The Fungal Iron Chelator Desferricoprogen Inhibits Atherosclerotic Plaque Formation

 , ,
, ,
Abstract
:
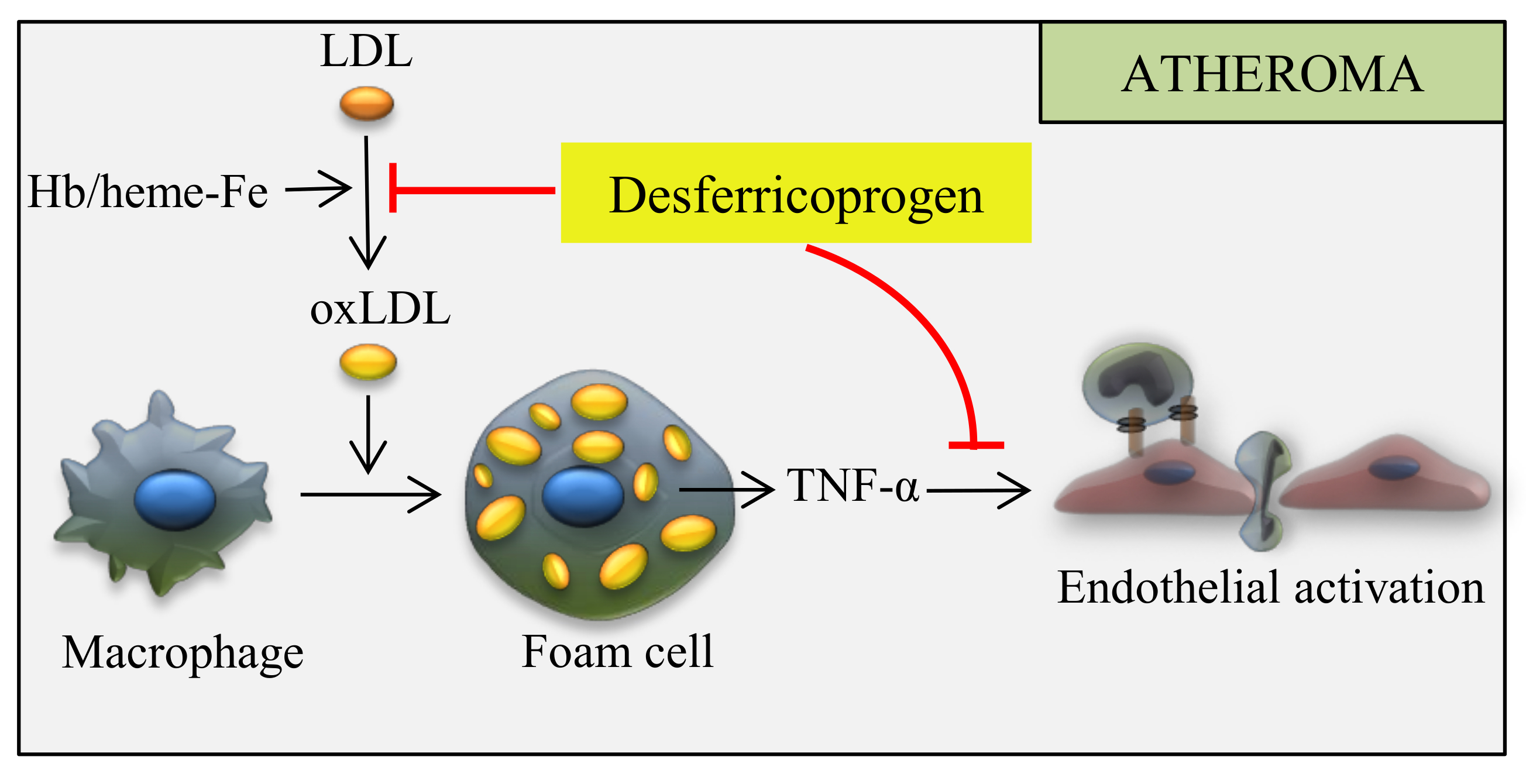{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. DFC Attenuates High Fat Diet-Induced Atherosclerotic Plaque Formation in ApoE−/− Mice
2.2. DFC Inhibits Lipid Peroxidation of Plaque Lipids and LDL in ApoE−/− Mice as well as Heme/Hemoglobin-Catalyzed Oxidation of Lipid Derived from Human Carotid Artery Plaque and LDL
2.3. DFC Prevents TNF-α-Induced Expressions of Adhesion Molecules as well as Endothelial Cell Activation
2.4. DFC Attenuates LDL-Induced Oxidative Stress Catalyzed by Heme
2.5. DFC Inhibits Foam Cell Formation and Suppresses Macrophage Activation Resulted from Heme-Catalyzed LDL Oxidation
2.6. DFC Accumulates within the Atheroma of the Aorta in ApoE−/− Mice
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Study Approval
4.3. Mice
4.4. Coprogen Production, Purification and Deferration
4.5. Quantification of Aortic Lesions in ApoE–/– Mice
4.6. Lipids Extraction from the Human Carotid Arteries
4.7. Cell Culture
4.8. Immunohistochemistry
4.9. Measurement of the Plasma Cholesterol and oxLDL Levels
4.10. Immunofluorescent Staining
4.11. Hemoglobin Preparation
4.12. Preparation and Oxidation of LDL
4.13. Measurement of the Lipid Peroxidation Products
4.14. Isolation and Oxidation of Human Plaque Lipids
4.15. Western Blot
4.16. Endothelial Cell Monolayer Integrity Assay
4.17. Monocyte Adhesion Assay
4.18. Viability Assay
4.19. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
4.20. Macrophages Lipid Uptake Measurement
4.21. Small Animal PET/MRI
4.22. Small Animal PET/MRI Imaging Using 68Ga-DFC
4.23. PET Data Analysis
4.24. Experimental Units
4.25. Statistics
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Insull, W., Jr. The pathology of atherosclerosis: Plaque development and plaque responses to medical treatment. Am. J. Med. 2009, 122, S3–S14. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.; Noels, H. Atherosclerosis: Current pathogenesis and therapeutic options. Nat. Med. 2011, 17, 1410–1422. [Google Scholar] [CrossRef] [PubMed]
- Balla, G.; Jacob, H.S.; Eaton, J.W.; Belcher, J.D.; Vercellotti, G.M. Hemin: A possible physiological mediator of low density lipoprotein oxidation and endothelial injury. Arterioscler. Thromb. Vasc. Biol. 1991, 11, 1700–1711. [Google Scholar] [CrossRef] [Green Version]
- Esterbauer, H.; Wag, G.; Puhl, H. Lipid peroxidation and its role in atherosclerosis. Br. Med. Bull. 1993, 49, 566–576. [Google Scholar] [CrossRef]
- Rong, J.X.; Shapiro, M.; Trogan, E.; Fisher, E.A. Transdifferentiation of mouse aortic smooth muscle cells to a macrophage-like state after cholesterol loading. Proc. Natl. Acad. Sci. USA 2003, 100, 13531–13536. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Dubland, J.A.; Allahverdian, S.; Asonye, E.; Sahin, B.; Jaw, J.E.; Sin, D.D.; Seidman, M.A.; Leeper, N.J.; Francis, G.A. Smooth Muscle Cells Contribute the Majority of Foam Cells in ApoE (Apolipoprotein E)-Deficient Mouse Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 876–887. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Kisugi, R. Mechanisms of LDL oxidation. Clin. Chim. Acta 2010, 411, 1875–1882. [Google Scholar] [CrossRef] [PubMed]
- Maor, I.; Hayek, T.; Coleman, R.; Aviram, M. Plasma LDL oxidation leads to its aggregation in the atherosclerotic apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 2995–3005. [Google Scholar] [CrossRef] [PubMed]
- Holvoet, P.; Perez, G.; Zhao, Z.; Brouwers, E.; Bernar, H.; Collen, D. Malondialdehyde-modified low density lipoproteins in patients with atherosclerotic disease. J. Clin. Investig. 1995, 95, 2611–2619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stadler, N.; Lindner, R.A.; Davies, M.J. Direct detection and quantification of transition metal ions in human atherosclerotic plaques: Evidence for the presence of elevated levels of iron and copper. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 949–954. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, J.L. Iron and the sex difference in heart disease risk. Lancet 1981, 1, 1293–1294. [Google Scholar] [CrossRef]
- Steinberg, D.; Parthasarathy, S.; Carew, T.E.; Khoo, J.C.; Witztum, J.L. Beyond cholesterol. Modifications of low-density lipoprotein that increase its atherogenicity. N. Engl. J. Med. 1989, 320, 915–924. [Google Scholar] [CrossRef]
- Henriksen, T.; Mahoney, E.M.; Steinberg, D. Enhanced macrophage degradation of biologically modified low density lipoprotein. Arteriosclerosis 1983, 3, 149–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esterbauer, H.; Puhl, H.; Dieber-Rotheneder, M.; Waeg, G.; Rabl, H. Effect of antioxidants on oxidative modification of LDL. Ann. Med. 1991, 23, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Blankenberg, S.; Barbaux, S.; Tiret, L. Adhesion molecules and atherosclerosis. Atherosclerosis 2003, 170, 191–203. [Google Scholar] [CrossRef]
- Yu, X.H.; Fu, Y.C.; Zhang, D.W.; Yin, K.; Tang, C.K. Foam cells in atherosclerosis. Clin. Chim. Acta 2013, 424, 245–252. [Google Scholar] [CrossRef] [Green Version]
- Libby, P.; Ridker, P.M.; Maseri, A. Inflammation and atherosclerosis. Circulation 2002, 105, 1135–1143. [Google Scholar] [CrossRef]
- Galkina, E.; Ley, K. Immune and inflammatory mechanisms of atherosclerosis *. Annu. Rev. Immunol. 2009, 27, 165–197. [Google Scholar] [CrossRef] [Green Version]
- Bekkering, S.; Quintin, J.; Joosten, L.A.; van der Meer, J.W.; Netea, M.G.; Riksen, N.P. Oxidized low-density lipoprotein induces long-term proinflammatory cytokine production and foam cell formation via epigenetic reprogramming of monocytes. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1731–1738. [Google Scholar] [CrossRef]
- Witztum, J.L.; Steinberg, D. Role of oxidized low density lipoprotein in atherogenesis. J. Clin. Investig. 1991, 88, 1785–1792. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- O’Donoghue, M.L.; Glaser, R.; Cavender, M.A.; Aylward, P.E.; Bonaca, M.P.; Budaj, A.; Davies, R.Y.; Dellborg, M.; Fox, K.A.; Gutierrez, J.A.; et al. Effect of Losmapimod on Cardiovascular Outcomes in Patients Hospitalized With Acute Myocardial Infarction: A Randomized Clinical Trial. Jama 2016, 315, 1591–1599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridker, P.M.; Everett, B.M.; Pradhan, A.; MacFadyen, J.G.; Solomon, D.H.; Zaharris, E.; Mam, V.; Hasan, A.; Rosenberg, Y.; Iturriaga, E.; et al. Low-Dose Methotrexate for the Prevention of Atherosclerotic Events. N. Engl. J. Med. 2019, 380, 752–762. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.L.; Martinez, C.M.; Gennis, P.; Gallagher, E.J. The cardiac toxicity of anabolic steroids. Prog. Cardiovasc. Dis. 1998, 41, 1–15. [Google Scholar] [CrossRef]
- Kokotou, M.G.; Limnios, D.; Nikolaou, A.; Psarra, A.; Kokotos, G. Inhibitors of phospholipase A(2) and their therapeutic potential: An update on patents (2012-2016). Expert Opin. Ther. Pat. 2017, 27, 217–225. [Google Scholar] [CrossRef]
- Nguyen, M.T.; Fernando, S.; Schwarz, N.; Tan, J.T.; Bursill, C.A.; Psaltis, P.J. Inflammation as a Therapeutic Target in Atherosclerosis. J. clin. Med. 2019, 8, 1109. [Google Scholar] [CrossRef] [Green Version]
- Charo, I.F.; Taub, R. Anti-inflammatory therapeutics for the treatment of atherosclerosis. Nat. Rev. Drug Discov. 2011, 10, 365–376. [Google Scholar] [CrossRef] [Green Version]
- Muñoz, M.; Villar, I.; García-Erce, J.A. An update on iron physiology. World J. Gastroenterol. 2009, 15, 4617–4626. [Google Scholar] [CrossRef]
- Kohgo, Y.; Ikuta, K.; Ohtake, T.; Torimoto, Y.; Kato, J. Body iron metabolism and pathophysiology of iron overload. Int. J. Hematol. 2008, 88, 7–15. [Google Scholar] [CrossRef] [Green Version]
- Pantopoulos, K.; Porwal, S.K.; Tartakoff, A.; Devireddy, L. Mechanisms of mammalian iron homeostasis. Biochemistry 2012, 51, 5705–5724. [Google Scholar] [CrossRef]
- Valko, M.; Jomova, K.; Rhodes, C.J.; Kuca, K.; Musilek, K. Redox- and non-redox-metal-induced formation of free radicals and their role in human disease. Arch. Toxicol. 2016, 90, 1–37. [Google Scholar] [CrossRef] [PubMed]
- Crichton, R.R.; Wilmet, S.; Legssyer, R.; Ward, R.J. Molecular and cellular mechanisms of iron homeostasis and toxicity in mammalian cells. J. Inorg. Biochem. 2002, 91, 9–18. [Google Scholar] [CrossRef]
- Sullivan, J.L. The iron paradigm of ischemic heart disease. Am. Heart. J. 1989, 117, 1177–1188. [Google Scholar] [CrossRef]
- Dabbagh, A.J.; Shwaery, G.T.; Keaney, J.F., Jr.; Frei, B. Effect of iron overload and iron deficiency on atherosclerosis in the hypercholesterolemic rabbit. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 2638–2645. [Google Scholar] [CrossRef] [PubMed]
- Kirk, E.A.; Heinecke, J.W.; LeBoeuf, R.C. Iron overload diminishes atherosclerosis in apoE-deficient mice. J. Clin. Investig. 2001, 107, 1545–1553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winterbourn, C.C. Toxicity of iron and hydrogen peroxide: The Fenton reaction. Toxicol. Lett. 1995, 82, 969–974. [Google Scholar] [CrossRef]
- Balla, G.; Vercellotti, G.M.; Muller-Eberhard, U.; Eaton, J.; Jacob, H.S. Exposure of endothelial cells to free heme potentiates damage mediated by granulocytes and toxic oxygen species. Lab. Investig. 1991, 64, 648–655. [Google Scholar]
- Michel, J.B.; Martin-Ventura, J.L.; Nicoletti, A.; Ho-Tin-Noe, B. Pathology of human plaque vulnerability: Mechanisms and consequences of intraplaque haemorrhages. Atherosclerosis 2014, 234, 311–319. [Google Scholar] [CrossRef]
- Schaer, D.J.; Buehler, P.W.; Alayash, A.I.; Belcher, J.D.; Vercellotti, G.M. Hemolysis and free hemoglobin revisited: Exploring hemoglobin and hemin scavengers as a novel class of therapeutic proteins. Blood 2013, 121, 1276–1284. [Google Scholar] [CrossRef] [Green Version]
- Nagy, E.; Eaton, J.W.; Jeney, V.; Soares, M.P.; Varga, Z.; Galajda, Z.; Szentmiklosi, J.; Mehes, G.; Csonka, T.; Smith, A.; et al. Red cells, hemoglobin, heme, iron, and atherogenesis. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1347–1353. [Google Scholar] [CrossRef]
- Potor, L.; Banyai, E.; Becs, G.; Soares, M.P.; Balla, G.; Balla, J.; Jeney, V. Atherogenesis may involve the prooxidant and proinflammatory effects of ferryl hemoglobin. Oxid. Med. Cell. Longev. 2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delbosc, S.; Bayles, R.G.; Laschet, J.; Ollivier, V.; Ho-Tin-Noe, B.; Touat, Z.; Deschildre, C.; Morvan, M.; Louedec, L.; Gouya, L.; et al. Erythrocyte Efferocytosis by the Arterial Wall Promotes Oxidation in Early-Stage Atheroma in Humans. Front. Cardiovasc. Med. 2017, 4, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neilands, J.B. Siderophores: Structure and function of microbial iron transport compounds. J. Biol. Chem. 1995, 270, 26723–26726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, E.; Holmström, S.J.M. Siderophores in environmental research: Roles and applications. Microb. Biotechnol. 2014, 7, 196–208. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Protection against tissue damage in vivo by desferrioxamine: What is its mechanism of action? Free. Radic. Biol. Med. 1989, 7, 645–651. [Google Scholar] [CrossRef]
- Hershko, C.; Pinson, A.; Link, G. Iron chelation. Blood Rev. 1990, 4, 1–8. [Google Scholar] [CrossRef]
- Zhang, W.J.; Frei, B. Intracellular metal ion chelators inhibit TNFalpha-induced SP-1 activation and adhesion molecule expression in human aortic endothelial cells. Free. Radic. Biol. Med. 2003, 34, 674–682. [Google Scholar] [CrossRef]
- Zhang, W.J.; Wei, H.; Frei, B. The iron chelator, desferrioxamine, reduces inflammation and atherosclerotic lesion development in experimental mice. Exp. Biol. Med. 2010, 235, 633–641. [Google Scholar] [CrossRef] [Green Version]
- Choi, E.Y.; Kim, E.C.; Oh, H.M.; Kim, S.; Lee, H.J.; Cho, E.Y.; Yoon, K.H.; Kim, E.A.; Han, W.C.; Choi, S.C.; et al. Iron chelator triggers inflammatory signals in human intestinal epithelial cells: Involvement of p38 and extracellular signal-regulated kinase signaling pathways. J. Immunol. 2004, 172, 7069–7077. [Google Scholar] [CrossRef] [Green Version]
- Hoffbrand, A.V. Prospects for oral iron chelation therapy. J. Lab. Clin. Med. 1994, 123, 492–494. [Google Scholar]
- Kicic, A.; Chua, A.C.G.; Baker, E. Effect of iron chelators on proliferation and iron uptake in hepatoma cells. Cancer 2001, 92, 3093–3110. [Google Scholar] [CrossRef]
- Pocsi, I.; Jeney, V.; Kertai, P.; Pocsi, I.; Emri, T.; Gyemant, G.; Fesus, L.; Balla, J.; Balla, G. Fungal siderophores function as protective agents of LDL oxidation and are promising anti-atherosclerotic metabolites in functional food. Mol. Nutr. Food. Res. 2008, 52, 1434–1447. [Google Scholar] [CrossRef] [PubMed]
- Pendse, A.A.; Arbones-Mainar, J.M.; Johnson, L.A.; Altenburg, M.K.; Maeda, N. Apolipoprotein E knock-out and knock-in mice: Atherosclerosis, metabolic syndrome, and beyond. J. Lipid Res. 2009, 50, S178–S182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, U.; Jialal, I. Oxidative stress and atherosclerosis. Pathophysiology 2006, 13, 129–142. [Google Scholar] [CrossRef]
- Friedl, J.; Puhlmann, M.; Bartlett, D.L.; Libutti, S.K.; Turner, E.N.; Gnant, M.F.; Alexander, H.R. Induction of permeability across endothelial cell monolayers by tumor necrosis factor (TNF) occurs via a tissue factor-dependent mechanism: Relationship between the procoagulant and permeability effects of TNF. Blood 2002, 100, 1334–1339. [Google Scholar] [CrossRef] [Green Version]
- Xia, P.; Gamble, J.R.; Rye, K.A.; Wang, L.; Hii, C.S.T.; Cockerill, P.; Khew-Goodall, Y.; Bert, A.G.; Barter, P.J.; Vadas, M.A. Tumor necrosis factor-α induces adhesion molecule expression through the sphingosine kinase pathway. Proc. Natl. Acad. Sci. USA 1998, 95, 14196–14201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mestas, J.; Ley, K. Monocyte-endothelial cell interactions in the development of atherosclerosis. Trends Cardiovasc Med. 2008, 18, 228–232. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, A.; Balla, J.; Balla, G.; Croatt, A.J.; Vercellotti, G.M.; Nath, K.A. Renal tubular epithelial cells mimic endothelial cells upon exposure to oxidized LDL. Am. J. Physiol. 1996, 271, 814–823. [Google Scholar] [CrossRef]
- Berliner, J.A.; Heinecke, J.W. The role of oxidized lipoproteins in atherogenesis. Free. Radic. Biol. Med. 1996, 20, 707–727. [Google Scholar] [CrossRef]
- Kaplan, M.; Aviram, M. Oxidized low density lipoprotein: Atherogenic and proinflammatory characteristics during macrophage foam cell formation. An inhibitory role for nutritional antioxidants and serum paraoxonase. Clin. Chem. Lab. Med. 1999, 37, 777–787. [Google Scholar] [CrossRef]
- Park, Y.M. CD36, a scavenger receptor implicated in atherosclerosis. Exp. Mol. Med. 2014, 46, e99. [Google Scholar] [CrossRef] [Green Version]
- Nagy, L.; Tontonoz, P.; Alvarez, J.G.; Chen, H.; Evans, R.M. Oxidized LDL regulates macrophage gene expression through ligand activation of PPARgamma. Cell 1998, 93, 229–240. [Google Scholar] [CrossRef] [Green Version]
- Jovinge, S.; Ares, M.P.; Kallin, B.; Nilsson, J. Human monocytes/macrophages release TNF-alpha in response to Ox-LDL. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 1573–1579. [Google Scholar] [CrossRef]
- Branen, L.; Hovgaard, L.; Nitulescu, M.; Bengtsson, E.; Nilsson, J.; Jovinge, S. Inhibition of tumor necrosis factor-alpha reduces atherosclerosis in apolipoprotein E knockout mice. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 2137–2142. [Google Scholar] [CrossRef] [Green Version]
- Jeney, V.; Balla, J.; Yachie, A.; Varga, Z.; Vercellotti, G.M.; Eaton, J.W.; Balla, G. Pro-oxidant and cytotoxic effects of circulating heme. Blood 2002, 100, 879–887. [Google Scholar] [CrossRef] [Green Version]
- Balla, J.; Vercellotti, G.M.; Jeney, V.; Yachie, A.; Varga, Z.; Jacob, H.S.; Eaton, J.W.; Balla, G. Heme, heme oxygenase, and ferritin: How the vascular endothelium survives (and dies) in an iron-rich environment. Antioxid. Redox. Signal. 2007, 9, 2119–2137. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J.M.; Cross, C.E. Free radicals, antioxidants, and human disease: Where are we now? J. Lab. Clin. Med. 1992, 119, 598–620. [Google Scholar]
- Tanner, M.A.; Galanello, R.; Dessi, C.; Smith, G.C.; Westwood, M.A.; Agus, A.; Roughton, M.; Assomull, R.; Nair, S.V.; Walker, J.M.; et al. A randomized, placebo-controlled, double-blind trial of the effect of combined therapy with deferoxamine and deferiprone on myocardial iron in thalassemia major using cardiovascular magnetic resonance. Circulation 2007, 115, 1876–1884. [Google Scholar] [CrossRef]
- Duffy, S.J.; Biegelsen, E.S.; Holbrook, M.; Russell, J.D.; Gokce, N.; Keaney, J.F., Jr.; Vita, J.A. Iron chelation improves endothelial function in patients with coronary artery disease. Circulation 2001, 103, 2799–2804. [Google Scholar] [CrossRef] [Green Version]
- Matthews, A.J.; Vercellotti, G.M.; Menchaca, H.J.; Bloch, P.H.; Michalek, V.N.; Marker, P.H.; Murar, J.; Buchwald, H. Iron and atherosclerosis: Inhibition by the iron chelator deferiprone (L1). J. Surg. Res. 1997, 73, 35–40. [Google Scholar] [CrossRef]
- Minqin, R.; Rajendran, R.; Pan, N.; Tan, B.K.; Ong, W.Y.; Watt, F.; Halliwell, B. The iron chelator desferrioxamine inhibits atherosclerotic lesion development and decreases lesion iron concentrations in the cholesterol-fed rabbit. Free Radic. Biol. Med. 2005, 38, 1206–1211. [Google Scholar] [CrossRef]
- Copper(II), nickel(II), zinc(II), and molybdenum(VI) complexes of desferrioxamine B in aqueous solution. J. Inorg. Biochem. 1997, 65, 281–286. [CrossRef]
- Franchini, M.; Gandini, G.; Veneri, D.; Aprili, G. Safety and efficacy of subcutaneous bolus injection of deferoxamine in adult patients with iron overload: An update. Blood 2004, 103, 747–748. [Google Scholar] [CrossRef]
- Ceci, A.; Felisi, M.; De Sanctis, V.; De Mattia, D. Pharmacotherapy of iron overload in thalassaemic patients. Expert. Opin. Pharmacother. 2003, 4, 1763–1774. [Google Scholar] [CrossRef]
- Yun, M.; Yeh, D.; Araujo, L.I.; Jang, S.; Newberg, A.; Alavi, A. F-18 FDG uptake in the large arteries: A new observation. Clin. Nucl. Med. 2001, 26, 314–319. [Google Scholar] [CrossRef] [PubMed]
- Laitinen, I.; Marjamaki, P.; Nagren, K.; Laine, V.J.; Wilson, I.; Leppanen, P.; Yla-Herttuala, S.; Roivainen, A.; Knuuti, J. Uptake of inflammatory cell marker [11C]PK11195 into mouse atherosclerotic plaques. Eur. J. Nucl. Med. Mol. Imaging 2009, 36, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Mojtahedi, A.; Alavi, A.; Thamake, S.; Amerinia, R.; Ranganathan, D.; Tworowska, I.; Delpassand, E.S. Assessment of vulnerable atherosclerotic and fibrotic plaques in coronary arteries using (68)Ga-DOTATATE PET/CT. Am. J. Nucl. Med. Mol. Imaging 2015, 5, 65–71. [Google Scholar] [PubMed]
- Linton, M.R.F.; Yancey, P.G.; Davies, S.S.; Jerome, W.G.; Linton, E.F.; Song, W.L.; Doran, A.C.; Vickers, K.C. The Role of Lipids and Lipoproteins in Atherosclerosis. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., Dungan, K., Grossman, A., Hershman, J.M., Kaltsas, G., Koch, C., Kopp, P., et al., Eds.; MDText.com, Inc.: South Dartmouth, UK, 2000. [Google Scholar]
- Newby, A.C.; Zaltsman, A.B. Fibrous cap formation or destruction--the critical importance of vascular smooth muscle cell proliferation, migration and matrix formation. Cardiovasc. Res. 1999, 41, 345–360. [Google Scholar] [CrossRef] [Green Version]
- Cybulsky, M.I.; Iiyama, K.; Li, H.; Zhu, S.; Chen, M.; Iiyama, M.; Davis, V.; Gutierrez-Ramos, J.C.; Connelly, P.W.; Milstone, D.S. A major role for VCAM-1, but not ICAM-1, in early atherosclerosis. J. Clin. Investig. 2001, 107, 1255–1262. [Google Scholar] [CrossRef] [Green Version]
- Felton, C.V.; Crook, D.; Davies, M.J.; Oliver, M.F. Relation of plaque lipid composition and morphology to the stability of human aortic plaques. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 1337–1345. [Google Scholar] [CrossRef]
- Negre-Salvayre, A.; Garoby-Salom, S.; Swiader, A.; Rouahi, M.; Pucelle, M.; Salvayre, R. Proatherogenic effects of 4-hydroxynonenal. Free Radic. Biol. Med. 2017, 111, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Gargiulo, S.; Gamba, P.; Testa, G.; Rossin, D.; Biasi, F.; Poli, G.; Leonarduzzi, G. Relation between TLR4/NF-kappaB signaling pathway activation by 27-hydroxycholesterol and 4-hydroxynonenal, and atherosclerotic plaque instability. Aging cell 2015, 14, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Hamilton, R.F., Jr.; Kirichenko, A.; Holian, A. 4-Hydroxynonenal-induced cell death in murine alveolar macrophages. Toxicol. Appl. Pharmacol. 1996, 139, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Tsimikas, S.; Bergmark, C.; Beyer, R.W.; Patel, R.; Pattison, J.; Miller, E.; Juliano, J.; Witztum, J.L. Temporal increases in plasma markers of oxidized low-density lipoprotein strongly reflect the presence of acute coronary syndromes. J. Am. Coll. Cardiol. 2003, 41, 360–370. [Google Scholar] [CrossRef] [Green Version]
- Holvoet, P.; Vanhaecke, J.; Janssens, S.; Van de Werf, F.; Collen, D. Oxidized LDL and malondialdehyde-modified LDL in patients with acute coronary syndromes and stable coronary artery disease. Circulation 1998, 98, 1487–1494. [Google Scholar] [CrossRef] [Green Version]
- Nishi, K.; Itabe, H.; Uno, M.; Kitazato, K.T.; Horiguchi, H.; Shinno, K.; Nagahiro, S. Oxidized LDL in carotid plaques and plasma associates with plaque instability. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1649–1654. [Google Scholar] [CrossRef] [Green Version]
- Ehara, S.; Ueda, M.; Naruko, T.; Haze, K.; Itoh, A.; Otsuka, M.; Komatsu, R.; Matsuo, T.; Itabe, H.; Takano, T.; et al. Elevated levels of oxidized low density lipoprotein show a positive relationship with the severity of acute coronary syndromes. Circulation 2001, 103, 1955–1960. [Google Scholar] [CrossRef]
- Kato, R.; Mori, C.; Kitazato, K.; Arata, S.; Obama, T.; Mori, M.; Takahashi, K.; Aiuchi, T.; Takano, T.; Itabe, H. Transient increase in plasma oxidized LDL during the progression of atherosclerosis in apolipoprotein E knockout mice. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 33–39. [Google Scholar] [CrossRef]
- Tavridou, A.; Efthimiadis, A.; Efthimiadis, I.; Paschalidou, H. Antioxidant effects of simvastatin in primary and secondary prevention of coronary heart disease. Eur. J. Clin. Pharmacol. 2006, 62, 485–489. [Google Scholar] [CrossRef]
- Aydin, M.U.; Aygul, N.; Altunkeser, B.B.; Unlu, A.; Taner, A. Comparative effects of high-dose atorvastatin versus moderate-dose rosuvastatin on lipid parameters, oxidized-LDL and inflammatory markers in ST elevation myocardial infarction. Atherosclerosis 2015, 239, 439–443. [Google Scholar] [CrossRef]
- Ndrepepa, G.; Braun, S.; von Beckerath, N.; Mehilli, J.; Gorchakova, O.; Vogt, W.; Schomig, A.; Kastrati, A. Oxidized low density lipoproteins, statin therapy and severity of coronary artery disease. Clin. Chim. Acta 2005, 360, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Endemann, G.; Stanton, L.W.; Madden, K.S.; Bryant, C.M.; White, R.T.; Protter, A.A. CD36 is a receptor for oxidized low density lipoprotein. J. Biol. Chem. 1993, 268, 11811–11816. [Google Scholar]
- Stewart, C.R.; Stuart, L.M.; Wilkinson, K.; van Gils, J.M.; Deng, J.; Halle, A.; Rayner, K.J.; Boyer, L.; Zhong, R.; Frazier, W.A.; et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 2010, 11, 155–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansson, G.K.; Robertson, A.K.; Soderberg-Naucler, C. Inflammation and atherosclerosis. Annu. Rev. Pathol. 2006, 1, 297–329. [Google Scholar] [CrossRef]
- Persson, J.; Nilsson, J.; Lindholm, M.W. Interleukin-1beta and tumour necrosis factor-alpha impede neutral lipid turnover in macrophage-derived foam cells. BMC Immunol. 2008, 9, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, K.J.; Sheedy, F.J.; Fisher, E.A. Macrophages in atherosclerosis: A dynamic balance. Nat. Rev. Immunol. 2013, 13, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Zapolska-Downar, D.; Zapolski-Downar, A.; Markiewski, M.; Ciechanowicz, A.; Kaczmarczyk, M.; Naruszewicz, M. Selective inhibition by probucol of vascular cell adhesion molecule-1 (VCAM-1) expression in human vascular endothelial cells. Atherosclerosis 2001, 155, 123–130. [Google Scholar] [CrossRef]
- Fruebis, J.; Silvestre, M.; Shelton, D.; Napoli, C.; Palinski, W. Inhibition of VCAM-1 expression in the arterial wall is shared by structurally different antioxidants that reduce early atherosclerosis in NZW rabbits. J. Lipid Res. 1999, 40, 1958–1966. [Google Scholar]
- Leiter, E.; Emri, T.; Gyemant, G.; Nagy, I.; Pocsi, I.; Winkelmann, G.; Pocsi, I. Penicillin V production by Penicillium chrysogenum in the presence of Fe3+ and in low-iron culture medium. Folia. Microbiol. 2001, 46, 127–132. [Google Scholar] [CrossRef]
- Balla, J.; Jacob, H.S.; Balla, G.; Nath, K.; Eaton, J.W.; Vercellotti, G.M. Endothelial-cell heme uptake from heme proteins: Induction of sensitization and desensitization to oxidant damage. Proc. Natl. Acad. Sci. USA 1993, 90, 9285–9289. [Google Scholar] [CrossRef] [Green Version]
- Winterbourn, C.C. Oxidative reactions of hemoglobin. Methods Enzymol. 1990, 186, 265–272. [Google Scholar] [PubMed]
- Barger, A.C.; Beeuwkes, R., 3rd; Lainey, L.L.; Silverman, K.J. Hypothesis: Vasa vasorum and neovascularization of human coronary arteries. A possible role in the pathophysiology of atherosclerosis. N. Engl. J. Med. 1984, 310, 175–177. [Google Scholar] [CrossRef] [PubMed]
- Nagy, E.; Jeney, V.; Yachie, A.; Szabo, R.P.; Wagner, O.; Vercellotti, G.M.; Eaton, J.W.; Balla, G.; Balla, J. Oxidation of hemoglobin by lipid hydroperoxide associated with low-density lipoprotein (LDL) and increased cytotoxic effect by LDL oxidation in heme oxygenase-1 (HO-1) deficiency. Cell. Mol. Biol. (Noisy-le-grand) 2005, 51, 377–385. [Google Scholar]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Potor, L.; Sikura, K.É.; Hegedűs, H.; Pethő, D.; Szabó, Z.; Szigeti, Z.M.; Pócsi, I.; Trencsényi, G.; Szikra, D.; Garai, I.; et al. The Fungal Iron Chelator Desferricoprogen Inhibits Atherosclerotic Plaque Formation. Int. J. Mol. Sci. 2020, 21, 4746. https://doi.org/10.3390/ijms21134746
Potor L, Sikura KÉ, Hegedűs H, Pethő D, Szabó Z, Szigeti ZM, Pócsi I, Trencsényi G, Szikra D, Garai I, et al. The Fungal Iron Chelator Desferricoprogen Inhibits Atherosclerotic Plaque Formation. International Journal of Molecular Sciences. 2020; 21(13):4746. https://doi.org/10.3390/ijms21134746
Chicago/Turabian StylePotor, László, Katalin Éva Sikura, Hajnalka Hegedűs, Dávid Pethő, Zsuzsa Szabó, Zsuzsa M Szigeti, István Pócsi, György Trencsényi, Dezső Szikra, Ildikó Garai, and et al. 2020. "The Fungal Iron Chelator Desferricoprogen Inhibits Atherosclerotic Plaque Formation" International Journal of Molecular Sciences 21, no. 13: 4746. https://doi.org/10.3390/ijms21134746