal, P.; Balasubramanian, S.; Totey, S.; Ta, M. Optimization and scale-up of wharton’s jelly-derived mesenchymal stem cells for clinical applications. Stem Cell Res. 2010, 5, 244–254. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.J.; Bae, Y.K.; Kim, M.; Kwon, S.J.; Jeon, H.B.; Choi, S.J.; Kim, S.W.; Yang, Y.S.; Oh, W.; Chang, J.W. Comparative analysis of human mesenchymal stem cells from bone marrow, adipose tissue, and umbilical cord blood as sources of cell therapy. Int. J. Mol. Sci. 2013, 14, 17986–18001. [Google Scholar] [CrossRef]
- Heo, J.S.; Choi, Y.; Kim, H.S.; Kim, H.O. Comparison of molecular profiles of human mesenchymal stem cells derived from bone marrow, umbilical cord blood, placenta and adipose tissue. Int. J. Mol. Med. 2016, 37, 115–125. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.S.; Ning, H.; Lin, G.; Lue, T.F. Is cd34 truly a negative marker for mesenchymal stromal cells? Cytotherapy 2012, 14, 1159–1163. [Google Scholar] [CrossRef] [Green Version]
- Sakaguchi, Y.; Sekiya, I.; Yagishita, K.; Muneta, T. Comparison of human stem cells derived from various mesenchymal tissues: Superiority of synovium as a cell source. Arthritis Rheum. 2005, 52, 2521–2529. [Google Scholar] [CrossRef]
- Rebelatto, C.K.; Aguiar, A.M.; Moretão, M.P.; Senegaglia, A.C.; Hansen, P.; Barchiki, F.; Oliveira, J.; Martins, J.; Kuligovski, C.; Mansur, F.; et al. Dissimilar differentiation of mesenchymal stem cells from bone marrow, umbilical cord blood, and adipose tissue. Exp. Biol. Med. 2008, 233, 901–913. [Google Scholar] [CrossRef]
- Saparov, A.; Ogay, V.; Nurgozhin, T.; Jumabay, M.; Chen, W.C. Preconditioning of human mesenchymal stem cells to enhance their regulation of the immune response. Stem Cells Int. 2016, 2016, 3924858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.W.; Ryu, S.; Kim, D.S.; Sung, K.W.; Koo, H.H.; Yoo, K.H. Strategies to improve the immunosuppressive properties of human mesenchymal stem cells. Stem Cell Res. Ther. 2015, 6, 179. [Google Scholar] [CrossRef] [Green Version]
- Amati, E.; Sella, S.; Perbellini, O.; Alghisi, A.; Bernardi, M.; Chieregato, K.; Lievore, C.; Peserico, D.; Rigno, M.; Zilio, A.; et al. Generation of mesenchymal stromal cells from cord blood: Evaluation of in vitro quality parameters prior to clinical use. Stem Cell Res. Ther. 2017, 8, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassi, G.; Guilloton, F.; Menard, C.; Di Trapani, M.; Deschaseaux, F.; Sensebé, L.; Schrezenmeier, H.; Giordano, R.; Bourin, P.; Dominici, M.; et al. Effects of a ceramic biomaterial on immune modulatory properties and differentiation potential of human mesenchymal stromal cells of different origin. Tissue Eng. Part. A 2015, 21, 767–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Su, J.; Roberts, A.I.; Shou, P.; Rabson, A.B.; Ren, G. How mesenchymal stem cells interact with tissue immune responses. Trends Immunol. 2012, 33, 136–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espagnolle, N.; Balguerie, A.; Arnaud, E.; Sensebé, L.; Varin, A. Cd54-mediated interaction with pro-inflammatory macrophages increases the immunosuppressive function of human mesenchymal stromal cells. Stem Cell Rep. 2017, 8, 961–976. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Huang, J.; Gong, L.; Yu, D.; An, C.; Bunpetch, V.; Dai, J.; Huang, H.; Zou, X.; Ouyang, H.; et al. The plasticity of mesenchymal stem cells in regulating surface hla-i. iScience 2019, 15, 66–78. [Google Scholar] [CrossRef] [Green Version]
- Fu, X.; Chen, Y.; Xie, F.N.; Dong, P.; Liu, W.B.; Cao, Y.; Zhang, W.J.; Xiao, R. Comparison of immunological characteristics of mesenchymal stem cells derived from human embryonic stem cells and bone marrow. Tissue Eng. Part. A 2015, 21, 616–626. [Google Scholar] [CrossRef] [Green Version]
- Yagi, H.; Soto-Gutierrez, A.; Parekkadan, B.; Kitagawa, Y.; Tompkins, R.G.; Kobayashi, N.; Yarmush, M.L. Mesenchymal stem cells: Mechanisms of immunomodulation and homing. Cell Transplant. 2010, 19, 667–679. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Xu, Z.; Bai, J.; Yang, S.; Zhao, S.; Zhang, Y.; Chen, X.; Wang, Y. Umbilical cord tissue-derived mesenchymal stem cells induce t lymphocyte apoptosis and cell cycle arrest by expression of indoleamine 2, 3-dioxygenase. Stem Cells Int. 2016, 2016, 7495135. [Google Scholar] [CrossRef] [Green Version]
- Dabrowski, F.A.; Burdzinska, A.; Kulesza, A.; Sladowska, A.; Zolocinska, A.; Gala, K.; Paczek, L.; Wielgos, M. Comparison of the paracrine activity of mesenchymal stem cells derived from human umbilical cord, amniotic membrane and adipose tissue. J. Obstet. Gynaecol. Res. 2017, 43, 1758–1768. [Google Scholar] [CrossRef] [PubMed]
- Li, C.Y.; Wu, X.Y.; Tong, J.B.; Yang, X.X.; Zhao, J.L.; Zheng, Q.F.; Zhao, G.B.; Ma, Z.J. Comparative analysis of human mesenchymal stem cells from bone marrow and adipose tissue under xeno-free conditions for cell therapy. Stem Cell Res. Ther. 2015, 6, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozaki, K.; Sato, K.; Oh, I.; Meguro, A.; Tatara, R.; Muroi, K.; Ozawa, K. Mechanisms of immunomodulation by mesenchymal stem cells. Int. J. Hematol. 2007, 86, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Groh, M.E.; Maitra, B.; Szekely, E.; Koç, O.N. Human mesenchymal stem cells require monocyte-mediated activation to suppress alloreactive t cells. Exp. Hematol. 2005, 33, 928–934. [Google Scholar] [CrossRef] [PubMed]
- Nasef, A.; Chapel, A.; Mazurier, C.; Bouchet, S.; Lopez, M.; Mathieu, N.; Sensebé, L.; Zhang, Y.; Gorin, N.C.; Thierry, D.; et al. Identification of il-10 and tgf-beta transcripts involved in the inhibition of t-lymphocyte proliferation during cell contact with human mesenchymal stem cells. Gene Expr. 2007, 13, 217–226. [Google Scholar] [CrossRef]
- Tomic, S.; Djokic, J.; Vasilijic, S.; Vucevic, D.; Todorovic, V.; Supic, G.; Colic, M. Immunomodulatory properties of mesenchymal stem cells derived from dental pulp and dental follicle are susceptible to activation by toll-like receptor agonists. Stem Cells Dev. 2011, 20, 695–708. [Google Scholar] [CrossRef]
- Ouchi, T.; Nakagawa, T. Mesenchymal stem cell-based tissue regeneration therapies for periodontitis. Regen. Ther. 2020, 14, 72–78. [Google Scholar] [CrossRef]
- Song, H.; Kwon, K.; Lim, S.; Kang, S.M.; Ko, Y.G.; Xu, Z.; Chung, J.H.; Kim, B.S.; Lee, H.; Joung, B.; et al. Transfection of mesenchymal stem cells with the fgf-2 gene improves their survival under hypoxic conditions. Mol. Cells 2005, 19, 402–407. [Google Scholar]
- Prasanna, S.J.; Gopalakrishnan, D.; Shankar, S.R.; Vasandan, A.B. Pro-inflammatory cytokines, ifngamma and tnfalpha, influence immune properties of human bone marrow and wharton jelly mesenchymal stem cells differentially. PLoS ONE 2010, 5, e9016. [Google Scholar] [CrossRef]
- Manochantr, S.; U-pratya, Y.; Kheolamai, P.; Rojphisan, S.; Chayosumrit, M.; Tantrawatpan, C.; Supokawej, A.; Issaragrisil, S. Immunosuppressive properties of mesenchymal stromal cells derived from amnion, placenta, wharton’s jelly and umbilical cord. Intern. Med. J. 2013, 43, 430–439. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MSC Marker Expression (%) | Adhesion and HLA Molecule Expression (%) | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CD73 | CD105 | CD90 | ICAM-1 | LFA-3 | VCAM-1 | HLA-ABC | HLA-DR | ||||||||||
| N | P | N | P | N | P | N | P | N | P | N | P | N | P | N | P | ||
| UC | M1 | 90.0 | 98.2 | 99.9 | 99.6 | 100.0 | 99.0 | 37.9 | 99.7 * | 58.8 | 72.6 * | 0.2 | 8.5 * | 99.9 | 99.9 | 0.3 | 9.8 * |
| M2 | 99.4 | 99.5 | 97.7 | 93.5 | 99.4 | 81.9 | 45.7 | 99.9 * | 29.2 | 23.5 | 0.3 | 4.5 * | 99.0 | 99.7 | 0.7 | 21.0 * | |
| M3 | 98.4 | 97.3 | 94.8 | 91.0 | 96.1 | 98.1 | 5.9 | 76.3 * | 16.9 | 39.5 * | 0.1 | 1.3 * | 96.6 | 99.0 | 0.0 | 29.4 * | |
| BM | M1 | 99.4 | 99.5 | 99.0 | 98.6 | 100.0 | 100.0 | 21.4 | 97.8 * | 46.3 | 78.2 * | 17.2 | 32.6 * | 97.9 | 98.6 | 0.0 | 82.9 * |
| M2 | 97.2 | 95.5 | 98.9 | 97.6 | 84.6 | 70.2 | 10.4 | 85.8 * | 19.6 | 22.9 | 5.8 | 5.5 | 69.9 | 74.2 | 0.7 | 18.9 * | |
| M3 | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | |
| SVF | M1 | 98.9 | 99.6 | 99.5 | 99.6 | 99.8 | 99.7 | 48.5 | 96.4 * | 61.0 | 88.8 * | 1.4 | 27.7 * | 96.7 | 95.2 | 0.5 | 90.6 * |
| M2 | 99.4 | 96.9 | 97.3 | 96.5 | 96.8 | 53.4 | 6.1 | 96.8 * | 35.4 | 32.9 | 0.2 | 0.6 * | 96.9 | 95.7 | 42.5 | 93.9 * | |
| M3 | 98.2 | 99.2 | 98.2 | 99.5 | 99.9 | 99.7 | 3.1 | 99.5 * | 24.6 | 36.8 | 0.1 | 0.5 * | 99.4 | 99.8 | 0.1 | 26.0 * | |
| LA | M1 | 98.7 | 99.5 | 99.5 | 99.5 | 99.5 | 99.7 | 43.3 | 99.4 * | 70.4 | 91.1 * | 1.8 | 10.1 * | 98.3 | 98.7 | 0.6 | 92.5 * |
| M2 | 99.8 | 98.8 | 99.3 | 98.5 | 99.0 | 59.4 | 4.9 | 97.4 * | 57.5 | 60.0 | 0.7 | 5.5 * | 99.1 | 98.8 | 56.9 | 98.2 * | |
| M3 | 98.0 | 99.4 | 99.3 | 99.7 | 99.8 | 99.9 | 14.3 | 99.9 * | 75.9 | 83.3 * | 0.4 | 0.7 | 99.8 | 99.7 | 0.1 | 18.5 * | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tesarova, L.; Jaresova, K.; Simara, P.; Koutna, I. Umbilical Cord-Derived Mesenchymal Stem Cells Are Able to Use bFGF Treatment and Represent a Superb Tool for Immunosuppressive Clinical Applications. Int. J. Mol. Sci. 2020, 21, 5366. https://doi.org/10.3390/ijms21155366
Tesarova L, Jaresova K, Simara P, Koutna I. Umbilical Cord-Derived Mesenchymal Stem Cells Are Able to Use bFGF Treatment and Represent a Superb Tool for Immunosuppressive Clinical Applications. International Journal of Molecular Sciences. 2020; 21(15):5366. https://doi.org/10.3390/ijms21155366
Chicago/Turabian StyleTesarova, Lenka, Klara Jaresova, Pavel Simara, and Irena Koutna. 2020. "Umbilical Cord-Derived Mesenchymal Stem Cells Are Able to Use bFGF Treatment and Represent a Superb Tool for Immunosuppressive Clinical Applications" International Journal of Molecular Sciences 21, no. 15: 5366. https://doi.org/10.3390/ijms21155366