Selective Autophagy Maintains the Aryl Hydrocarbon Receptor Levels in HeLa Cells: A Mechanism That Is Dependent on the p23 Co-Chaperone

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Autophagy Activity in Wild Type and p23 Stable Knockdown HeLa Cells Governs the Levels of AHR
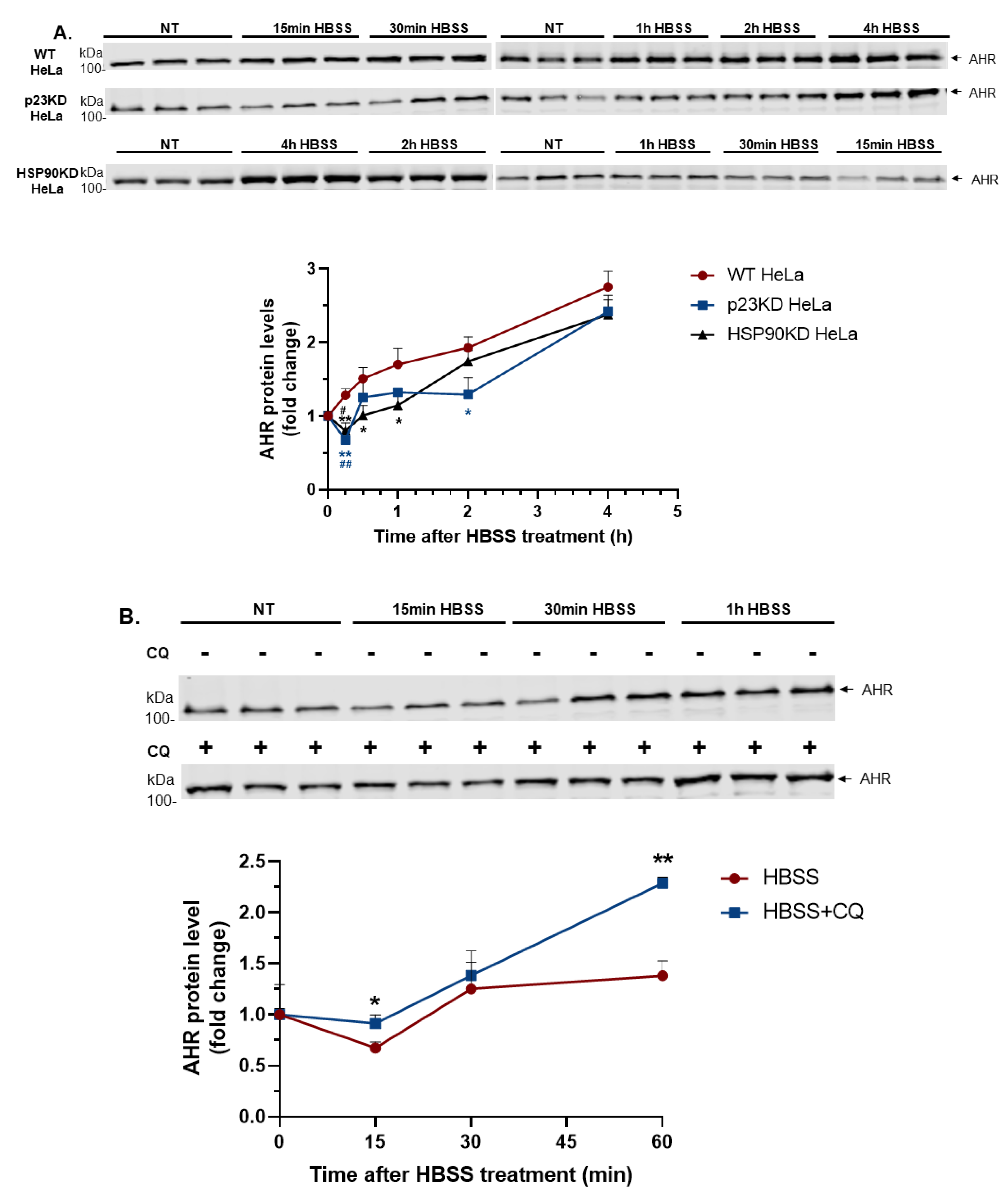
2.2. Short-Term Nutrient Deprivation Triggers Degradation of AHR in HeLa Cells When Either p23 or HSP90 is Down-Regulated
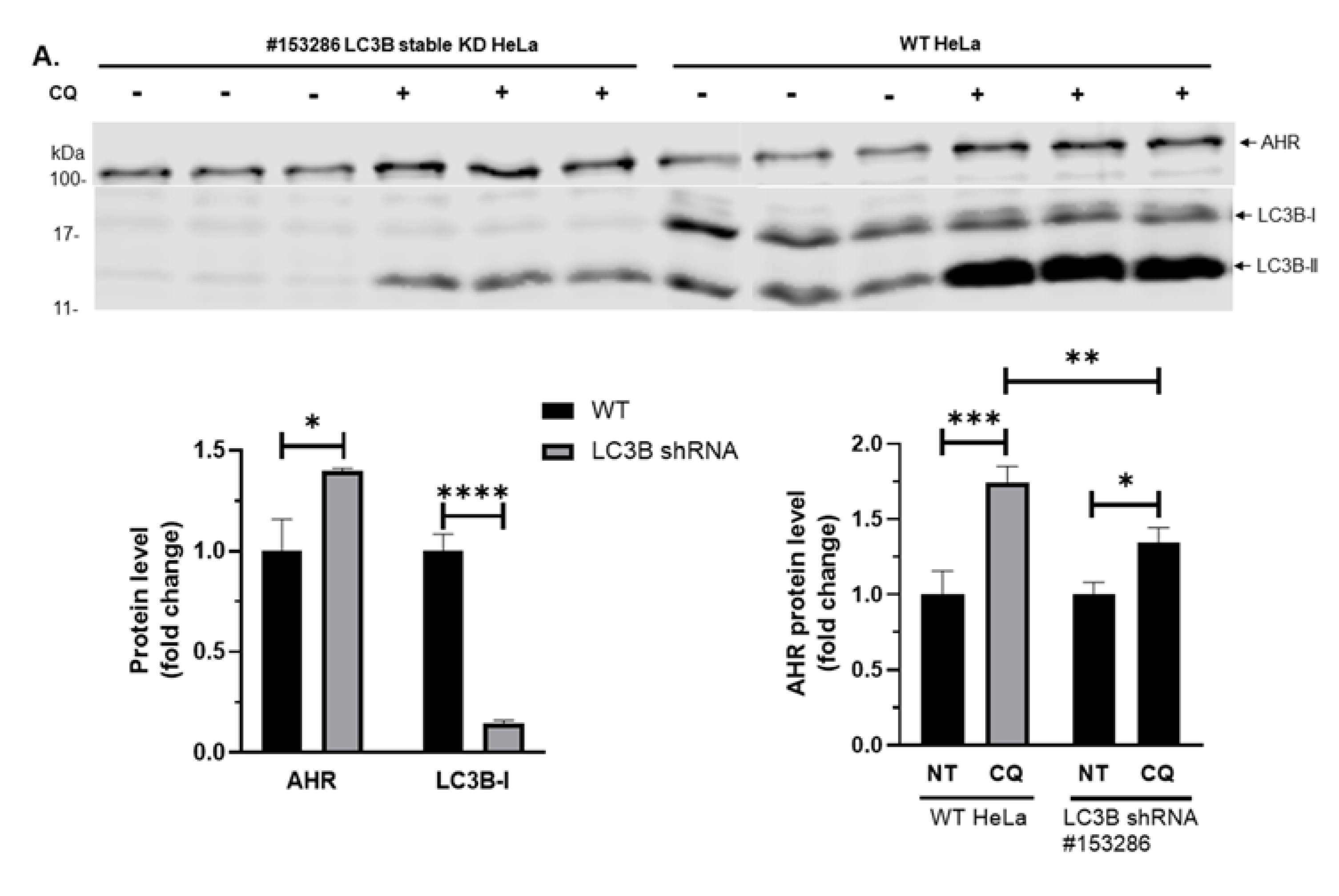
2.3. Down-Regulation of p23 in HeLa Cells Exhibits Higher Autophagic Flux
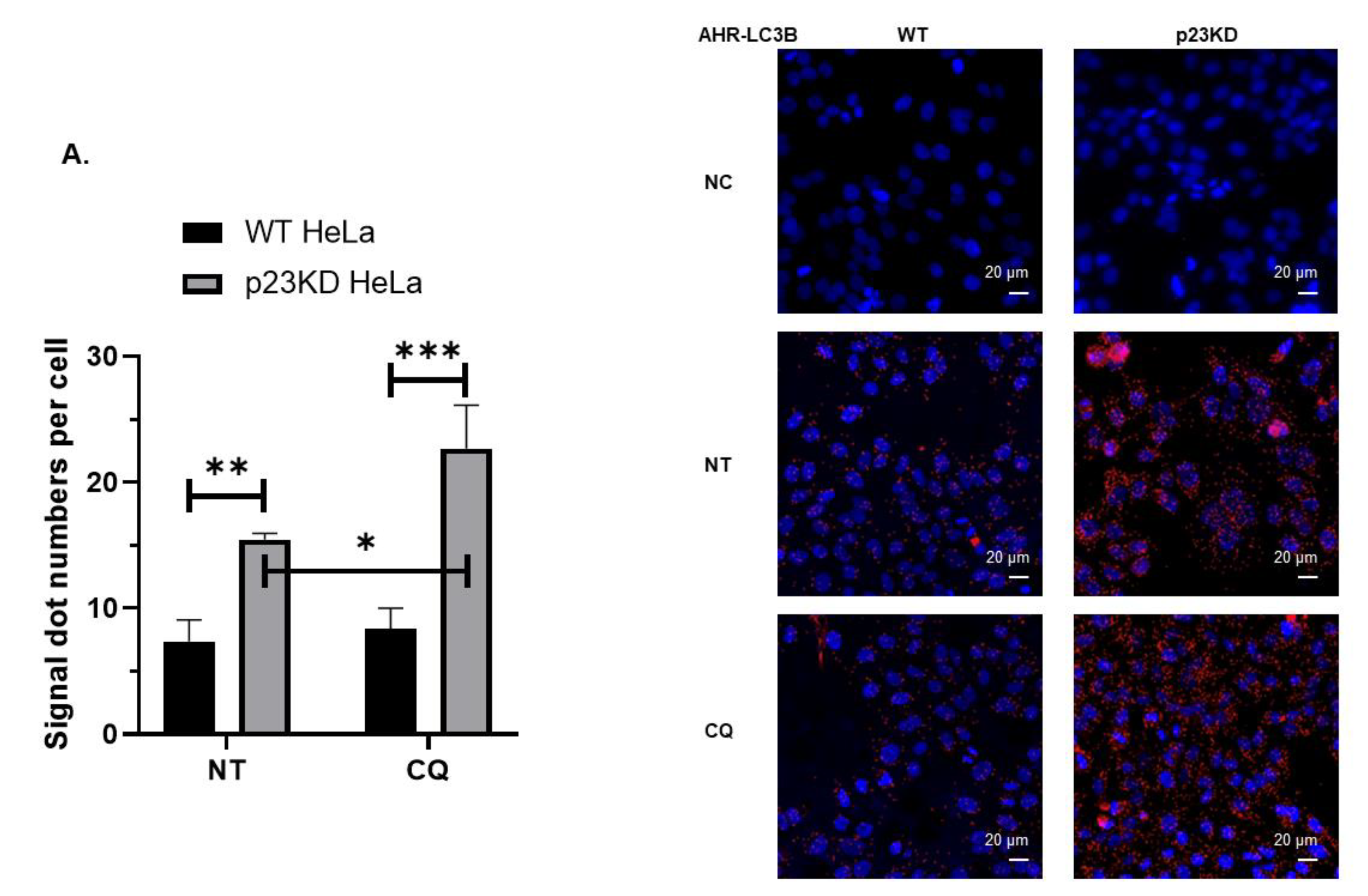
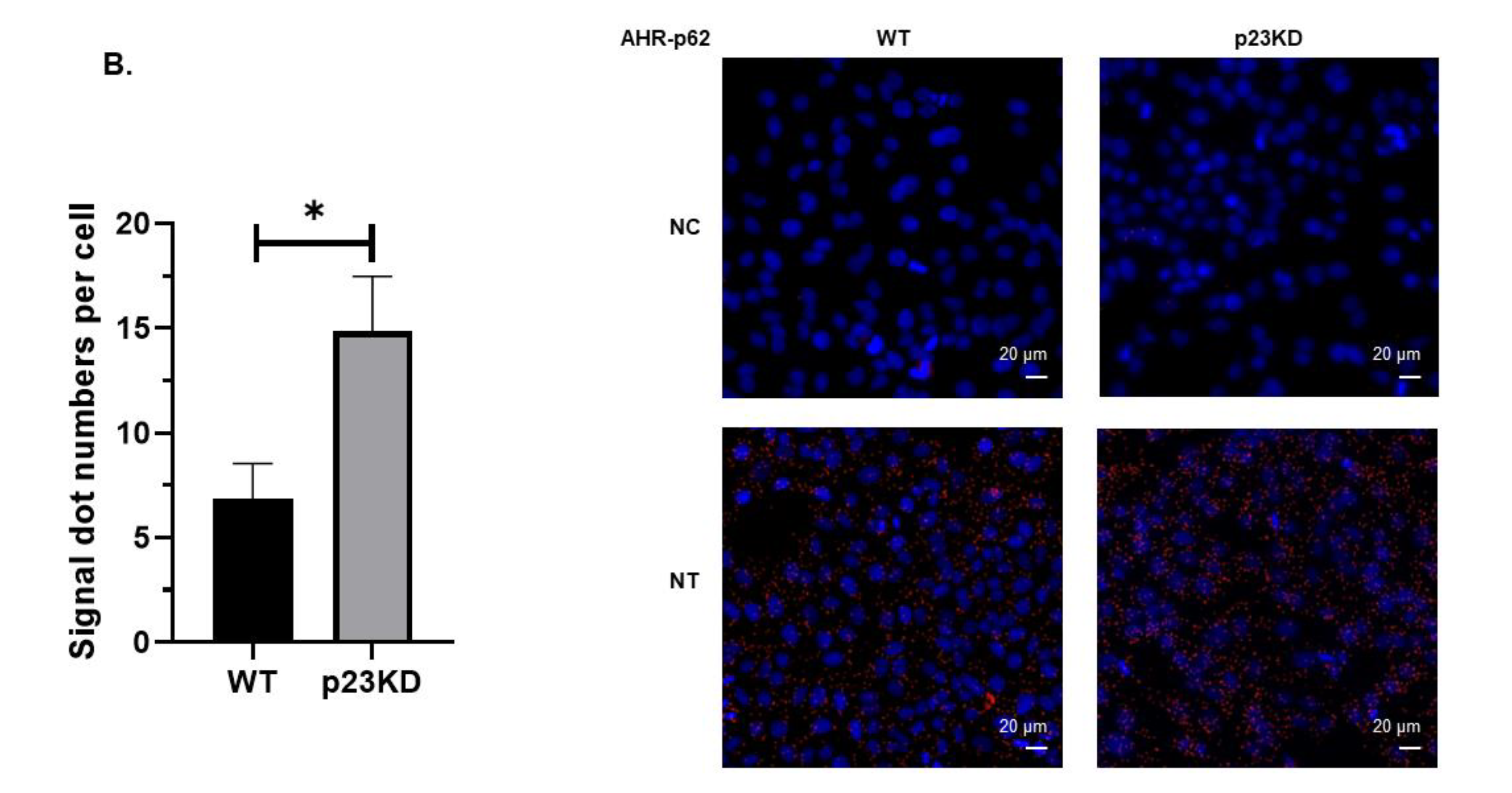
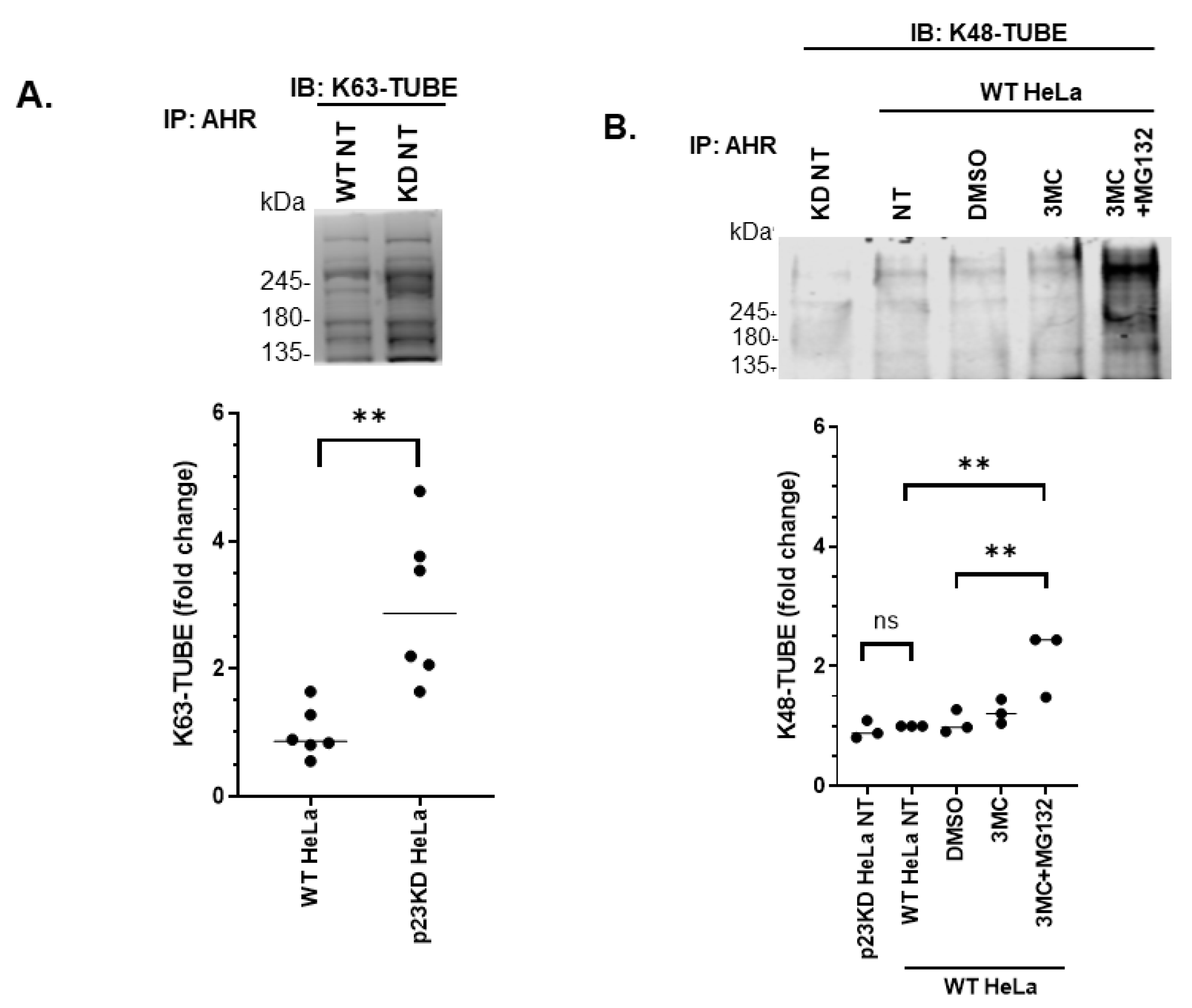
2.4. AHR Undergoes Selective Autophagy in p23 Knockdown and Wild Type HeLa Cells
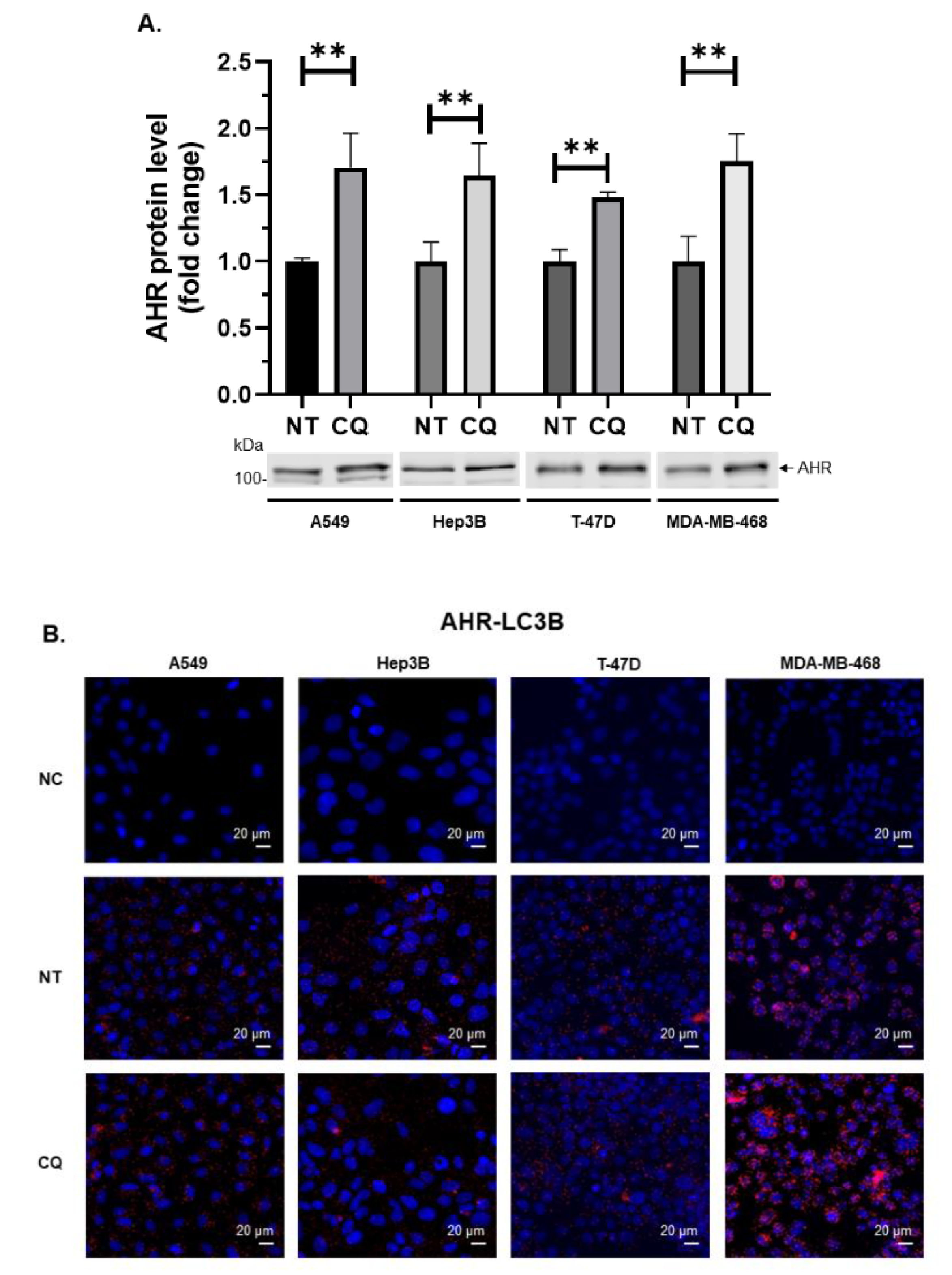
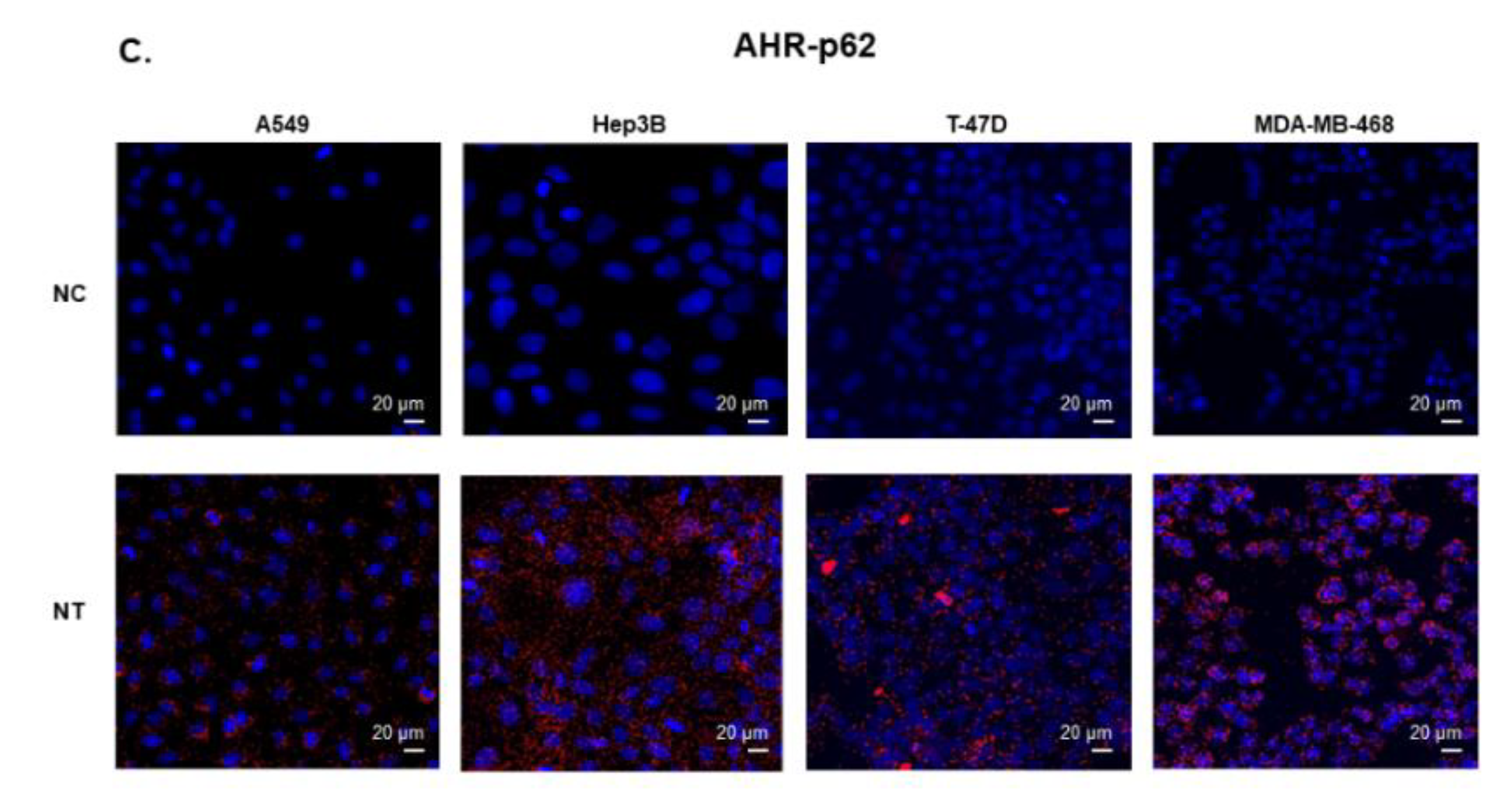
2.5. AHR is Degraded Via Autophagy in Human Lung, Liver, and Breast Cancer Cell Lines
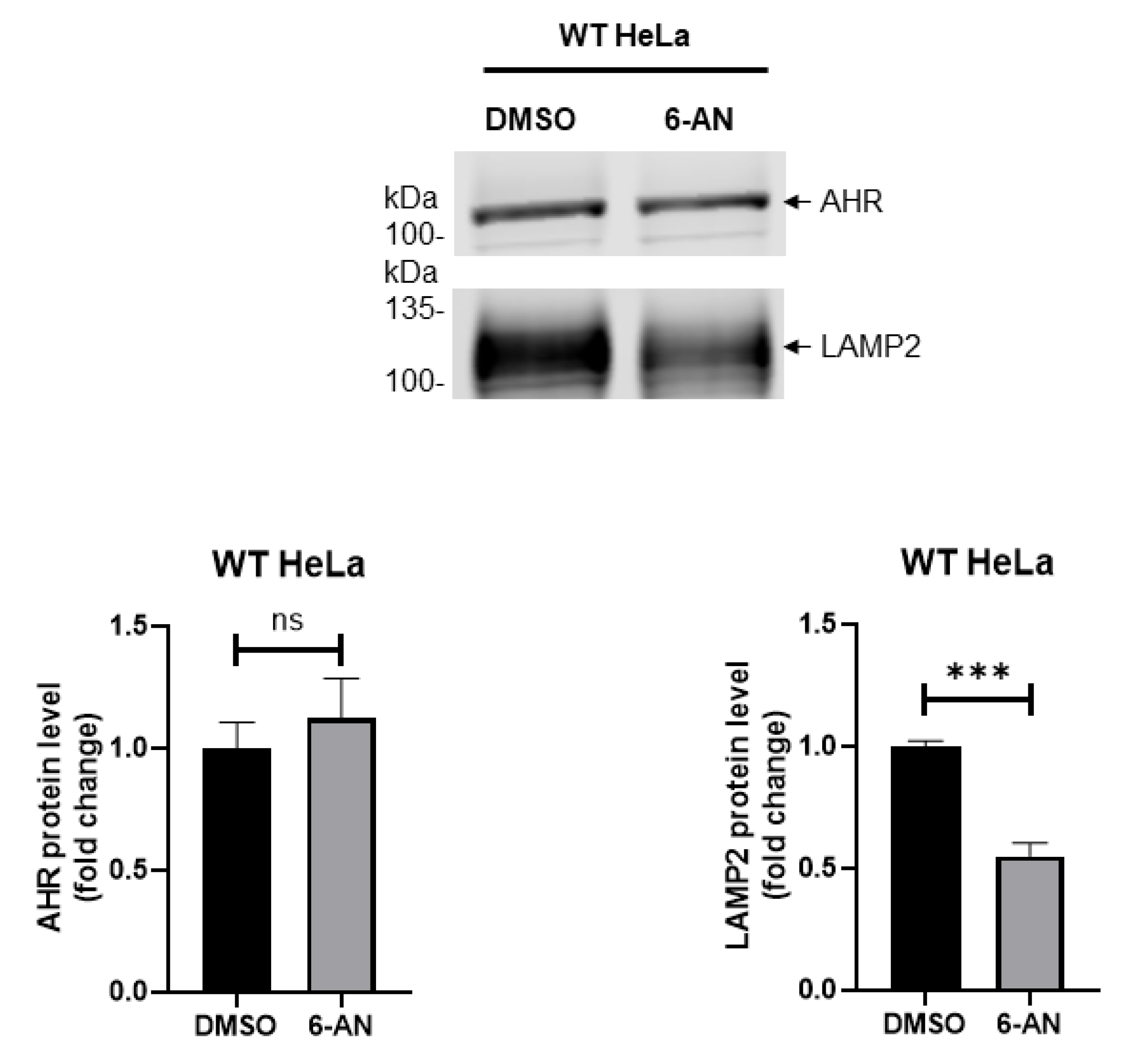
2.6. Chaperone-Mediated Autophagy Is Unlikely Involved in the Degradation of AHR in HeLa Cells
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. Generation of HSP90, p23, and LC3B Stable Knockdown Cells
4.4. Transient Transfection
4.5. Whole-Cell Lysate Preparation and Western Analysis
4.6. RT-qPCR
4.7. Immunoprecipitation and Co-Immunoprecipitation Experiments
4.8. K48- and K63-TUBE and Far-Western Analysis
4.9. Proximity Ligation Assay
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AHR | aryl hydrocarbon receptor |
| CQ | chloroquine |
| Baf A1 | bafilomycin A1 |
| 3MA | 3-methyladenine |
| Met p23 | metformin cytosolic prostaglandin E2 synthase |
| p23KD p62 LC3B | p23 knockdown SQSTM1 (sequestosome-1) microtubule-associated protein 1 light chain 3B |
| WT | wild type |
| CHX | cycloheximide |
| ActD | actinomycin D |
| 3MC | 3-methylcholanthrene |
| WCL | whole-cell lysate |
| PLA | proximity ligation assay |
| NC | negative control |
| NT | no treatment |
| CMA | chaperone-mediated autophagy |
| 6-AN | 6-aminonicotinamide |
| HBSS | Hank’s balanced salt solution |
| HIF-1α | hypoxia-inducible factor 1-alpha |
| HSC70 | heat shock cognate protein of 70KDa |
| LAMP-2A | lysosomal associated membrane protein 2A |
| HSP90 | heat shock protein 90 |
| XAP2 | Hepatitis B Virus-X associated protein 2 |
| KFERQ NEM | lysine-phenylalanine-glutamate-arginine-glutamine N-ethylmaleimide |
References
- Poland, A.; Glover, E.; Kende, A.S. Stereospecific, high affinity binding of 2,3,7,8-tetrachlorodibenzo-p-dioxin by hepatic cytosol. J. Biol. Chem. 1976, 251, 4936–4946. [Google Scholar]
- Schmidt, J.V.; Su, G.H.; Reddy, J.K.; Simon, M.C.; Bradfield, C.A. Characterization of a murine Ahr null allele: Involvement of the Ah receptor in hepatic growth and development. Proc. Natl. Acad. Sci. USA 1996, 93, 6731–6736. [Google Scholar] [CrossRef] [Green Version]
- Quintana, F.J.; Basso, A.S.; Iglesias, A.H.; Korn, T.; Farez, M.F.; Bettelli, E.; Caccamo, M.; Oukka, M.; Weiner, H.L. Control of Treg and TH17 cell differentiation by the aryl hydrocarbon receptor. Nature 2008, 453, 65–71. [Google Scholar] [CrossRef]
- Ye, J.; Qiu, J.; Bostick, J.W.; Ueda, A.; Schjerven, H.; Li, S.; Jobin, C.; Chen, Z.E.; Zhou, L. The aryl hydrocarbon receptor preferentially marks and promotes gut regulatory T cells. Cell Rep. 2017, 21, 2277–2290. [Google Scholar] [CrossRef] [Green Version]
- Juricek, L.; Coumoul, X. The aryl hydrocarbon receptor and the nervous system. Int. J. Mol. Sci. 2018, 19, 2504. [Google Scholar] [CrossRef] [Green Version]
- Opitz, C.A.; Litzenburger, U.M.; Sahm, F.; Ott, M.; Tritschler, I.; Trump, S.; Schumacher, T.; Jestaedt, L.; Schrenk, D.; Weller, M.; et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 2011, 478, 197–203. [Google Scholar] [CrossRef]
- Kolluri, S.K.; Jin, U.H.; Safe, S. Role of the aryl hydrocarbon receptor in carcinogenesis and potential as an anti-cancer drug target. Arch. Toxicol. 2017, 91, 2497–2513. [Google Scholar] [CrossRef]
- Holt, S.E.; Aisner, D.L.; Baur, J.; Tesmer, V.M.; Dy, M.; Ouellette, M.; Trager, J.B.; Morin, G.B.; Toft, D.O.; Shay, J.W.; et al. Functional requirement of p23 and Hsp90 in telomerase complexes. Genes Dev. 1999, 13, 817–826. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.L.; Toft, D.O. Binding of p23 and hsp90 during assembly with the progesterone receptor. Mol. Endocrinol. 1995, 9, 670–678. [Google Scholar] [PubMed] [Green Version]
- Knoblauch, R.; Garabedian, M.J. Role for Hsp90-associated cochaperone p23 in estrogen receptor signal transduction. Mol. Cell. Biol. 1999, 19, 3748–3759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, S.C.; Toran, E.J.; Rimerman, R.A.; Hjermstad, S.; Smithgall, T.E.; Smith, D.F. A pathway of multi-chaperone interactions common to diverse regulatory proteins: Estrogen receptor, Fes tyrosine kinase, heat shock transcription factor Hsf1, and the aryl hydrocarbon receptor. Cell Stress Chaperones 1996, 1, 237–250. [Google Scholar] [CrossRef] [Green Version]
- Cox, M.B.; Miller, C.A., 3rd. Cooperation of heat shock protein 90 and p23 in aryl hydrocarbon receptor signaling. Cell Stress Chaperones 2004, 9, 4–20. [Google Scholar] [CrossRef]
- Young, J.C.; Moarefi, I.; Hartl, F.U. Hsp90: A specialized but essential protein-folding tool. J. Cell Biol. 2001, 154, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Shetty, P.V.; Bhagwat, B.Y.; Chan, W.K. p23 enhances the formation of the aryl hydrocarbon receptor-DNA complex. Biochem. Pharmacol. 2003, 65, 941–948. [Google Scholar] [CrossRef]
- Nguyen, P.M.; Wang, D.; Wang, Y.; Li, Y.; Uchizono, J.A.; Chan, W.K. p23 co-chaperone protects the aryl hydrocarbon receptor from degradation in mouse and human cell lines. Biochem. Pharmacol. 2012, 84, 838–850. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Yakkundi, P.; Chan, W.K. Down-regulation of p23 in normal lung epithelial cells reduces toxicities from exposure to benzo[a]pyrene and cigarette smoke condensate via an aryl hydrocarbon receptor-dependent mechanism. Toxicol. Sci. 2019, 167, 239–248. [Google Scholar] [CrossRef]
- Flaveny, C.; Perdew, G.H.; Miller, C.A., 3rd. The aryl-hydrocarbon receptor does not require the p23 co-chaperone for ligand binding and target gene expression in vivo. Toxicol. Lett. 2009, 189, 57–62. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [Green Version]
- Korolchuk, V.I.; Menzies, F.M.; Rubinsztein, D.C. Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett. 2010, 584, 1393–1398. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.-K.; Lee, J.-A. Role of the mammalian ATG8/LC3 family in autophagy: Differential and compensatory roles in the spatiotemporal regulation of autophagy. BMB Rep. 2016, 49, 424–430. [Google Scholar] [CrossRef] [Green Version]
- Zaffagnini, G.; Martens, S. Mechanisms of selective autophagy. J. Mol. Biol. 2016, 428, 1714–1724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Q.; Baldwin, K.T. 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced degradation of aryl hydrocarbon receptor (AhR) by the ubiquitin-proteasome pathway. Role of the transcription activaton and DNA binding of AhR. J. Biol. Chem. 2000, 275, 8432–8438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pappas, B.; Yang, Y.; Wang, Y.; Kim, K.; Chung, H.J.; Cheung, M.; Ngo, K.; Shinn, A.; Chan, W.K. p23 protects the human aryl hydrocarbon receptor from degradation via a heat shock protein 90-independent mechanism. Biochem. Pharmacol. 2018, 152, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Korolchuk, V.I.; Mansilla, A.; Menzies, F.M.; Rubinsztein, D.C. Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol. Cell 2009, 33, 517–527. [Google Scholar] [CrossRef]
- Nam, T.; Han, J.H.; Devkota, S.; Lee, H.W. Emerging paradigm of crosstalk between autophagy and the ubiquitin-proteasome system. Mol. Cells. 2017, 40, 897–905. [Google Scholar] [PubMed]
- Zhu, K.; Dunner, K., Jr.; McConkey, D.J. Proteasome inhibitors activate autophagy as a cytoprotective response in human prostate cancer cells. Oncogene 2010, 29, 451–462. [Google Scholar] [CrossRef] [Green Version]
- Harhouri, K.; Navarro, C.; Depetris, D.; Mattei, M.G.; Nissan, X.; Cau, P.; De Sandre-Giovannoli, A.; Levy, N. MG132-induced progerin clearance is mediated by autophagy activation and splicing regulation. EMBO Mol. Med. 2017, 9, 1294–1313. [Google Scholar] [CrossRef]
- Jiang, P.; Mizushima, N. LC3- and p62-based biochemical methods for the analysis of autophagy progression in mammalian cells. Methods 2015, 75, 13–18. [Google Scholar] [CrossRef]
- Danieli, A.; Martens, S. p62-mediated phase separation at the intersection of the ubiquitin- proteasome system and autophagy. J. Cell Sci. 2018, 131, jcs214304. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, D.; Riezman, H. Proteasome-independent functions of ubiquitin in endocytosis and signaling. Science 2007, 315, 201–205. [Google Scholar] [CrossRef] [Green Version]
- Steinman, R.M.; Mellman, I.S.; Muller, W.A.; Cohn, Z.A. Endocytosis and the recycling of plasma membrane. J. Cell Biol. 1983, 96, 1–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.T.; Tan, H.L.; Shui, G.; Bauvy, C.; Huang, Q.; Wenk, M.R.; Ong, C.N.; Codogno, P.; Shen, H.M. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J. Biol. Chem. 2010, 285, 10850–10861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, C.; Cao, W.; Bao, L.; Zuo, W.; Xie, G.; Cai, T.; Fu, W.; Zhang, J.; Wu, W.; Zhang, X.; et al. Autophagy negatively regulates Wnt signalling by promoting Dishevelled degradation. Nature Cell Biol. 2010, 12, 781–790. [Google Scholar] [CrossRef]
- Rivera Vargas, T.; Cai, Z.; Shen, Y.; Dosset, M.; Benoit-Lizon, I.; Martin, T.; Roussey, A.; Flavell, R.A.; Ghiringhelli, F.; Apetoh, L. Selective degradation of PU.1 during autophagy represses the differentiation and antitumour activity of TH9 cells. Nature Commun. 2017, 8, 559. [Google Scholar] [CrossRef] [Green Version]
- Kwon, Y.T.; Ciechanover, A. The ubiquitin code in the ubiquitin-proteasome system and autophagy. Trends Biochem. Sci. 2017, 42, 873–886. [Google Scholar] [CrossRef]
- Ohtake, F.; Tsuchiya, H. The emerging complexity of ubiquitin architecture. J. Biochem. 2017, 161, 125–133. [Google Scholar] [CrossRef] [Green Version]
- Kazlauskas, A.; Poellinger, L.; Pongratz, I. Evidence that the co-chaperone p23 regulates ligand responsiveness of the dioxin receptor. J. Biol. Chem. 1999, 274, 13519–13524. [Google Scholar] [CrossRef] [Green Version]
- Ravikumar, B.; Duden, R.; Rubinsztein, D.C. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Human Mol. Genetics 2002, 11, 1107–1117. [Google Scholar] [CrossRef] [Green Version]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H.; Overvatn, A.; Bjorkoy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, J.V.; Soares, A.R.; Ramalho, J.S.; Pereira, P.; Girao, H. K63 linked ubiquitin chain formation is a signal for HIF1A degradation by Chaperone-Mediated Autophagy. Sci. Rep. 2015, 5, 10210. [Google Scholar] [CrossRef] [Green Version]
- Cuervo, A.M.; Knecht, E.; Terlecky, S.R.; Dice, J.F. Activation of a selective pathway of lysosomal proteolysis in rat liver by prolonged starvation. Am. J. Physiol. 1995, 269, C1200–C1208. [Google Scholar] [CrossRef] [PubMed]
- Kiffin, R.; Christian, C.; Knecht, E.; Cuervo, A.M. Activation of chaperone-mediated autophagy during oxidative stress. Mol. Biol. Cell 2004, 15, 4829–4840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogov, V.; Dotsch, V.; Johansen, T.; Kirkin, V. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol. Cell 2014, 53, 167–178. [Google Scholar] [CrossRef] [Green Version]
- Jinwal, U.K.; Abisambra, J.F.; Zhang, J.; Dharia, S.; O’Leary, J.C.; Patel, T.; Braswell, K.; Jani, T.; Gestwicki, J.E.; Dickey, C.A. Cdc37/Hsp90 protein complex disruption triggers an autophagic clearance cascade for TDP-43 protein. J. Biol. Chem. 2012, 287, 24814–24820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qing, G.; Yan, P.; Xiao, G. Hsp90 inhibition results in autophagy-mediated proteasome-independent degradation of IkappaB kinase (IKK). Cell Res. 2006, 16, 895–901. [Google Scholar] [CrossRef]
- Qing, G.; Yan, P.; Qu, Z.; Liu, H.; Xiao, G. Hsp90 regulates processing of NF-kappa B2 p100 involving protection of NF-kappa B-inducing kinase (NIK) from autophagy-mediated degradation. Cell Res. 2007, 17, 520–530. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.S.; Singh, S.S.; Perdew, G.H. The Ah receptor is a sensitive target of geldanamycin-induced protein turnover. Arch. Biochem. Biophys. 1997, 348, 190–198. [Google Scholar] [CrossRef]
- Xie, J.; Huang, X.; Park, M.S.; Pham, H.M.; Chan, W.K. Differential suppression of the aryl hydrocarbon receptor nuclear translocator-dependent function by an aryl hydrocarbon receptor PAS-A-derived inhibitory molecule. Biochem. Pharmacol. 2014, 88, 253–265. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Y.; Chan, W.K. Selective Autophagy Maintains the Aryl Hydrocarbon Receptor Levels in HeLa Cells: A Mechanism That Is Dependent on the p23 Co-Chaperone. Int. J. Mol. Sci. 2020, 21, 3449. https://doi.org/10.3390/ijms21103449
Yang Y, Chan WK. Selective Autophagy Maintains the Aryl Hydrocarbon Receptor Levels in HeLa Cells: A Mechanism That Is Dependent on the p23 Co-Chaperone. International Journal of Molecular Sciences. 2020; 21(10):3449. https://doi.org/10.3390/ijms21103449
Chicago/Turabian StyleYang, Yujie, and William K. Chan. 2020. "Selective Autophagy Maintains the Aryl Hydrocarbon Receptor Levels in HeLa Cells: A Mechanism That Is Dependent on the p23 Co-Chaperone" International Journal of Molecular Sciences 21, no. 10: 3449. https://doi.org/10.3390/ijms21103449