β-Catenin/Smad3 Interaction Regulates Transforming Growth Factor-β-Induced Epithelial to Mesenchymal Transition in the Lens


Abstract
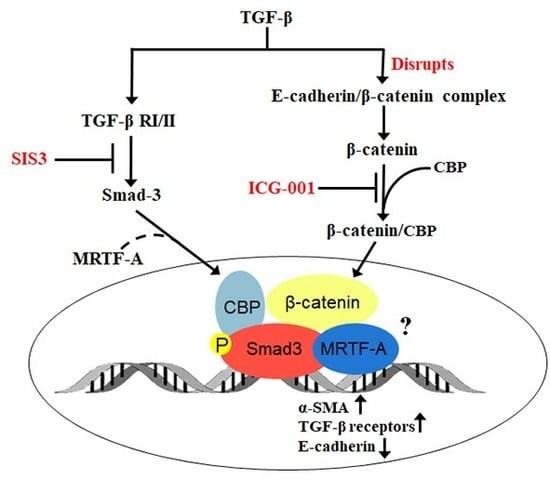
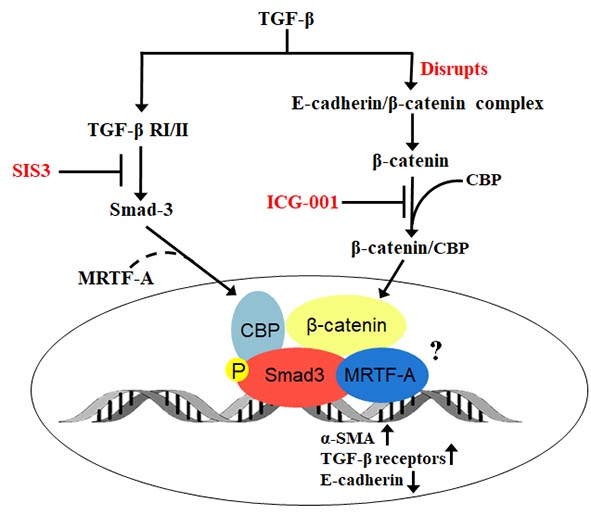
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
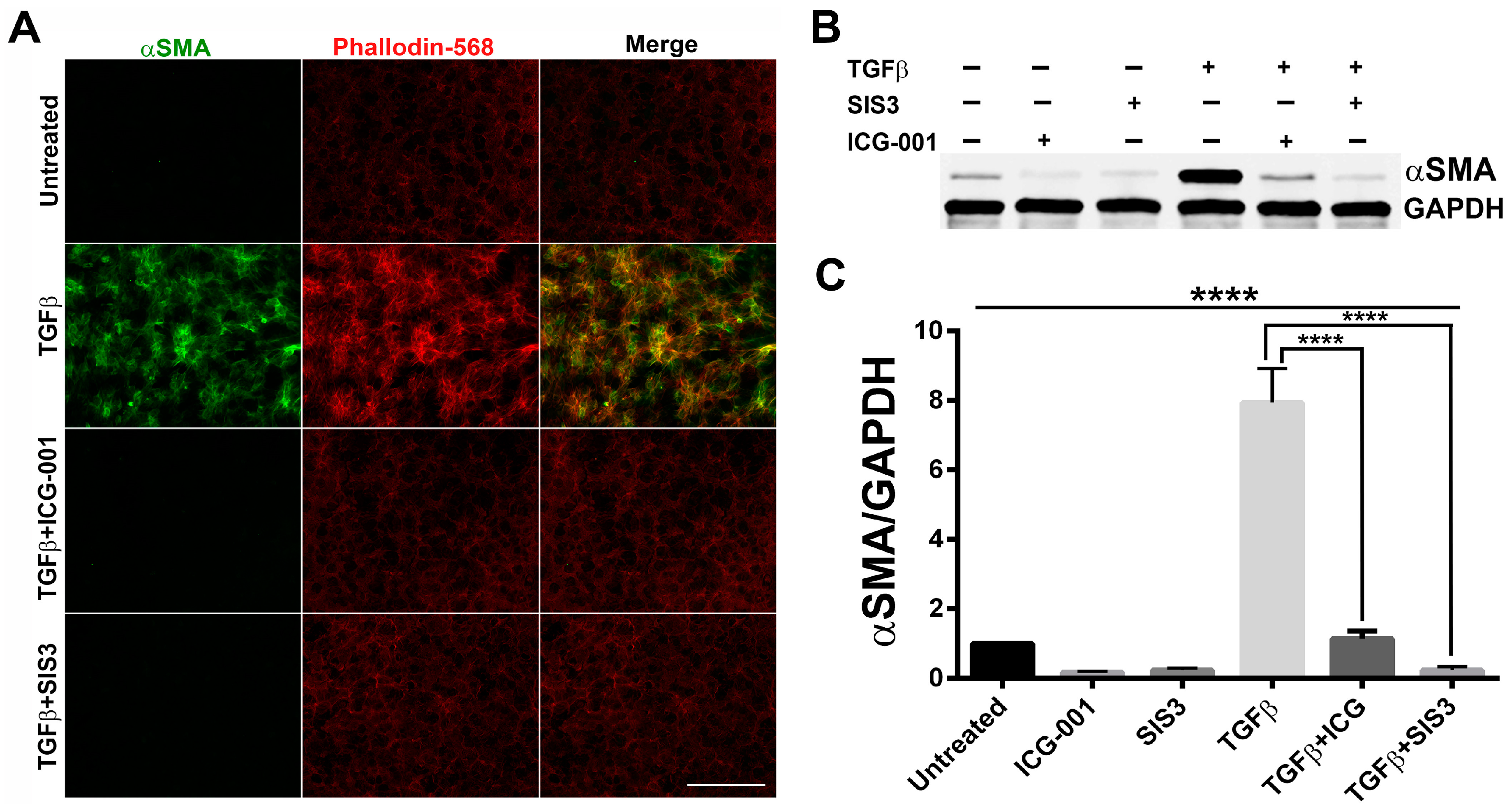
2.1. Smad3 Signaling Inhibition Blocks TGF-B-Induced EMT in Lens Explants
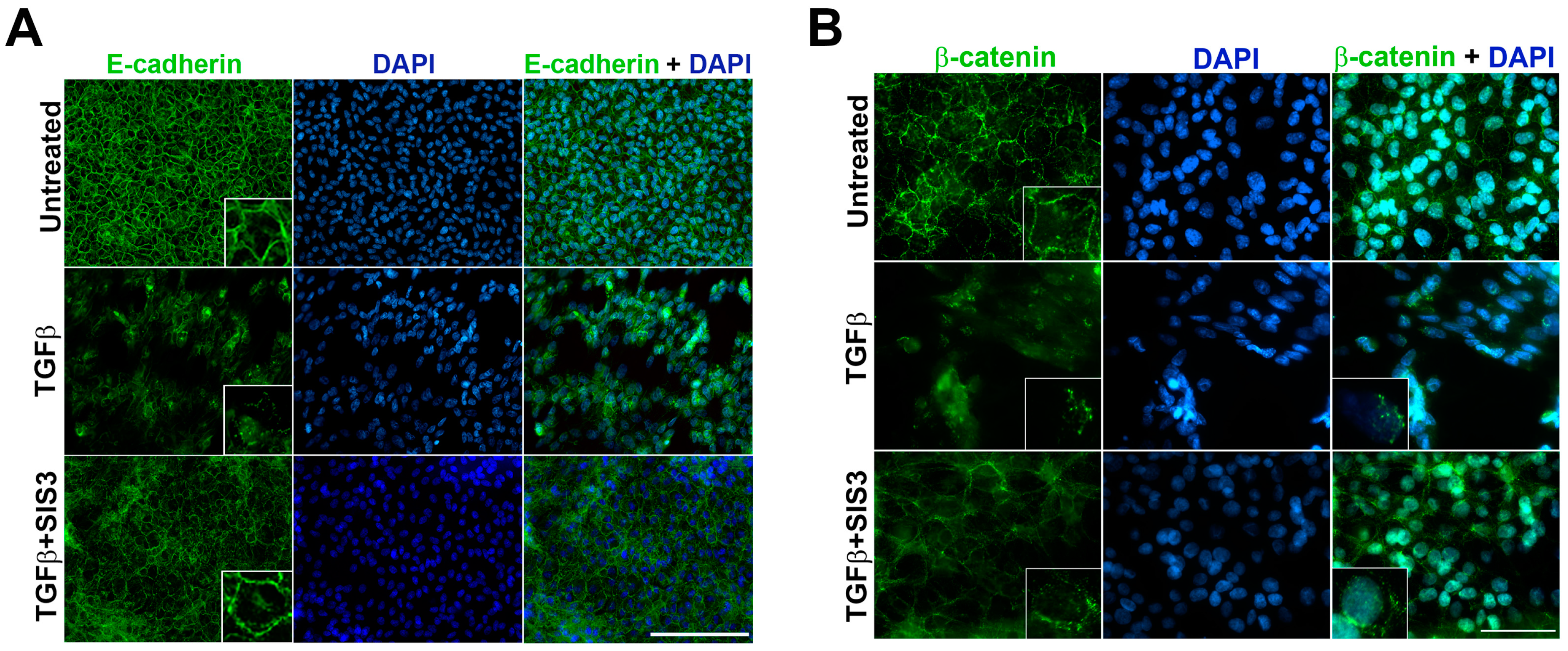
2.2. Smad3 Signaling Inhibition Blocks B-Catenin and E-Cadherin Delocalization
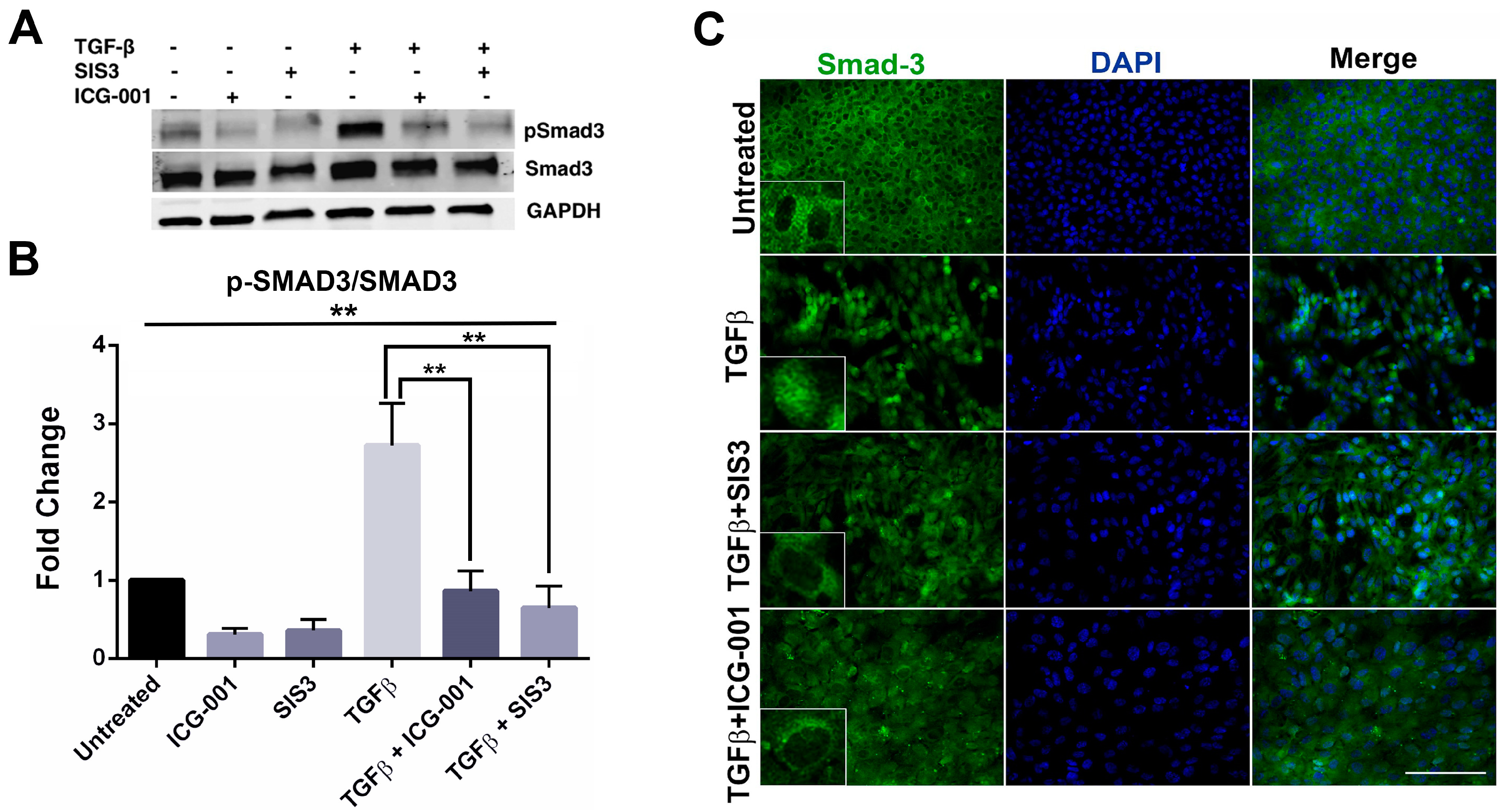
2.3. β-Catenin Inhibition Blocks Smad3 Activation and Nuclear Localization
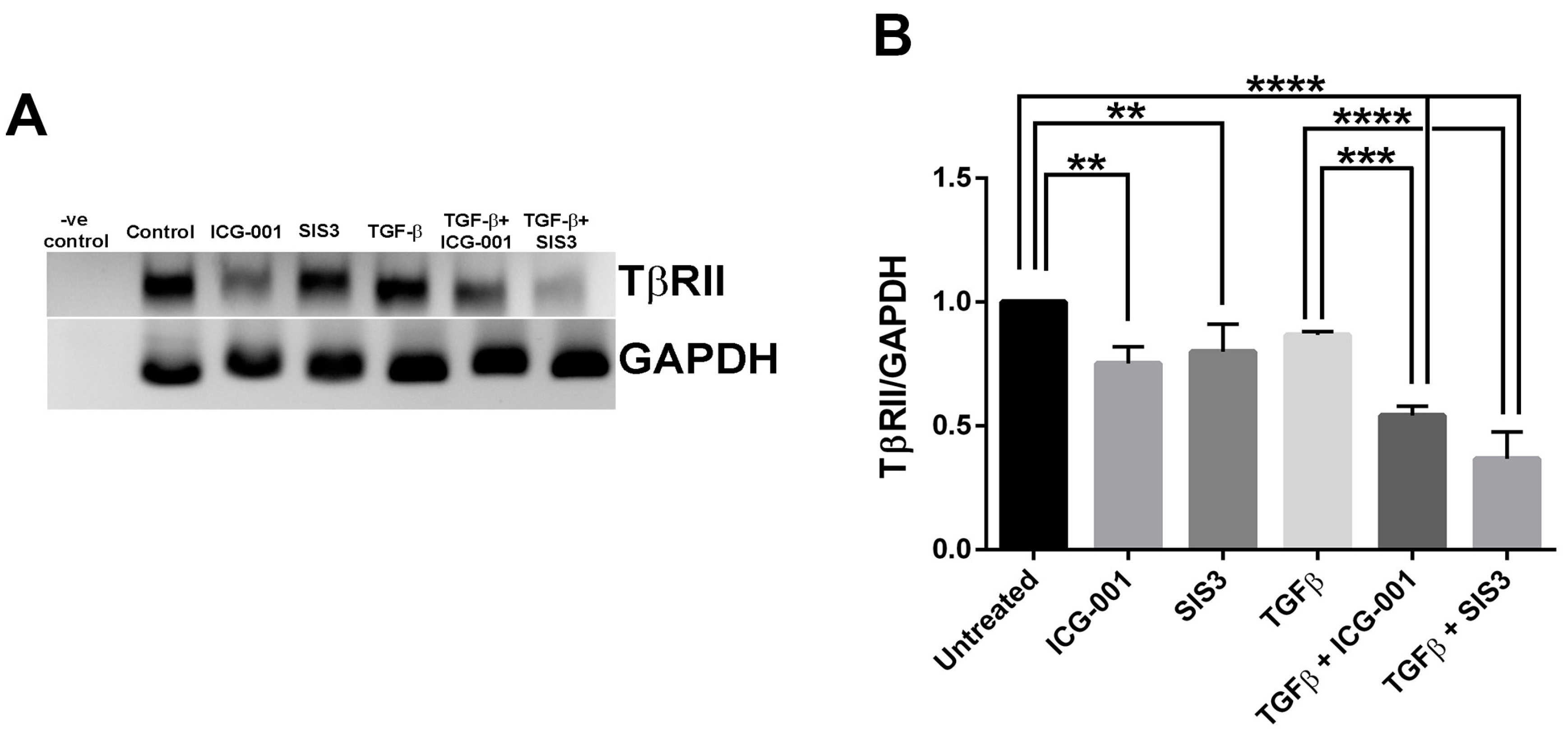
2.4. β-Catenin and Smad3 Inhibition Decreases TGF-B Receptor II mRNA Levels
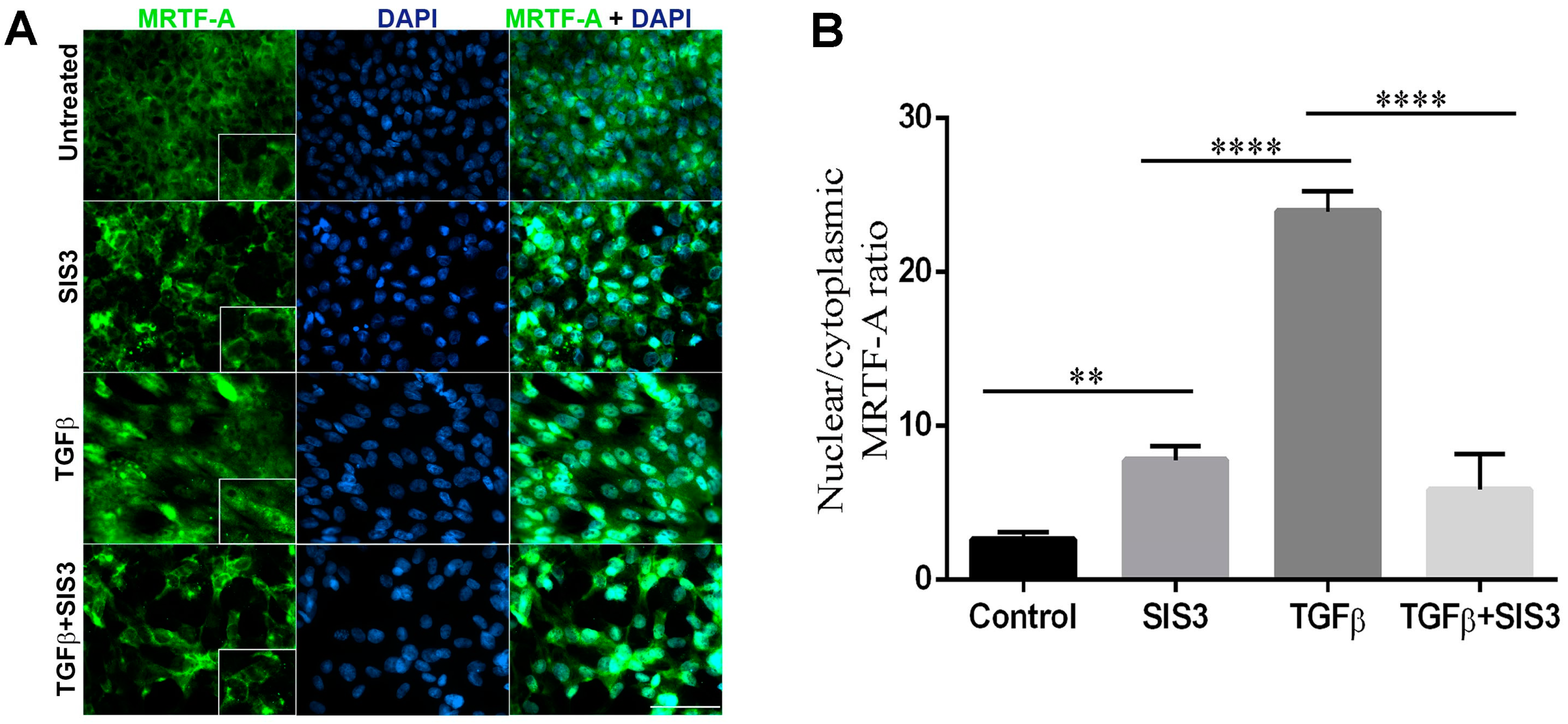
2.5. Smad3 Activation Inhibition Blocks MRTF-A Delocalization
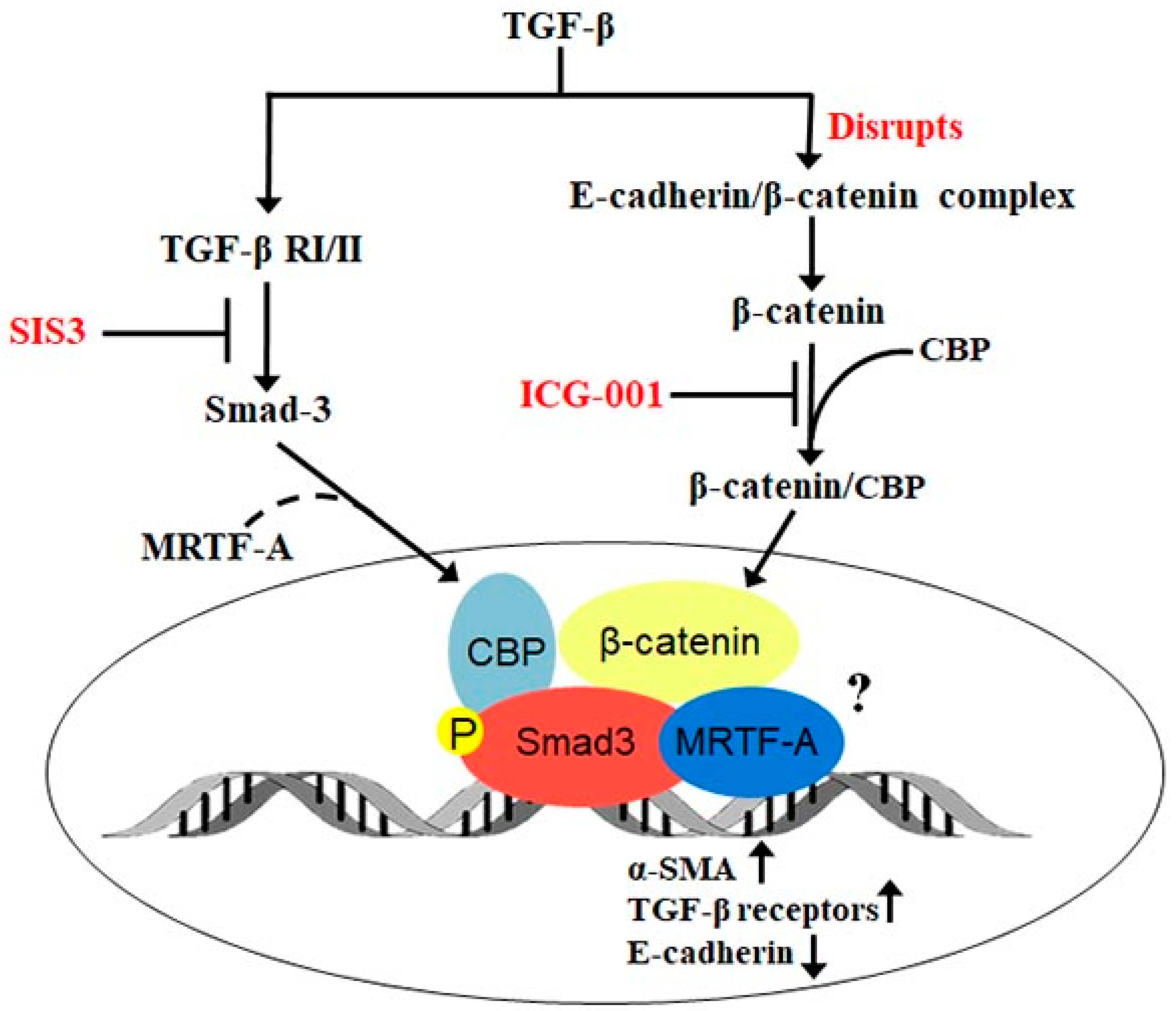
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Culturing Rat Lens Epithelial Explants
4.3. Treatment of Lecs with TGF-B and Inhibitors
4.4. Western Blot Analyses
4.5. Immunocytochemistry
4.6. Reverse Transcription (RT)–PCR Assay
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Harocopos, G.J.; Alvares, K.M.; Kolker, A.E.; Beebe, D.C. Human age-related cataract and lens epithelial cell death. Investig. Ophthalmol. Vis. Sci. 1998, 39, 2696–2706. [Google Scholar] [PubMed]
- Pascolini, D.; Mariotti, S.P. Global estimates of visual impairment: 2010. Br. J. Ophthalmol. 2012, 96, 614–618. [Google Scholar] [CrossRef] [PubMed]
- WHO. Visual Impairment and Blindness. Available online: http://www.who.int/mediacentre/factsheets/fs282/en/index.html (accessed on 23 July 2018).
- Awasthi, N.; Guo, S.; Wagner, B.J. Posterior capsular opacification: A problem reduced but not yet eradicated. Arch. Ophthalmol. 2009, 127, 555–562. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Marcantonio, J.M.; Vrensen, G.F. Cell biology of posterior capsular opacification. Eye (Lond) 1999, 13 Pt 3b, 484–488. [Google Scholar] [CrossRef]
- Lee, J.M.; Dedhar, S.; Kalluri, R.; Thompson, E.W. The epithelial-mesenchymal transition: New insights in signaling, development, and disease. J. Cell Biol. 2006, 172, 973–981. [Google Scholar] [CrossRef]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Yilmaz, M.; Christofori, G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 2009, 28, 15–33. [Google Scholar] [CrossRef]
- Vyalov, S.L.; Gabbiani, G.; Kapanci, Y. Rat alveolar myofibroblasts acquire alpha-smooth muscle actin expression during bleomycin-induced pulmonary fibrosis. Am. J. Pathol. 1993, 143, 1754–1765. [Google Scholar]
- Sato, M.; Muragaki, Y.; Saika, S.; Roberts, A.B.; Ooshima, A. Targeted disruption of TGF-beta1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J. Clin. Investig. 2003, 112, 1486–1494. [Google Scholar] [CrossRef]
- Koitabashi, N.; Danner, T.; Zaiman, A.L.; Pinto, Y.M.; Rowell, J.; Mankowski, J.; Zhang, D.; Nakamura, T.; Takimoto, E.; Kass, D.A. Pivotal role of cardiomyocyte TGF-beta signaling in the murine pathological response to sustained pressure overload. J. Clin. Investig. 2011, 121, 2301–2312. [Google Scholar] [CrossRef]
- De Iongh, R.U.; Wederell, E.; Lovicu, F.J.; McAvoy, J.W. Transforming growth factor-beta-induced epithelial-mesenchymal transition in the lens: A model for cataract formation. Cells Tissues Organs 2005, 179, 43–55. [Google Scholar] [CrossRef]
- Gordon-Thomson, C.; de Iongh, R.U.; Hales, A.M.; Chamberlain, C.G.; McAvoy, J.W. Differential cataractogenic potency of TGF-beta1, -beta2, and -beta3 and their expression in the postnatal rat eye. Invest. Ophthalmol. Vis. Sci. 1998, 39, 1399–1409. [Google Scholar] [PubMed]
- Cousins, S.W.; McCabe, M.M.; Danielpour, D.; Streilein, J.W. Identification of transforming growth factor-beta as an immunosuppressive factor in aqueous humor. Investig. Ophthalmol. Vis. Sci. 1991, 32, 2201–2211. [Google Scholar] [PubMed]
- Lovicu, F.J.; Schulz, M.W.; Hales, A.M.; Vincent, L.N.; Overbeek, P.A.; Chamberlain, C.G.; McAvoy, J.W. TGFbeta induces morphological and molecular changes similar to human anterior subcapsular cataract. Br. J. Ophthalmol. 2002, 86, 220–226. [Google Scholar] [CrossRef]
- Robertson, J.V.; Nathu, Z.; Najjar, A.; Dwivedi, D.; Gauldie, J.; West-Mays, J.A. Adenoviral gene transfer of bioactive TGFbeta1 to the rodent eye as a novel model for anterior subcapsular cataract. Mol. Vis. 2007, 13, 457–469. [Google Scholar]
- Saika, S.; Kono-Saika, S.; Ohnishi, Y.; Sato, M.; Muragaki, Y.; Ooshima, A.; Flanders, K.C.; Yoo, J.; Anzano, M.; Liu, C.Y.; et al. Smad3 signaling is required for epithelial-mesenchymal transition of lens epithelium after injury. Am. J. Pathol. 2004, 164, 651–663. [Google Scholar] [CrossRef]
- Wallentin, N.; Wickstrom, K.; Lundberg, C. Effect of cataract surgery on aqueous TGF-beta and lens epithelial cell proliferation. Investig. Ophthalmol. Vis. Sci. 1998, 39, 1410–1418. [Google Scholar] [PubMed]
- Jampel, H.D.; Roche, N.; Stark, W.J.; Roberts, A.B. Transforming growth factor-beta in human aqueous humor. Curr. Eye Res. 1990, 9, 963–969. [Google Scholar] [CrossRef]
- Hales, A.M.; Chamberlain, C.G.; McAvoy, J.W. Cataract induction in lenses cultured with transforming growth factor-beta. Invest. Ophthalmol. Vis. Sci. 1995, 36, 1709–1713. [Google Scholar] [PubMed]
- Dwivedi, D.J.; Pino, G.; Banh, A.; Nathu, Z.; Howchin, D.; Margetts, P.; Sivak, J.G.; West-Mays, J.A. Matrix Metalloproteinase Inhibitors Suppress Transforming Growth Factor-β-Induced Subcapsular Cataract Formation. Am. J. Pathol. 2006, 168, 69–79. [Google Scholar] [CrossRef]
- Banh, A.; Deschamps, P.A.; Vijayan, M.M.; Sivak, J.G.; West-Mays, J.A. The role of Hsp70 and Hsp90 in TGF-beta-induced epithelial-to-mesenchymal transition in rat lens epithelial explants. Mol. Vis. 2007, 13, 2248–2262. [Google Scholar]
- Korol, A.; Pino, G.; Dwivedi, D.; Robertson, J.V.; Deschamps, P.A.; West-Mays, J.A. Matrix metalloproteinase-9-null mice are resistant to TGF-beta-induced anterior subcapsular cataract formation. Am. J. Pathol. 2014, 184, 2001–2012. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Hales, A.M.; Chamberlain, C.G.; McAvoy, J.W. Induction of cataract-like changes in rat lens epithelial explants by transforming growth factor beta. Investig. Ophthalmol. Vis. Sci. 1994, 35, 388–401. [Google Scholar] [PubMed]
- Shi, Y.; Massague, J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef]
- Massague, J.; Seoane, J.; Wotton, D. Smad transcription factors. Genes Dev. 2005, 19, 2783–2810. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.C.; Phillips, A.O. Interaction between the transforming growth factor-beta type II receptor/Smad pathway and beta-catenin during transforming growth factor-beta1-mediated adherens junction disassembly. Am. J. Pathol. 2002, 160, 1619–1628. [Google Scholar] [CrossRef]
- Saika, S. TGFbeta pathobiology in the eye. Lab. Investig. 2006, 86, 106–115. [Google Scholar] [CrossRef]
- Banh, A.; Deschamps, P.A.; Gauldie, J.; Overbeek, P.A.; Sivak, J.G.; West-Mays, J.A. Lens-specific expression of TGF-beta induces anterior subcapsular cataract formation in the absence of Smad3. Investig. Ophthalmol Vis. Sci. 2006, 47, 3450–3460. [Google Scholar] [CrossRef]
- Korol, A.; Taiyab, A.; West-Mays, J.A. RhoA/ROCK signaling regulates TGFbeta-induced epithelial-mesenchymal transition of lens epithelial cells through MRTF-A. Mol. Med. 2016, 22. [Google Scholar] [CrossRef]
- Taiyab, A.; Korol, A.; Deschamps, P.A.; West-Mays, J.A. beta-Catenin/CBP-Dependent Signaling Regulates TGF-beta-Induced Epithelial to Mesenchymal Transition of Lens Epithelial Cells. Investig. Ophthalmol. Vis. Sci. 2016, 57, 5736–5747. [Google Scholar] [CrossRef]
- Charbonney, E.; Speight, P.; Masszi, A.; Nakano, H.; Kapus, A. beta-catenin and Smad3 regulate the activity and stability of myocardin-related transcription factor during epithelial-myofibroblast transition. Mol. Biol. Cell 2011, 22, 4472–4485. [Google Scholar] [CrossRef]
- Zhou, B.; Liu, Y.; Kahn, M.; Ann, D.K.; Han, A.; Wang, H.; Nguyen, C.; Flodby, P.; Zhong, Q.; Krishnaveni, M.S.; et al. Interactions between beta-catenin and transforming growth factor-beta signaling pathways mediate epithelial-mesenchymal transition and are dependent on the transcriptional co-activator cAMP-response element-binding protein (CREB)-binding protein (CBP). J. Biol. Chem. 2012, 287, 7026–7038. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Zhang, J.; Tan, T.K.; Lyons, J.G.; Zhao, H.; Niu, B.; Lee, S.R.; Tsatralis, T.; Zhao, Y.; Wang, Y.; et al. Association of beta-catenin with P-Smad3 but not LEF-1 dissociates in vitro profibrotic from anti-inflammatory effects of TGF-beta1. J. Cell. Sci. 2013, 126, 67–76. [Google Scholar] [CrossRef]
- Massague, J. TGFbeta signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Jinnin, M.; Ihn, H.; Tamaki, K. Characterization of SIS3, a novel specific inhibitor of Smad3, and its effect on transforming growth factor-beta1-induced extracellular matrix expression. Mol. Pharmacol. 2006, 69, 597–607. [Google Scholar] [CrossRef]
- Liu, X.; Sun, Y.; Constantinescu, S.N.; Karam, E.; Weinberg, R.A.; Lodish, H.F. Transforming growth factor beta-induced phosphorylation of Smad3 is required for growth inhibition and transcriptional induction in epithelial cells. Proc. Natl. Acad. Sci. USA 1997, 94, 10669–10674. [Google Scholar] [CrossRef]
- Attisano, L.; Wrana, J.L. Signal integration in TGF-beta, WNT, and Hippo pathways. F1000Prime Rep. 2013, 5, 17. [Google Scholar] [CrossRef]
- Gupta, M.; Korol, A.; West-Mays, J.A. Nuclear translocation of myocardin-related transcription factor-A during transforming growth factor beta-induced epithelial to mesenchymal transition of lens epithelial cells. Mol. Vis. 2013, 19, 1017–1028. [Google Scholar]
- Saika, S.; Miyamoto, T.; Ishida, I.; Shirai, K.; Ohnishi, Y.; Ooshima, A.; McAvoy, J.W. TGFbeta-Smad signalling in postoperative human lens epithelial cells. Br. J. Ophthalmol. 2002, 86, 1428–1433. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.B.; Tian, F.; Byfield, S.D.; Stuelten, C.; Ooshima, A.; Saika, S.; Flanders, K.C. Smad3 is key to TGF-beta-mediated epithelial-to-mesenchymal transition, fibrosis, tumor suppression and metastasis. Cytokine Growth Factor Rev. 2006, 17, 19–27. [Google Scholar] [CrossRef]
- Han, G.; Li, A.G.; Liang, Y.Y.; Owens, P.; He, W.; Lu, S.; Yoshimatsu, Y.; Wang, D.; Ten Dijke, P.; Lin, X.; et al. Smad7-induced beta-catenin degradation alters epidermal appendage development. Dev. Cell 2006, 11, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Fishilevich, S.; Nudel, R.; Rappaport, N.; Hadar, R.; Plaschkes, I.; Iny Stein, T.; Rosen, N.; Kohn, A.; Twik, M.; Safran, M.; et al. GeneHancer: Genome-wide integration of enhancers and target genes in GeneCards. Database (Oxford) 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Henderson, W.R., Jr.; Chi, E.Y.; Ye, X.; Nguyen, C.; Tien, Y.T.; Zhou, B.; Borok, Z.; Knight, D.A.; Kahn, M. Inhibition of Wnt/beta-catenin/CREB binding protein (CBP) signaling reverses pulmonary fibrosis. Proc. Natl. Acad. Sci. USA 2010, 107, 14309–14314. [Google Scholar] [CrossRef]
- Hao, S.; He, W.; Li, Y.; Ding, H.; Hou, Y.; Nie, J.; Hou, F.F.; Kahn, M.; Liu, Y. Targeted inhibition of beta-catenin/CBP signaling ameliorates renal interstitial fibrosis. J. Am. Soc. Nephrol. 2011, 22, 1642–1653. [Google Scholar] [CrossRef]
- Radhakrishnan, I.; Perez-Alvarado, G.C.; Parker, D.; Dyson, H.J.; Montminy, M.R.; Wright, P.E. Structural analyses of CREB-CBP transcriptional activator-coactivator complexes by NMR spectroscopy: Implications for mapping the boundaries of structural domains. J. Mol. Biol. 1999, 287, 859–865. [Google Scholar] [CrossRef] [PubMed]
- Ernst, P.; Wang, J.; Huang, M.; Goodman, R.H.; Korsmeyer, S.J. MLL and CREB bind cooperatively to the nuclear coactivator CREB-binding protein. Mol. Cell. Biol. 2001, 21, 2249–2258. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Guo, S.; Wang, C.C.; Sun, X.; Wang, D.; Xu, N.; Jin, S.F.; Li, K.Z. Cross-talk between TGF-beta/Smad pathway and Wnt/beta-catenin pathway in pathological scar formation. Int. J. Clin. Exp. Pathol. 2015, 8, 7631–7639. [Google Scholar]
- Nawshad, A.; Medici, D.; Liu, C.C.; Hay, E.D. TGFbeta3 inhibits E-cadherin gene expression in palate medial-edge epithelial cells through a Smad2-Smad4-LEF1 transcription complex. J. Cell. Sci 2007, 120, 1646–1653. [Google Scholar] [CrossRef]
- Schindelin, J.; Rueden, C.T.; Hiner, M.C.; Eliceiri, K.W. The ImageJ ecosystem: An open platform for biomedical image analysis. Mol. Reprod. Dev. 2015, 82, 518–529. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taiyab, A.; Holms, J.; West-Mays, J.A. β-Catenin/Smad3 Interaction Regulates Transforming Growth Factor-β-Induced Epithelial to Mesenchymal Transition in the Lens. Int. J. Mol. Sci. 2019, 20, 2078. https://doi.org/10.3390/ijms20092078
Taiyab A, Holms J, West-Mays JA. β-Catenin/Smad3 Interaction Regulates Transforming Growth Factor-β-Induced Epithelial to Mesenchymal Transition in the Lens. International Journal of Molecular Sciences. 2019; 20(9):2078. https://doi.org/10.3390/ijms20092078
Chicago/Turabian StyleTaiyab, Aftab, Julie Holms, and Judith A. West-Mays. 2019. "β-Catenin/Smad3 Interaction Regulates Transforming Growth Factor-β-Induced Epithelial to Mesenchymal Transition in the Lens" International Journal of Molecular Sciences 20, no. 9: 2078. https://doi.org/10.3390/ijms20092078