Multifactorial Activation of NLRP3 Inflammasome: Relevance for a Precision Approach to Atherosclerotic Cardiovascular Risk and Disease



Abstract
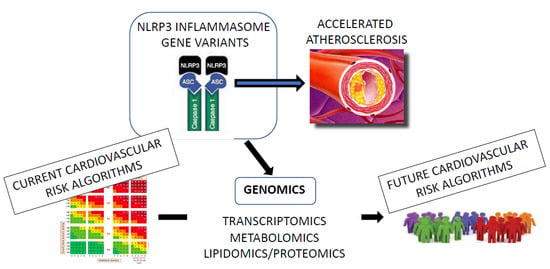
:
1. Chronic Low-Grade Inflammation and Atherosclerotic Cardiovascular Risk and Disease: Moving Above and Beyond the Lipid Profile?
- To discuss the role of different factors activating the NLRP3 inflammasome-IL-1β pathway in the progression of ASCVD, highlighting the related molecular mechanisms leading to low-grade inflammation;
- To report the current evidence about the individual impact of such pro-inflammatory mechanisms on the personal ASCVD risk, in the context of a precision medicine approach to CVD risk assessment and prevention, based on genomic analysis.
2. NLRP3 Inflammasome Structure and Function
2.1. NLRP3 Inflammasome Functions
2.2. Molecular Structure of NLRP3 Inflammasome
3. NLRP3 Inflammasome Activation by Common Ligands
4. Multiple Pathogenic Factors Converge to Promote ASCVD through Modulation of NLRP3 Inflammasome
4.1. Nutrition
4.2. Microbiota
4.3. Cholesterol
5. Role of NLRP3 Inflammasome Genetic Variants in ASCVD Pathophysiology
6. Identification of NLRP3-Related Gene Variants: A Useful Tool to Refine CVD Risk Assessment?
Author Contributions
Funding
Conflicts of Interest
References
- Libby, P.; Hansson, G.K. From Focal Lipid Storage to Systemic Inflammation: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2019, 74, 1594–1607. [Google Scholar] [CrossRef]
- Zhang, W.; Song, M.; Qu, J.; Liu, G.H. Epigenetic Modifications in Cardiovascular Aging and Diseases. Circ. Res. 2018, 123, 773–786. [Google Scholar] [CrossRef] [PubMed]
- Ference, B.A.; Ginsberg, H.N.; Graham, I.; Ray, K.K.; Packard, C.J.; Bruckert, E.; Hegele, R.A.; Krauss, R.M.; Raal, F.J.; Schunkert, H.; et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2017, 38, 2459–2472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Heart J. 2019, 290, 140–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahlin, G.; Hilgendorf, C.; Karlsson, J.; Szigyarto, C.A.; Uhlén, M.; Artursson, P. Endogenous gene and protein expression of drug-transporting proteins in cell lines routinely used in drug discovery programs. Drug Metab. Dispos. 2009, 37, 2275–2283. [Google Scholar] [CrossRef] [Green Version]
- D’Agostino, R.B., Sr.; Vasan, R.S.; Pencina, M.J.; Wolf, P.A.; Cobain, M.; Massaro, J.M.; Kannel, W.B. General cardiovascular risk profile for use in primary care: The Framingham Heart Study. Circulation 2008, 117, 743–753. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Fulcher, J.; Abeysuriya, N.; Park, L.; Kumar, S.; Di Tanna, G.L.; Wilcox, I.; Keech, A.; Rodgers, A.; Lal, S. Intensive LDL cholesterol-lowering treatment beyond current recommendations for the prevention of major vascular events: A systematic review and meta-analysis of randomised trials including 327,037 participants. Lancet Diabetes Endocrinol. 2020, 8, 36–49. [Google Scholar] [CrossRef]
- Neeland, I.J.; Ross, R.; Després, J.P.; Matsuzawa, Y.; Yamashita, S.; Shai, I.; Seidell, J.; Magni, P.; Santos, R.D.; Arsenault, B.; et al. Visceral and ectopic fat, atherosclerosis, and cardiometabolic disease: A position statement. Lancet Diabetes Endocrinol. 2019, 7, 715–725. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Baldrighi, M.; Mallat, Z.; Li, X. NLRP3 inflammasome pathways in atherosclerosis. Atherosclerosis 2017, 267, 127–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridker, P.M. Anti-inflammatory therapy for atherosclerosis: Interpreting divergent results from the CANTOS and CIRT clinical trials. J. Intern. Med. 2019, 285, 503–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Kayagaki, N.; Wong, M.T.; Stowe, I.B.; Ramani, S.R.; Gonzalez, L.C.; Akashi-Takamura, S.; Miyake, K.; Zhang, J.; Lee, W.P.; Muszynski, A.; et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 2013, 341, 1246–1249. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Dixit, V.M. Mechanisms and functions of inflammasomes. Cell 2014, 157, 1013–1022. [Google Scholar] [CrossRef]
- Duncan, J.A.; Bergstralh, D.T.; Wang, Y.; Willingham, S.B.; Ye, Z.; Zimmermann, A.G.; Ting, J.P. Cryopyrin/NALP3 binds ATP/dATP, is an ATPase, and requires ATP binding to mediate inflammatory signaling. Proc. Natl. Acad. Sci. USA 2007, 104, 8041–8046. [Google Scholar] [CrossRef] [Green Version]
- Masumoto, J.; Taniguchi, S.; Ayukawa, K.; Sarvotham, H.; Kishino, T.; Niikawa, N.; Hidaka, E.; Katsuyama, T.; Higuchi, T.; Sagara, J. ASC, a novel 22-kDa protein, aggregates during apoptosis of human promyelocytic leukemia HL-60 cells. J. Biol. Chem. 1999, 274, 33835–33838. [Google Scholar] [CrossRef] [Green Version]
- Srinivasula, S.M.; Poyet, J.L.; Razmara, M.; Datta, P.; Zhang, Z.; Alnemri, E.S. The PYRIN-CARD protein ASC is an activating adaptor for caspase-1. J. Biol. Chem. 2002, 277, 21119–21122. [Google Scholar] [CrossRef] [Green Version]
- Agostini, L.; Martinon, F.; Burns, K.; McDermott, M.F.; Hawkins, P.N.; Tschopp, J. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity 2004, 20, 319–325. [Google Scholar] [CrossRef] [Green Version]
- Fernandes-Alnemri, T.; Kang, S.; Anderson, C.; Sagara, J.; Fitzgerald, K.A.; Alnemri, E.S. Cutting edge: TLR signaling licenses IRAK1 for rapid activation of the NLRP3 inflammasome. J. Immunol. 2013, 191, 3995–3999. [Google Scholar] [CrossRef]
- Lin, K.M.; Hu, W.; Troutman, T.D.; Jennings, M.; Brewer, T.; Li, X.; Nanda, S.; Cohen, P.; Thomas, J.A.; Pasare, C. IRAK-1 bypasses priming and directly links TLRs to rapid NLRP3 inflammasome activation. Proc. Natl. Acad. Sci. USA 2014, 111, 775–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauernfeind, F.; Bartok, E.; Rieger, A.; Franchi, L.; Nunez, G.; Hornung, V. Cutting edge: Reactive oxygen species inhibitors block priming, but not activation, of the NLRP3 inflammasome. J. Immunol. 2011, 187, 613–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munoz-Planillo, R.; Kuffa, P.; Martinez-Colon, G.; Smith, B.L.; Rajendiran, T.M.; Nunez, G. K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 2013, 38, 1142–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hornung, V.; Bauernfeind, F.; Halle, A.; Samstad, E.O.; Kono, H.; Rock, K.L.; Fitzgerald, K.A.; Latz, E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 2008, 9, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Misawa, T.; Takahama, M.; Kozaki, T.; Lee, H.; Zou, J.; Saitoh, T.; Akira, S. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat. Immunol. 2013, 14, 454–460. [Google Scholar] [CrossRef]
- Subramanian, N.; Natarajan, K.; Clatworthy, M.R.; Wang, Z.; Germain, R.N. The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell 2013, 153, 348–361. [Google Scholar] [CrossRef] [Green Version]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [Green Version]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [Green Version]
- Iyer, S.S.; He, Q.; Janczy, J.R.; Elliott, E.I.; Zhong, Z.; Olivier, A.K.; Sadler, J.J.; Knepper-Adrian, V.; Han, R.; Qiao, L.; et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 2013, 39, 311–323. [Google Scholar] [CrossRef] [Green Version]
- Juliana, C.; Fernandes-Alnemri, T.; Kang, S.; Farias, A.; Qin, F.; Alnemri, E.S. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J. Biol. Chem. 2012, 287, 36617–36622. [Google Scholar] [CrossRef] [Green Version]
- Groslambert, M.; Py, B.F. Spotlight on the NLRP3 inflammasome pathway. J. Inflamm. Res. 2018, 11, 359–374. [Google Scholar] [CrossRef] [Green Version]
- Schroder, K.; Sagulenko, V.; Zamoshnikova, A.; Richards, A.A.; Cridland, J.A.; Irvine, K.M.; Stacey, K.J.; Sweet, M.J. Acute lipopolysaccharide priming boosts inflammasome activation independently of inflammasome sensor induction. Immunobiology 2012, 217, 1325–1329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satish, M.; Agrawal, D.K. Atherothrombosis and the NLRP3 inflammasome - endogenous mechanisms of inhibition. Transl. Res. 2020, 215, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Joosten, L.A.; Latz, E.; Mills, K.H.; Natoli, G.; Stunnenberg, H.G.; O’Neill, L.A.; Xavier, R.J. Trained immunity: A program of innate immune memory in health and disease. Science 2016, 352, aaf1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iqbal, R.; Anand, S.; Ounpuu, S.; Islam, S.; Zhang, X.; Rangarajan, S.; Chifamba, J.; Al-Hinai, A.; Keltai, M.; Yusuf, S.; et al. Dietary patterns and the risk of acute myocardial infarction in 52 countries: Results of the INTERHEART study. Circulation 2008, 118, 1929–1937. [Google Scholar] [CrossRef] [Green Version]
- Pavillard, L.E.; Marín-Aguilar, F.; Bullon, P.; Cordero, M.D. Cardiovascular diseases, NLRP3 inflammasome, and western dietary patterns. Pharmacol. Res. 2018, 131, 44–50. [Google Scholar] [CrossRef]
- Legrand-Poels, S.; Esser, N.; L’homme, L.; Scheen, A.; Paquot, N.; Piette, J. Free fatty acids as modulators of the NLRP3 inflammasome in obesity/type 2 diabetes. Biochem. Pharmacol. 2014, 92, 131–141. [Google Scholar] [CrossRef]
- Norata, G.D.; Raselli, S.; Grigore, L.; Garlaschelli, K.; Dozio, E.; Magni, P.; Catapano, A.L. Leptin:adiponectin ratio is an independent predictor of intima media thickness of the common carotid artery. Stroke 2007, 38, 2844–2846. [Google Scholar] [CrossRef] [Green Version]
- Esser, N.; L’homme, L.; De Roover, A.; Kohnen, L.; Scheen, A.J.; Moutschen, M.; Piette, J.; Legrand-Poels, S.; Paquot, N. Obesity phenotype is related to NLRP3 inflammasome activity and immunological profile of visceral adipose tissue. Diabetologia 2013, 56, 2487–2497. [Google Scholar] [CrossRef] [Green Version]
- Ross, R.; Neeland, I.J.; Yamashita, S.; Shai, I.; Seidell, J.; Magni, P.; Santos, R.D.; Arsenault, B.; Cuevas, A.; Hu, F.B.; et al. Waist circumference as a vital sign in clinical practice: A Consensus Statement from the IAS and ICCR Working Group on Visceral Obesity. Nat. Rev. Endocrinol. 2020, 16, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Bertoli, S.; Magni, P.; Krogh, V.; Ruscica, M.; Dozio, E.; Testolin, G.; Battezzati, A. Is ghrelin a signal of decreased fat-free mass in elderly subjects? Eur. J. Endocrinol. 2006, 155, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Comassi, M.; Santini, E.; Rossi, C.; Vitolo, E.; Seghieri, M.; Tocchini, L.; Franzoni, F.; Solini, A. The level of physical training modulates cytokine levels through P2X7 receptor in healthy subjects. Eur. J. Clin. Invest. 2018, 48. [Google Scholar] [CrossRef] [PubMed]
- Branchereau, M.; Burcelin, R.; Heymes, C. The gut microbiome and heart failure: A better gut for a better heart. Rev. Endocr. Metab. Dis. 2019, 20, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, C.; Antonioli, L.; Lopez-Castejon, G.; Blandizzi, C.; Fornai, M. Canonical and Non-Canonical Activation of NLRP3 Inflammasome at the Crossroad between Immune Tolerance and Intestinal Inflammation. Front. Immunol. 2017, 8, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. A core gut microbiome in obese and lean twins. Nature 2008, 457, 480–484. [Google Scholar] [CrossRef] [Green Version]
- Beer, K. The Gut Microbiome in Type 2 Diabetes. Clin. Rev. 2018, 28, 13–16. [Google Scholar]
- Lau, K.; Srivatsav, V.; Rizwan, A.; Nashed, A.; Liu, R.; Shen, R.; Akhtar, M. Bridging the Gap between Gut Microbial Dysbiosis and Cardiovascular Diseases. Nutrients 2017, 9, 859. [Google Scholar] [CrossRef] [Green Version]
- Lopetuso, L.R.; Scaldaferri, F.; Bruno, G.; Petito, V.; Franceschi, F.; Gasbarrini, A. The therapeutic management of gut barrier leaking: The emerging role for mucosal barrier protectors. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 1068–1076. [Google Scholar]
- Spadoni, I.; Zagato, E.; Bertocchi, A.; Paolinelli, R.; Hot, E.; Di Sabatino, A.; Caprioli, F.; Bottiglieri, L.; Oldani, A.; Viale, G.; et al. A gut-vascular barrier controls the systemic dissemination of bacteria. Science 2015, 350, 830–834. [Google Scholar] [CrossRef]
- Liu, X.R.; Xu, Q.; Xiao, J.; Deng, Y.M.; Tang, Z.H.; Tang, Y.L.; Liu, L.S. Role of oral microbiota in atherosclerosis. Clin. Chim. Acta. 2020, 506, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Karasawa, T.; Takahashi, M. Role of NLRP3 Inflammasomes in Atherosclerosis. J. Atheroscler. Thromb. 2017, 24, 443–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendrikx, T.; Jeurissen, M.L.; van Gorp, P.J.; Gijbels, M.J.; Walenbergh, S.M.; Houben, T.; van Gorp, R.; Pottgens, C.C.; Stienstra, R.; Netea, M.G.; et al. Bone marrow-specific caspase-1/11 deficiency inhibits atherosclerosis development in Ldlr(-/-) mice. Febs. J. 2015, 282, 2327–2338. [Google Scholar] [CrossRef]
- Christ, A.; Gunther, P.; Lauterbach, M.A.R.; Duewell, P.; Biswas, D.; Pelka, K.; Scholz, C.J.; Oosting, M.; Haendler, K.; Bassler, K.; et al. Western Diet Triggers NLRP3-Dependent Innate Immune Reprogramming. Cell 2018, 172, 162–175. [Google Scholar] [CrossRef] [Green Version]
- Pirillo, A.; Bonacina, F.; Norata, G.D.; Catapano, A.L. The Interplay of Lipids, Lipoproteins, and Immunity in Atherosclerosis. Curr. Atheroscler. Rep. 2018, 20, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paramel, G.V.; Sirsjö, A.; Fransén, K. Role of genetic alterations in the NLRP3 and CARD8 genes in health and disease. Mediat. Inflamm. 2015, 2015, 846782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, H.; Okayama, N.; Yamaguchi, N.; Miyahara, Y.; Morishima, Y.; Suehiro, Y.; Yamasaki, T.; Tamada, K.; Takahashi, S.; Tojo, A.; et al. Associations of interactions between NLRP3 SNPs and HLA mismatch with acute and extensive chronic graft-versus-host diseases. Sci. Rep. 2017, 7, 13097. [Google Scholar] [CrossRef] [Green Version]
- Klen, J.; Goričar, K.; Janež, A.; Dolžan, V. NLRP3 Inflammasome Polymorphism and Macrovascular Complications in Type 2 Diabetes Patients. J. Diabetes Res. 2015, 2015, 616747. [Google Scholar] [CrossRef] [Green Version]
- Cordero, M.D.; Alcocer-Gómez, E.; Ryffel, B. Gain of function mutation and inflammasome driven diseases in human and mouse models. J. Autoimmun. 2018, 91, 13–22. [Google Scholar] [CrossRef]
- Hoffman, H.M.; Mueller, J.L.; Broide, D.H.; Wanderer, A.A.; Kolodner, R.D. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat. Genet. 2001, 29, 301–305. [Google Scholar] [CrossRef] [Green Version]
- Dowds, T.A.; Masumoto, J.; Zhu, L.; Inohara, N.; Núñez, G. Cryopyrin-induced interleukin 1beta secretion in monocytic cells: Enhanced activity of disease-associated mutants and requirement for ASC. J. Biol. Chem. 2004, 279, 21924–21928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcocer-Gómez, E.; Castejón-Vega, B.; López-Sánchez, M.; Cordero, M.D. Inflammasomes in Clinical Practice: A Brief Introduction. Exp. Suppl. 2018, 108, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Varghese, G.P.; Fransén, K.; Hurtig-Wennlöf, A.; Bengtsson, T.; Jansson, J.H.; Sirsjö, A. Q705K variant in NLRP3 gene confers protection against myocardial infarction in female individuals. Biomed. Rep. 2013, 1, 879–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, R.L.; Van Rij, A.M.; Phillips, L.V.; Young, S.; McCormick, S.P.; Merriman, T.R.; Jones, G.T. Interaction of the inflammasome genes CARD8 and NLRP3 in abdominal aortic aneurysms. Atherosclerosis 2011, 218, 123–126. [Google Scholar] [CrossRef]
- Paramel, G.V.; Folkersen, L.; Strawbridge, R.J.; Elmabsout, A.A.; Särndahl, E.; Lundman, P.; Jansson, J.H.; Hansson, G.K.; Sirsjö, A.; Fransén, K. CARD8 gene encoding a protein of innate immunity is expressed in human atherosclerosis and associated with markers of inflammation. Clin. Sci. 2013, 125, 401–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, Y.; Nie, S.; Jiang, G.; Zhou, Y.; Zhou, M.; Zhao, Y.; Li, S.; Wang, F.; Lv, Q.; Huang, Y.; et al. Regulation of CARD8 expression by ANRIL and association of CARD8 single nucleotide polymorphism rs2043211 (p.C10X) with ischemic stroke. Stroke 2014, 45, 383–388. [Google Scholar] [CrossRef] [Green Version]
- García-Bermúdez, M.; López-Mejías, R.; González-Juanatey, C.; Corrales, A.; Castañeda, S.; Ortiz, A.M.; Miranda-Filloy, J.A.; Gómez-Vaquero, C.; Fernández-Gutiérrez, B.; Balsa, A.; et al. CARD8 rs2043211 (p.C10X) polymorphism is not associated with disease susceptibility or cardiovascular events in Spanish rheumatoid arthritis patients. DNA Cell Biol. 2013, 32, 28–33. [Google Scholar] [CrossRef] [Green Version]
- Raal, F.J.; Hovingh, G.K.; Catapano, A.L. Familial hypercholesterolemia treatments: Guidelines and new therapies. Atherosclerosis 2018, 277, 483–492. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Dron, J.S.; Ban, M.R.; Robinson, J.F.; McIntyre, A.D.; Alazzam, M.; Zhao, P.J.; Dilliott, A.A.; Cao, H.; Huff, M.W.; et al. Polygenic Versus Monogenic Causes of Hypercholesterolemia Ascertained Clinically. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2439–2445. [Google Scholar] [CrossRef] [Green Version]
- Zannad, F.; De Backer, G.; Graham, I.; Lorenz, M.; Mancia, G.; Morrow, D.A.; Reiner, Z.; Koenig, W.; Dallongeville, J.; Macfadyen, R.J.; et al. Risk stratification in cardiovascular disease primary prevention - scoring systems, novel markers, and imaging techniques. Fund. Clin. Pharmacol. 2012, 26, 163–174. [Google Scholar] [CrossRef]
- Ference, B.A.; Bhatt, D.L.; Catapano, A.L.; Packard, C.J.; Graham, I.; Kaptoge, S.; Ference, T.B.; Guo, Q.; Laufs, U.; Ruff, C.T.; et al. Association of Genetic Variants Related to Combined Exposure to Lower Low-Density Lipoproteins and Lower Systolic Blood Pressure With Lifetime Risk of Cardiovascular Disease. JAMA 2019, 322, 1381–1391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klarin, D.; Damrauer, S.M.; Cho, K.; Sun, Y.V.; Teslovich, T.M.; Honerlaw, J.; Gagnon, D.R.; DuVall, S.L.; Li, J.; Peloso, G.M.; et al. Genetics of blood lipids among ~300,000 multi-ethnic participants of the Million Veteran Program. Nat. Genet. 2018, 50, 1514–1523. [Google Scholar] [CrossRef]
- Dainis, A.M.; Ashley, E.A. Cardiovascular Precision Medicine in the Genomics Era. JACC Basic Transl. Sci. 2018, 3, 313–326. [Google Scholar] [CrossRef]
- Leopold, J.A.; Loscalzo, J. Emerging Role of Precision Medicine in Cardiovascular Disease. Circ. Res. 2018, 122, 1302–1315. [Google Scholar] [CrossRef] [PubMed]
- Chun, S.; Imakaev, M.; Hui, D.; Patsopoulos, N.A.; Neale, B.M.; Kathiresan, S.; Stitziel, N.O.; Sunyaev, S.R. Non-parametric Polygenic Risk Prediction via Partitioned GWAS Summary Statistics. Am. J. Hum. Genet. 2020. [Google Scholar] [CrossRef]
- Dron, J.S.; Hegele, R.A. The evolution of genetic-based risk scores for lipids and cardiovascular disease. Curr. Opin. Lipidol. 2019, 30, 71–81. [Google Scholar] [CrossRef]
- Wierzbicki, A.S.; Reynolds, T.M. Genetic risk scores in lipid disorders. Curr. Opin. Cardiol. 2019, 34, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Shao, B.Z.; Xu, Z.Q.; Han, B.Z.; Su, D.F.; Liu, C. NLRP3 inflammasome and its inhibitors: A review. Front. Pharmacol. 2015, 6, 262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zahid, A.; Li, B.; Kombe, A.J.K.; Jin, T.; Tao, J. Pharmacological Inhibitors of the NLRP3 Inflammasome. Front. Immunol. 2019, 10, 2538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbate, A.; Toldo, S.; Marchetti, C.; Kron, J.; Van Tassell, B.W.; Dinarello, C.A. Interleukin-1 and the Inflammasome as Therapeutic Targets in Cardiovascular Disease. Circ. Res. 2020, 126, 1260–1280. [Google Scholar] [CrossRef]
- Koushki, K.; Shahbaz, S.K.; Mashayekhi, K.; Sadeghi, M.; Zayeri, Z.D.; Taba, M.Y.; Banach, M.; Al-Rasadi, K.; Johnston, T.P.; Sahebkar, A. Anti-inflammatory Action of Statins in Cardiovascular Disease: The Role of Inflammasome and Toll-Like Receptor Pathways. Clin. Rev. Allergy Immunol. 2020, 32. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Gene | Single Nucleotide Polymorphism |
|---|---|
| NACHT leucine-rich repeat- and PYD-containing (NLRP)3 inflammasome | rs4353135, rs4266924, rs6672995, rs10733113, rs107635144, rs55646866, rs35829419 |
| Caspase recruitment domain–containing protein (CARD)8 | rs2043211, rs1972619 |
| Caspase-1 | rs501192, rs556205 and rs530537 |
| IL-1 β | IL-1β (-511 and +3954) |
| IL-6 | (-G174C); rs1800795) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baragetti, A.; Catapano, A.L.; Magni, P. Multifactorial Activation of NLRP3 Inflammasome: Relevance for a Precision Approach to Atherosclerotic Cardiovascular Risk and Disease. Int. J. Mol. Sci. 2020, 21, 4459. https://doi.org/10.3390/ijms21124459
Baragetti A, Catapano AL, Magni P. Multifactorial Activation of NLRP3 Inflammasome: Relevance for a Precision Approach to Atherosclerotic Cardiovascular Risk and Disease. International Journal of Molecular Sciences. 2020; 21(12):4459. https://doi.org/10.3390/ijms21124459
Chicago/Turabian StyleBaragetti, Andrea, Alberico Luigi Catapano, and Paolo Magni. 2020. "Multifactorial Activation of NLRP3 Inflammasome: Relevance for a Precision Approach to Atherosclerotic Cardiovascular Risk and Disease" International Journal of Molecular Sciences 21, no. 12: 4459. https://doi.org/10.3390/ijms21124459