Synthesis and In Vitro Antitumor Activity of Naringenin Oxime and Oxime Ether Derivatives

 and
and
Abstract
:1. Introduction
2. Results and Discussion
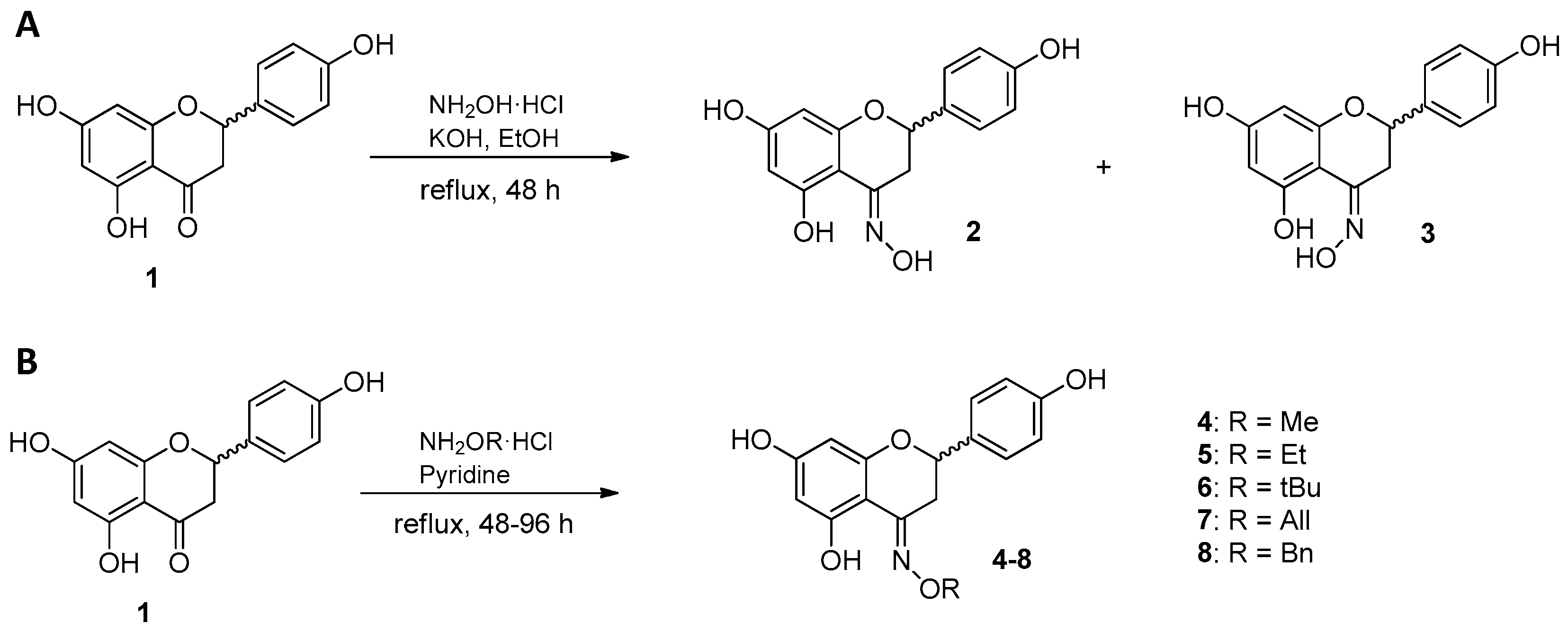
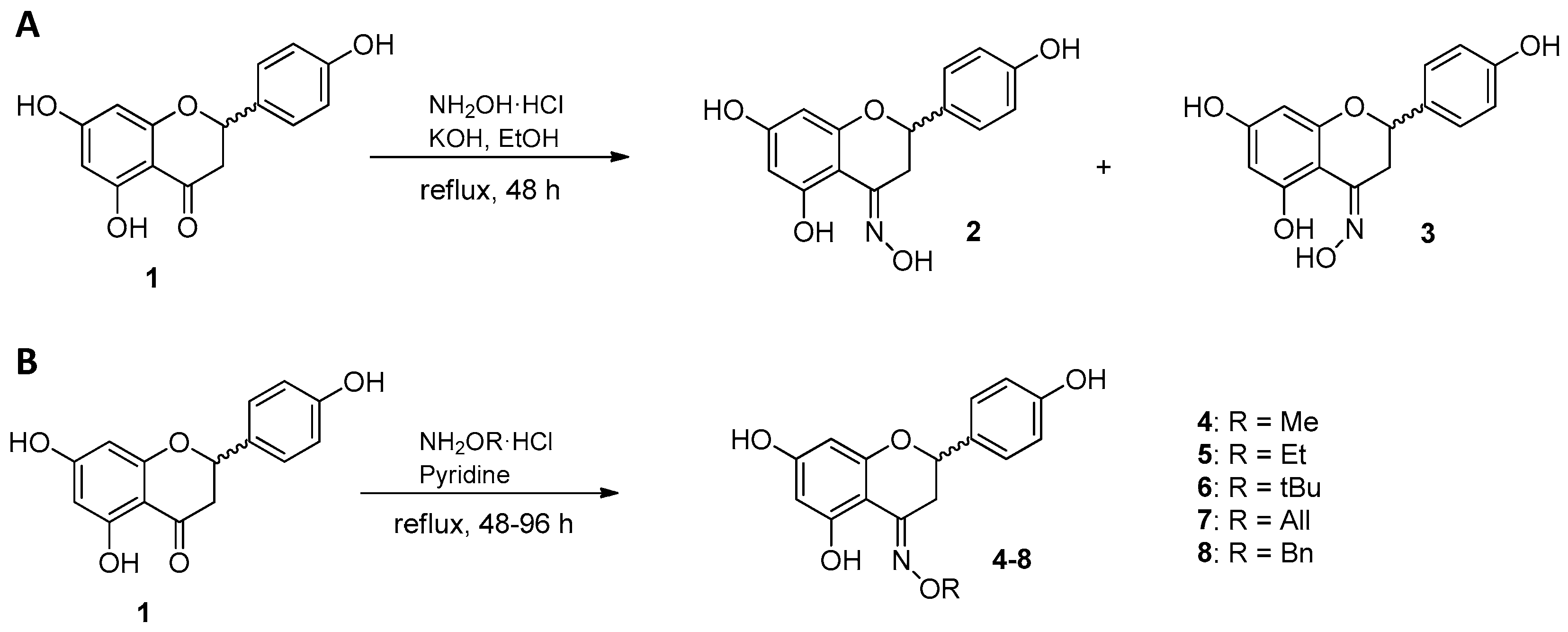
2.1. Synthesis of Naringenin Oximes and Naringenin Oxime Ethers
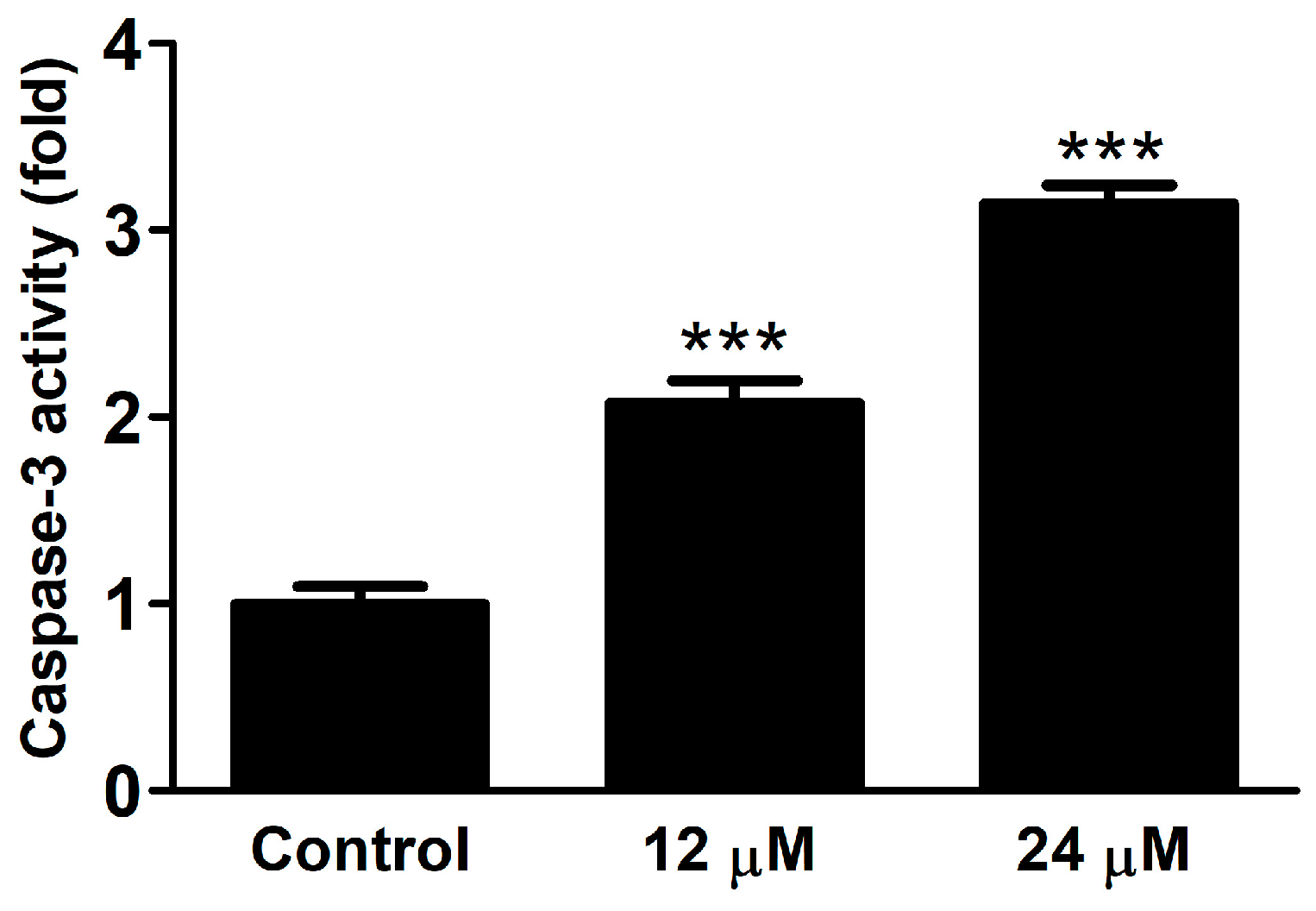
2.2. Biological Activity
2.2.1. Antiproliferative Assay
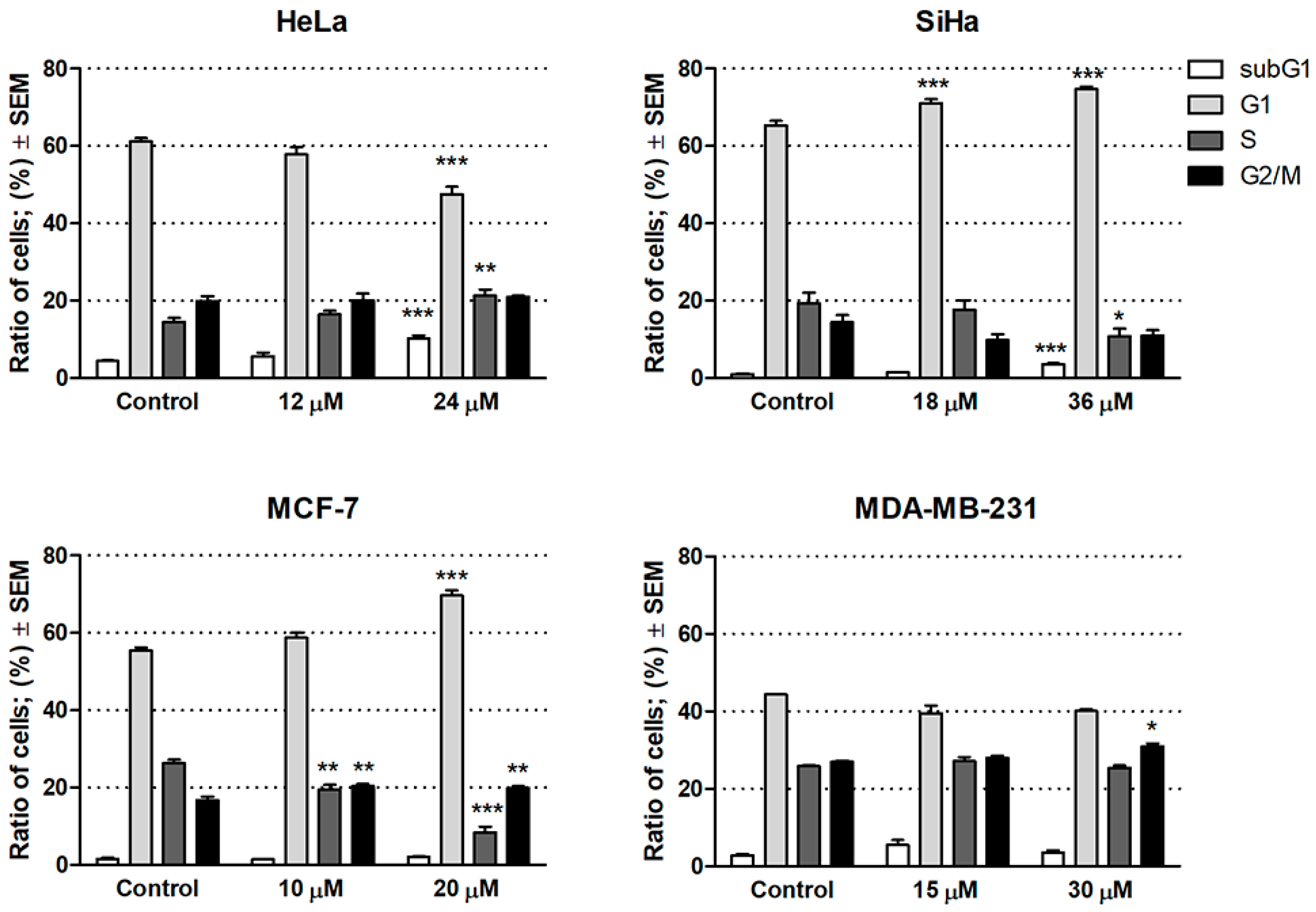
2.2.2. Cell Cycle Analysis
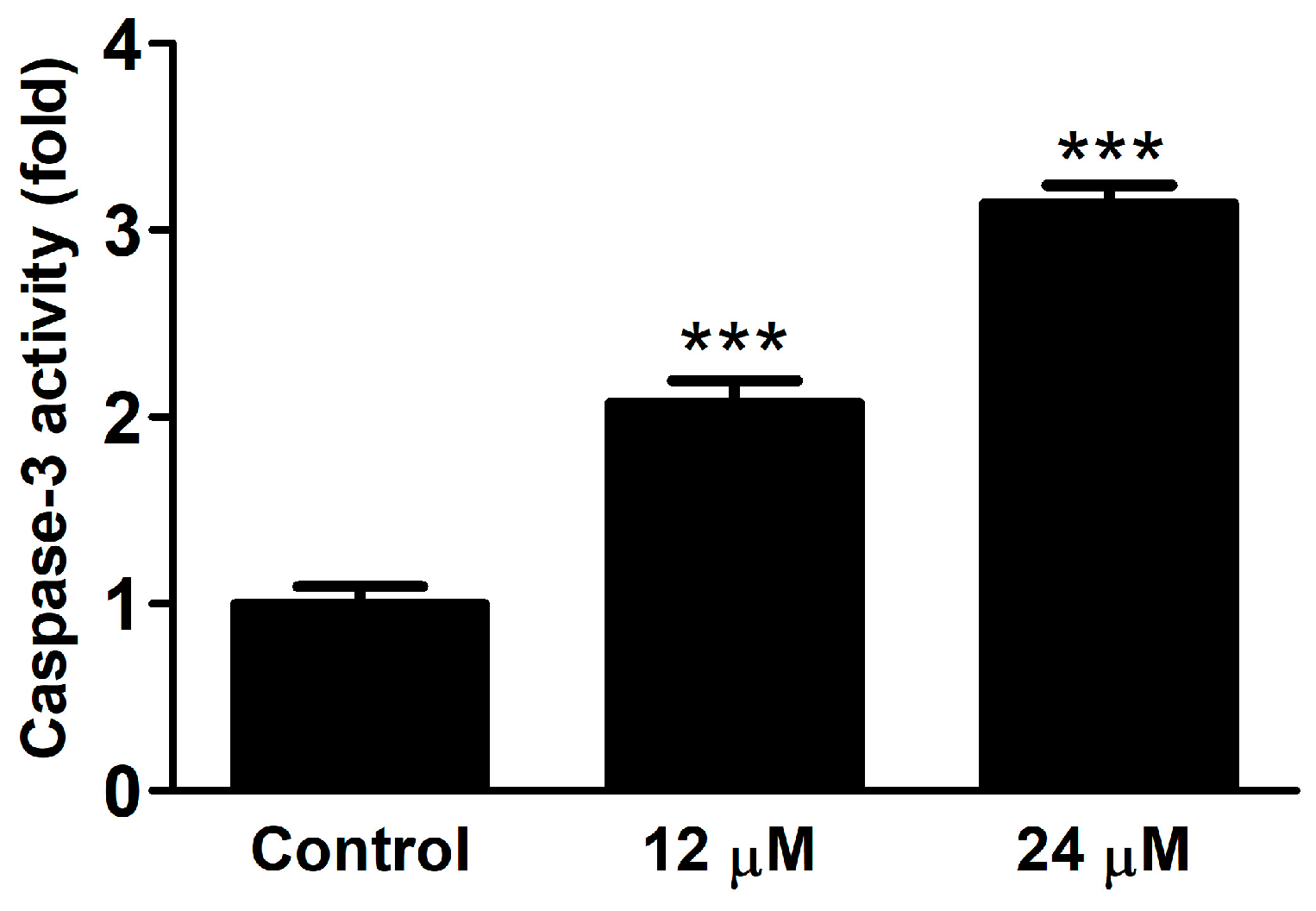
2.2.3. Caspase Activity
2.2.4. Antioxidant Activity
3. Materials and Methods
3.1. Chemical Methods
3.2. Cell Culture
3.3. Antiproliferative Assay
3.4. Cell Cycle Analysis
3.5. Caspase Activity
3.6. Antioxidant Activity
3.6.1. Diphenyl-2-picrylhydrazyl (DPPH) Assay
3.6.2. Oxygen Radical Absorbance Capacity (ORAC) Assay
3.6.3. Xantine-Oxidase Inhibitory Assay
4. Experimental
4.1. Procedure for the Synthesis of Naringenin-Derived Oximes
4.2. General Procedure for the Synthesis of Naringenin Oxime Ethers
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Erlund, I. Review of the flavonoids quercetin, hesperetin and naringenin. Dietary sources, bioactivities, bioavailability and epidemiology. Nutr. Res. 2004, 24, 851–874. [Google Scholar] [CrossRef]
- Khan, M.K.; Zill-E-Huma; Dangles, O. A comprehensive review on flavanones, the major citrus polyphenols. J. Food Compos. Anal. 2014, 33, 85–104. [Google Scholar] [CrossRef]
- Testai, L.; Da Pozzo, E.; Piano, I.; Pistelli, L.; Gargini, C.; Breschi, M.C.; Braca, A.; Martini, C.; Martelli, A.; Calderone, V. The citrus flavanone naringenin produces cardioprotective effects in hearts from 1 year old rat, through activation of mitoBK channels. Front. Pharmacol. 2017, 8. [Google Scholar] [CrossRef]
- Liao, A.C.H.; Kuo, C.-C.; Huang, Y.-C.; Yeh, C.-W.; Hseu, Y.-C.; Liu, J.-Y.; Hsu, L.-S. Naringenin inhibits migration of bladder cancer cells through downregulation of AKT and MMP-2. Mol. Med. Rep. 2014, 10, 1531–1536. [Google Scholar] [CrossRef]
- Hatkevich, T.; Ramos, J.; Santos-Sanchez, I.; Patel, Y.M. A naringenin-tamoxifen combination impairs cell proliferation and survival of MCF-7 breast cancer cells. Exp. Cell Res. 2014, 327, 331–339. [Google Scholar] [CrossRef]
- Ahamad, M.S.; Siddiqui, S.; Jafri, A.; Ahmed, S.; Afzal, M. Induction of apoptosis and antiproliferative activity naringenin in human epidermoid carcinoma cell through ROS generation and cell cycle arrest. PLoS ONE 2014, 10, e110003. [Google Scholar] [CrossRef]
- Arul, D.; Subramanian, P. Naringenin (citrus flavonone) induces growth inhibition, cell cycle arrest and apoptosis in human hepatocellular carcinoma cells. Pathol. Oncol. Res. 2013, 19, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Bao, L.; Liu, F.; Guo, H.; Li, Y.; Tan, B.; Zhang, W. Naringenin inhibits proliferation, migration, and invasion as well as induces apoptosis of gastric cancer SGC7901 cell line by downregulation of AKT pathway. Tumor Biol. 2016, 11365–11374. [Google Scholar] [CrossRef]
- Abaza, M.S.I.; Orabi, K.Y.; Al-Quattan, E.; Al-Attiyah, R.J. Growth inhibitory and chemo-sensitization effects of naringenin, a natural flavanone purified from Thymus vulgaris, on human breast and colorectal cancer. Cancer Cell Int. 2015, 15, 46–65. [Google Scholar] [CrossRef]
- Hee, J.; Jin, C.; Kyu, B.; Kim, G.; Hyun, Y.; Kee, Y. Naringenin induces apoptosis through downregulation of Akt and caspase-3 activation in human leukemia THP-1 cells. Food Chem. Toxicol. 2008, 46, 3684–3690. [Google Scholar] [CrossRef]
- Wang, K.; Chen, Z.; Huang, J.; Huang, L.; Luo, N.; Liang, X.; Liang, M.; Xie, W. Naringenin prevents ischaemic stroke damage via anti-apoptotic and anti-oxidant effects. Clin. Exp. Pharmacol. Physiol. 2017, 44, 862–871. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.; Kim, T.W.; Shin, S.Y.; Park, M.J.; Yong, Y.; Kim, D.W.; Islam, T.; Lee, Y.H.; Jung, K.-Y.; Lim, Y. Design, synthesis and inhibitory activities of naringenin derivatives on human colon cancer cells. Bioorg. Med. Chem. Lett. 2003, 23, 232–238. [Google Scholar] [CrossRef]
- Lee, S.; Lee, C.-H.; Moon, S.-S.; Kim, E.; Kim, C.-T.; Kim, B.-H.; Bok, S.-H.; Jeong, T.S. Naringenin derivatives as anti.atherogenic agents. Bioorg. Med. Chem. Lett. 2003, 13, 3901–3903. [Google Scholar] [CrossRef] [PubMed]
- Copmans, D.; Orellana-Paucar, A.M.; Steurs, G.; Zhang, Y.; Ny, A.; Foubert, K.; Exarchou, V.; Siekierska, A.; Kim, Y.; de Borggraeve, W.; et al. Methylated flavonoids as anti-seizure agents: Naringenin 4′,7-dimethyl ether attenuates epileptic siezures in zebrafish and mouse models. Neurochem. Int. 2018, 112, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Brodowska, K.; Sykula, A.; Garribba, E.; Lodyga-Chruscinska, E.; Sójka, M. Naringenin Schiff base: Antioxidant activity, acid-base profile, and interactions with DNA. Transit. Met. Chem. 2016, 41, 179–189. [Google Scholar] [CrossRef]
- Türkkan, B.; Özyürek, M.; Bener, M.; Güclü, K.; Apak, R. Synthesis, characterization and antioxidant capacity of naringenin-oxime. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2012, 85, 235–240. [Google Scholar] [CrossRef]
- Potaniec, B.; Grabarczyk, M.; Stompor, M.; Szumny, A.; Zielinsky, P.; Zolnierczyk, A.K.; Aniol, M. Antioxidant activity and spectroscopic data of isoxanthohomol oxime and related compounds. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2014, 118, 716–723. [Google Scholar] [CrossRef] [PubMed]
- Kocyigit, A.; Koyuncu, I.; Taskin, A.; Dikilitas, M.; Bahadori, F.; Turkkan, B. Antigenotoxic and antioxidant potentials of newly derivatized compound naringenin-oxime relative to naringenin on human mononuclear cells. Drug Chem. Toxicol. 2016, 39, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Koyuncu, I.; Kocyigit, A.; Gonel, A.; Arslan, E.; Durgun, M. The protective effect of naringenin-oxime on cisplatin induced toxicity in rats. Biochem. Res. Int. 2017, 9478958. [Google Scholar] [CrossRef] [PubMed]
- Kocyigit, A.; Koyuncu, I.; Dikilitas, M.; Bahadori, F.; Turkkan, B. Cytotoxic, genotoxic and apoptotic effects of naringenin-oxime relative to naringenin on normal and cancer cell lines. Asian Pac. J. Trop. Biomed. 2016, 6, 872–880. [Google Scholar] [CrossRef] [Green Version]
- Song, J.-Y.; Liu, Y.; Zhao, H.-Y.; Han, H.-T.; Li, Z.-F.; Guo, W.-H.; Chu, W.-Y.; Sun, Z.-Z. Efficient nickel(II) naringenin-oxime complex catalyzed Mizoroki-Heck cross-coupling reaction in the presence of hydrazine hydrate. New J. Chem. 2017, 41, 12288–12292. [Google Scholar] [CrossRef]
- Kozłowska, J.; Grela, E.; Baczyńska, D.; Grabowiecka, A.; Anioł, M. Novel O-alkyl Derivatives of Naringenin and Their Oximes with Antimicrobial and Anticancer Activity. Molecules 2019, 24, 679. [Google Scholar] [CrossRef]
- Liu, Z.; Wei, W.; Gan, C.; Huang, Y.; Liu, S.; Zhou, M.; Cui, J. Semisynthesis and cytotoxicity of E-Naringenin oximes from Naringin. Chin. J. Org. Chem. 2013, 33, 2551–2558. [Google Scholar] [CrossRef]
- Hawkes, G.E.; Herwig, K.; Roberts, J.D. Nuclear magnetic resonance spectroscopy. Use of 13C NMR spectra to establish configurations of oximes. J. Org. Chem. 1974, 39, 1017–1028. [Google Scholar] [CrossRef]
- Molnár, J.; Szebeni, G.J.; Csupor-Löffler, B.; Hajdú, Z. Investigation of the antiproliferative properties of natural sesquiterpenes from Artemisia asiatica and Onopordum acanthium on HL-60 cells in vitro. Int. J. Mol. Sci. 2016, 17, 83. [Google Scholar] [CrossRef]
- Zupkó, I.; Molnár, J.; Réthy, B.; Minorics, R.; Frank, É.; Wölfling, J.; Molnár, J.; Ocsovszki, I.; Topcu, Z.; Bitó, T.; et al. Anticancer and multidrug resistance-reversal effects of solanidine analogs synthetized from pregnadienolone acetate. Molecules 2014, 19, 2061–2076. [Google Scholar] [CrossRef]
- Szabó, J.; Jerkovics, N.; Schneider, G.; Wölfling, J.; Bózsity, N.; Minorics, R.; Zupkó, I.; Mernyák, E. Synthesis and in vitro antiproliferative evaluation of C-13 epimers of triazolyl-d-Secoestrone alcohols: The first potent 13α-D-Secoestrone derivative. Molecules 2016, 21, 611. [Google Scholar] [CrossRef]
- Réthy, B.; Hohmann, J.; Minorics, R.; Varga, A.; Ocsovszki, I.; Molnár, J.; Juhász, K.; Falkay, G.; Zupkó, I. Antitumour properties of acridone alkaloids on a murine lymphoma cell line. Anticancer Res. 2008, 28, 2737–2743. [Google Scholar]
- Baji, Á.; Kovács, F.; Schneider, G.; Wöl, J.; Sinka, I. Investigation of pH and substituent effects on the distribution ratio of novel steroidal ring D- and A-fused arylpyrazole regioisomers and evaluation of their cell-growth inhibitory effects in vitro. Steroids 2017, 126, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Bózsity, N.; Minorics, R.; Szabó, J.; Mernyák, E.; Schneider, G.; Wöl, J.; Wang, H.; Wu, C.; Ocsovszki, I.; Zupkó, I. Mechanism of antiproliferative action of a new D-secoestrone-triazole derivative in cervical cancer cells and its effect on cancer cell motility. J. Steroid Biochem. Mol. Biol. 2017, 165, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Fukumoto, L.; Mazza, G. Assessing Antioxidant and Prooxidant Activities of Phenolic Compounds. J. Agric. Food. Chem. 2000, 48, 3597–3604. [Google Scholar] [CrossRef] [PubMed]
- Mielnik, M.B.; Rzeszutek, A.; Triumf, E.C.; Egelandsdal, B. Antioxidant and other quality properties of reindeer muscle from two different Norwegian regions. Meat Sci. 2011, 89, 526–532. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Compound | Conc. (µM) | Growth Inhibition (%) ± SEM (Calculated IC50 Value (µM)) | ||||
|---|---|---|---|---|---|---|
| HeLa | SiHa | MCF-7 | MDA-MB-231 | HL-60 | ||
| (1) | 25 | <20 | <20 | <20 | <20 | <20 |
| 50 | 23.9 ± 2.09 | <20 | <20 | <20 | <20 | |
| (2) | 25 | <20 | <20 | <20 | <20 | <20 |
| 50 | 28.75 ± 2.44 | <20 | 21.83 ± 3.92 | <20 | 43.40 ± 2.81 | |
| (3) | 25 | <20 | <20 | <20 | <20 | <20 |
| 50 | <20 | <20 | <20 | <20 | <20 | |
| (4) | 25 | <20 | <20 | <20 | <20 | 37.67 ± 1.29 |
| 50 | 31.36 ± 2.97 | <20 | 48.44 ± 3.27 | 24.35 ± 1.88 | 57.89 ± 1.13 | |
| (5) | 25 | <20 | <20 | <20 | <20 | <20 |
| 50 | 29.36 ± 1.42 | <20 | 44.06 ± 2.18 | <20 | 44.89 ± 0.48 | |
| (6) | 25 | 52.37 ± 2.32 | <20 | 61.41 ± 1.93 | 27.19 ± 1.78 | 37.31 ± 3.65 |
| 50 | 92.22 ± 1.03 | 88.54 ± 1.51 | 87.00 ± 0.61 | 90.33 ± 0.58 | 88.07 ± 0.10 | |
| [23.49] | [35.41] | [19.46] | [29.74] | [31.76] | ||
| (7) | 25 | <20 | <20 | <20 | <20 | <20 |
| 50 | 25.04 ± 2.4 | <20 | 33.75 ± 2.45 | <20 | <20 | |
| (8) | 25 | 22.63 ± 0.63 | <20 | 24.29 ± 1.86 | <20 | <20 |
| 50 | 37.67 ± 2.01 | <20 | 64.47 ± 2.12 | 24.87 ± 3.47 | <20 | |
| Cisplatin | 25 | 98.71 ± 0.21 | 86.40 ± 1.02 | 90.81 ± 0.22 | 41.37 ± 1.05 | 64.03 ± 0.43 |
| 50 | 99.09 ± 0.24 | 96.72 ± 0.36 | 98.49 ± 0.11 | 84.43 ± 0.4 | 84.88 ± 0.41 | |
| [11.79] | [13.63] | [5.15] | [25.82] | [5.75] | ||
| Compound | Antioxidant Activity ± SD | ||
|---|---|---|---|
| DPPH EC50 (μM) | ORAC (μmolTE/μmol) | XO inh (%) | |
| 1 | - a | 11.18 ± 0.46 | 12.31 ± 4.60 b |
| 2 | 243.45 ± 4.88 | 8.88 ± 0.23 | 7.35 ± 1.32 |
| 3 | 1776.00 ± 123.71 | 6.95 ± 0.12 | 2.13 ± 0.78 |
| 4 | 212.20 ± 32.59 | 16.63 ± 1.68 | 4.00 ± 1.81 |
| 5 | 1437.50 ± 36.06 | 5.54 ± 0.41 | 8.13 ± 2.02 |
| 6 | - | 3.89 ± 0.87 | 6.95 ± 2.31 |
| 7 | 1164.00 ± 226.27 | 6.03 ± 2.79 | 12.84 ± 3.01 |
| 8 | - | 1.38 ± 0.41 | 9.06 ± 0.79 |
| Rutin | 39.88 ± 1.34 | 12.35 ± 0.38 | n.d. |
| Allopurinol | n.d. | n.d. | 98.23 ± 3.29 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Latif, A.D.; Gonda, T.; Vágvölgyi, M.; Kúsz, N.; Kulmány, Á.; Ocsovszki, I.; Zomborszki, Z.P.; Zupkó, I.; Hunyadi, A. Synthesis and In Vitro Antitumor Activity of Naringenin Oxime and Oxime Ether Derivatives. Int. J. Mol. Sci. 2019, 20, 2184. https://doi.org/10.3390/ijms20092184
Latif AD, Gonda T, Vágvölgyi M, Kúsz N, Kulmány Á, Ocsovszki I, Zomborszki ZP, Zupkó I, Hunyadi A. Synthesis and In Vitro Antitumor Activity of Naringenin Oxime and Oxime Ether Derivatives. International Journal of Molecular Sciences. 2019; 20(9):2184. https://doi.org/10.3390/ijms20092184
Chicago/Turabian StyleLatif, Ahmed Dhahir, Tímea Gonda, Máté Vágvölgyi, Norbert Kúsz, Ágnes Kulmány, Imre Ocsovszki, Zoltán Péter Zomborszki, István Zupkó, and Attila Hunyadi. 2019. "Synthesis and In Vitro Antitumor Activity of Naringenin Oxime and Oxime Ether Derivatives" International Journal of Molecular Sciences 20, no. 9: 2184. https://doi.org/10.3390/ijms20092184