LKB1 and Tumor Metabolism: The Interplay of Immune and Angiogenic Microenvironment in Lung Cancer

 , ,
, ,
Abstract
:1. Introduction
2. LKB1 and Tumor Metabolism
2.1. Inhibition of Anabolic Pathways
2.2. Activation of Catabolic Pathways
3. LKB1 and Angiogenesis
4. LKB1 and Response to Non-Immunotherapy Treatments
4.1. Platinum-Based Chemotherapy
4.2. Anti-Angiogenic Treatment
4.3. Other Non-Chemotherapy Agents
5. LKB1 and Immunotherapy
5.1. LKB1 and Immune Microenvironment
5.2. LKB1 and Response to Immunotherapy
6. The Interplay between Immune and Angiogenic Microenvironment
7. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Shackelford, D.B.; Shaw, R.J. The LKB1-AMPK pathway: Metabolism and growth control in tumour suppression. Nat. Rev. Cancer 2009, 9, 563–575. [Google Scholar] [CrossRef]
- Lizcano, J.M.; Göransson, O.; Toth, R.; Deak, M.; Morrice, N.A.; Boudeau, J.; Hawley, S.A.; Udd, L.; Mäkelä, T.P.; Hardie, D.G.; et al. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J. 2004, 23, 833–843. [Google Scholar] [CrossRef]
- Wang, Y.-S.; Chen, J.; Cui, F.; Wang, H.; Wang, S.; Hang, W.; Zeng, Q.; Quan, C.-S.; Zhai, Y.-X.; Wang, J.-W.; et al. LKB1 is a DNA damage response protein that regulates cellular sensitivity to PARP inhibitors. Oncotarget 2016, 7, 73389–73401. [Google Scholar] [CrossRef]
- Liang, X.; Wang, P.; Gao, Q.; Tao, X. Exogenous activation of LKB1/AMPK signaling induces G1 arrest in cells with endogenous LKB1 expression. Mol. Med. Rep. 2014, 9, 1019–1024. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Wang, P.; Gao, Q.; Xiang, T.; Tao, X. Endogenous LKB1 knockdown accelerates G1/S transition through p53 and p16 pathways. Cancer Biol. Ther. 2010, 9, 156–160. [Google Scholar] [CrossRef] [PubMed]
- Hezel, A.F.; Bardeesy, N. LKB1; linking cell structure and tumor suppression. Oncogene 2008, 27, 6908–6919. [Google Scholar] [CrossRef] [PubMed]
- Skoulidis, F.; Goldberg, M.E.; Greenawalt, D.M.; Hellmann, M.D.; Awad, M.M.; Gainor, J.F.; Schrock, A.B.; Hartmaier, R.J.; Trabucco, S.E.; Gay, L.; et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS -Mutant Lung Adenocarcinoma. Cancer Discov. 2018, 8, 822–835. [Google Scholar] [CrossRef]
- Kitajima, S.; Ivanova, E.; Guo, S.; Yoshida, R.; Campisi, M.; Sundararaman, S.K.; Tange, S.; Mitsuishi, Y.; Thai, T.C.; Masuda, S.; et al. Suppression of STING Associated with LKB1 Loss in KRAS-Driven Lung Cancer. Cancer Discov. 2019, 9, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Hemminki, A. The molecular basis and clinical aspects of Peutz–Jeghers syndrome. Cell. Mol. Life Sci. 1999, 55, 735–750. [Google Scholar] [CrossRef] [PubMed]
- Gill, R.K.; Yang, S.-H.; Meerzaman, D.; Mechanic, L.E.; Bowman, E.D.; Jeon, H.-S.; Roy Chowdhuri, S.; Shakoori, A.; Dracheva, T.; Hong, K.-M.; et al. Frequent homozygous deletion of the LKB1/STK11 gene in non-small cell lung cancer. Oncogene 2011, 30, 3784–3791. [Google Scholar] [CrossRef]
- Wingo, S.N.; Gallardo, T.D.; Akbay, E.A.; Liang, M.-C.; Contreras, C.M.; Boren, T.; Shimamura, T.; Miller, D.S.; Sharpless, N.E.; Bardeesy, N.; et al. Somatic LKB1 Mutations Promote Cervical Cancer Progression. PLoS ONE 2009, 4, e5137. [Google Scholar] [CrossRef]
- Tanwar, P.S.; Mohapatra, G.; Chiang, S.; Engler, D.A.; Zhang, L.; Kaneko-Tarui, T.; Ohguchi, Y.; Birrer, M.J.; Teixeira, J.M. Loss of LKB1 and PTEN tumor suppressor genes in the ovarian surface epithelium induces papillary serous ovarian cancer. Carcinogenesis 2014, 35, 546–553. [Google Scholar] [CrossRef]
- Morton, J.P.; Jamieson, N.B.; Karim, S.A.; Athineos, D.; Ridgway, R.A.; Nixon, C.; McKay, C.J.; Carter, R.; Brunton, V.G.; Frame, M.C.; et al. LKB1 Haploinsufficiency Cooperates With Kras to Promote Pancreatic Cancer Through Suppression of p21-Dependent Growth Arrest. Gastroenterology 2010, 139, 586.e6–597.e6. [Google Scholar] [CrossRef]
- Guldberg, P.; Straten, P.T.; Ahrenkiel, V.; Seremet, T.; Kirkin, A.F.; Zeuthen, J. Somatic mutation of the Peutz–Jeghers syndrome gene, LKB1/STK11, in malignant melanoma. Oncogene 1999, 18, 1777–1780. [Google Scholar] [CrossRef]
- Shen, Z.; Wen, X.F.; Lan, F.; Shen, Z.Z.; Shao, Z.M. The tumor suppressor gene LKB1 is associated with prognosis in human breast carcinoma. Clin. Cancer Res. 2002, 8, 2085–2090. [Google Scholar]
- Sengupta, S.; Nagalingam, A.; Muniraj, N.; Bonner, M.Y.; Mistriotis, P.; Afthinos, A.; Kuppusamy, P.; Lanoue, D.; Cho, S.; Korangath, P.; et al. Activation of tumor suppressor LKB1 by honokiol abrogates cancer stem-like phenotype in breast cancer via inhibition of oncogenic Stat3. Oncogene 2017, 36, 5709–5721. [Google Scholar] [CrossRef]
- Skoulidis, F.; Byers, L.A.; Diao, L.; Papadimitrakopoulou, V.A.; Tong, P.; Izzo, J.; Behrens, C.; Kadara, H.; Parra, E.R.; Canales, J.R.; et al. Co-occurring Genomic Alterations Define Major Subsets of KRAS-Mutant Lung Adenocarcinoma with Distinct Biology, Immune Profiles, and Therapeutic Vulnerabilities. Cancer Discov. 2015, 5, 860–877. [Google Scholar] [CrossRef]
- Shackelford, D.B.; Mihaylova, M.M.; Mery, A.; Shaw, R.J.; Egan, D.F.; Gwinn, D.M.; Turk, B.E.; Vasquez, D.S. AMPK Phosphorylation of Raptor Mediates a Metabolic Checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar]
- Neumann, D.; Fryer, L.G.D.; Leiper, F.C.; Carling, D.; Schlattner, U.; Johnstone, S.R.; Dickerson, K.; Carlson, M.; Wallimann, T.; Woods, A. LKB1 Is the Upstream Kinase in the AMP-Activated Protein Kinase Cascade. Curr. Biol. 2003, 13, 2004–2008. [Google Scholar]
- Shaw, R.J.; Kosmatka, M.; Bardeesy, N.; Hurley, R.L.; Witters, L.A.; DePinho, R.A.; Cantley, L.C. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc. Natl. Acad. Sci. USA 2004, 101, 3329–3335. [Google Scholar] [CrossRef]
- Carling, D.; Zammit, V.A.; Hardie, D.G. A common bicyclic protein kinase cascade inactivates the regulatory enzymes of fatty acid and cholesterol biosynthesis. FEBS Lett. 1987, 223, 217–222. [Google Scholar] [CrossRef]
- Munday, M.R.; Carling, D.; Hardie, D.G. Negative interactions between phosphorylation of acetyl-CoA carboxylase by the cyclic AMP-dependent and AMP-activated protein kinases. FEBS Lett. 1988, 235, 144–148. [Google Scholar] [CrossRef]
- Beckers, A.; Organe, S.; Timmermans, L.; Scheys, K.; Peeters, A.; Brusselmans, K.; Verhoeven, G.; Swinnen, J.V. Chemical Inhibition of Acetyl-CoA Carboxylase Induces Growth Arrest and Cytotoxicity Selectively in Cancer Cells. Cancer Res. 2007, 67, 8180–8187. [Google Scholar] [CrossRef]
- Orita, H.; Coulter, J.; Lemmon, C.; Tully, E.; Vadlamudi, A.; Medghalchi, S.M.; Kuhajda, F.P.; Gabrielson, E. Selective Inhibition of Fatty Acid Synthase for Lung Cancer Treatment. Clin. Cancer Res. 2007, 23, 7139–7145. [Google Scholar] [CrossRef]
- Bultot, L.; Guigas, B.; Von Wilamowitz-Moellendorff, A.; Maisin, L.; Vertommen, D.; Hussain, N.; Beullens, M.; Guinovart, J.J.; Foretz, M.; Viollet, B.; et al. AMP-activated protein kinase phosphorylates and inactivates liver glycogen synthase. Biochem. J. 2012, 443, 193–203. [Google Scholar] [CrossRef]
- Li, Y.; Xu, S.; Mihaylova, M.M.; Zheng, B.; Hou, X.; Jiang, B.; Park, O.; Luo, Z.; Lefai, E.; Shyy, J.Y.-J.; et al. AMPK Phosphorylates and Inhibits SREBP Activity to Attenuate Hepatic Steatosis and Atherosclerosis in Diet-Induced Insulin-Resistant Mice. Cell Metab. 2011, 13, 376–388. [Google Scholar] [CrossRef]
- Kawaguchi, T.; Osatomi, K.; Yamashita, H.; Kabashima, T.; Uyeda, K. Mechanism for Fatty Acid “Sparing” Effect on Glucose-induced Transcription. J. Biol. Chem. 2002, 277, 3829–3835. [Google Scholar] [CrossRef]
- Hong, Y.H.; Varanasi, U.S.; Yang, W.; Leff, T. AMP-activated protein kinase regulates HNF4α transcriptional activity by inhibiting dimer formation and decreasing protein stability. J. Biol. Chem. 2003, 278, 27495–27501. [Google Scholar] [CrossRef]
- Jeon, S.M.; Chandel, N.S.; Hay, N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature 2012, 485, 661–665. [Google Scholar] [CrossRef]
- Leprivier, G.; Remke, M.; Rotblat, B.; Dubuc, A.; Mateo, A.R.F.; Kool, M.; Agnihotri, S.; El-Naggar, A.; Yu, B.; Prakash Somasekharan, S.; et al. The eEF2 kinase confers resistance to nutrient deprivation by blocking translation elongation. Cell 2013, 153, 1064–1079. [Google Scholar] [CrossRef]
- Faller, W.J.; Jackson, T.J.; Knight, J.R.P.; Ridgway, R.A.; Jamieson, T.; Karim, S.A.; Jones, C.; Radulescu, S.; Huels, D.J.; Myant, K.B.; et al. mTORC1-mediated translational elongation limits intestinal tumour initiation and growth. Nature 2015, 517, 497–500. [Google Scholar] [CrossRef]
- Samborska, B.; Faubert, B.; Avizonis, D.; Izreig, S.; DeBerardinis, R.J.; Viollet, B.; Jones, R.G.; Dupuy, F.; Dong, Z.; Griss, T.; et al. AMPK Is a Negative Regulator of the Warburg Effect and Suppresses Tumor Growth In Vivo. Cell Metab. 2012, 17, 113–124. [Google Scholar]
- Kim, J.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef]
- Denko, N.C. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer 2008, 8, 705–713. [Google Scholar] [CrossRef]
- Chavez, J.A.; Roach, W.G.; Keller, S.R.; Lane, W.S.; Lienhard, G.E. Inhibition of GLUT4 translocation by Tbc1d1, a Rab GTPase-activating protein abundant in skeletal muscle, is partially relieved by AMP-activated protein kinase activation. J. Biol. Chem. 2008, 283, 9187–9195. [Google Scholar] [CrossRef]
- Tamargo-Gómez, I.; Mariño, G. AMPK: Regulation of Metabolic Dynamics in the Context of Autophagy. Int. J. Mol. Sci. 2018, 19, 3812. [Google Scholar] [CrossRef]
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; Gelino, S.; Kohnz, R.A.; Mair, W.; Vasquez, D.S.; Joshi, A.; Gwinn, D.M.; Taylor, R.; et al. Phosphorylation of ULK1 (hATG1) by AMP-Activated Protein Kinase Connects Energy Sensing to Mitophagy. Science (80-) 2011, 331, 456–461. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Zhuang, Z.-G.; Di, G.-H.; Shen, Z.-Z.; Ding, J.; Shao, Z.-M. Enhanced Expression of LKB1 in Breast Cancer Cells Attenuates Angiogenesis, Invasion, and Metastatic Potential. Mol. Cancer Res. 2006, 4, 843–849. [Google Scholar] [CrossRef]
- Okon, I.S.; Coughlan, K.A.; Zhang, C.; Moriasi, C.; Ding, Y.; Song, P.; Zhang, W.; Li, G.; Zou, M.-H. Protein kinase LKB1 promotes RAB7-mediated neuropilin-1 degradation to inhibit angiogenesis. J. Clin. Investig. 2014, 124, 4590–4602. [Google Scholar] [CrossRef]
- Miao, H.Q.; Klagsbrun, M. Neuropilin is a mediator of angiogenesis. Cancer Metastasis Rev. 2000, 19, 29–37. [Google Scholar] [CrossRef]
- Zulato, E.; Ciccarese, F.; Nardo, G.; Pinazza, M.; Agnusdei, V.; Silic-Benussi, M.; Ciminale, V.; Indraccolo, S. Involvement of NADPH Oxidase 1 in Liver Kinase B1-Mediated Effects on Tumor Angiogenesis and Growth. Front. Oncol. 2018, 8, 195. [Google Scholar] [CrossRef]
- Kamata, T. Roles of Nox1 and other Nox isoforms in cancer development. Cancer Sci. 2009, 100, 1382–1388. [Google Scholar] [CrossRef]
- Xia, C.; Meng, Q.; Liu, L.Z.; Rojanasakul, Y.; Wang, X.R.; Jiang, B.H. Reactive oxygen species regulate angiogenesis and tumor growth through vascular endothelial growth factor. Cancer Res. 2007, 67, 10823–10830. [Google Scholar] [CrossRef]
- Arbiser, J.L.; Brat, D.; Hunter, S.; D’Armiento, J.; Henske, E.P.; Arbiser, Z.K.; Bai, X.; Goldberg, G.; Cohen, C.; Weiss, S.W. Tuberous sclerosis-associated lesions of the kidney, brain, and skin are angiogenic neoplasms. J. Am. Acad. Dermatol. 2002, 46, 376–380. [Google Scholar] [CrossRef]
- Fernandes, N.; Sun, Y.; Chen, S.; Paul, P.; Shaw, R.J.; Cantley, L.C.; Price, B.D. DNA Damage-induced Association of ATM with Its Target Proteins Requires a Protein Interaction Domain in the N Terminus of ATM. J. Biol. Chem. 2005, 280, 15158–15164. [Google Scholar] [CrossRef]
- Sapkota, G.P.; Deak, M.; Kieloch, A.; Morrice, N.; Goodarzi, A.A.; Smythe, C.; Shiloh, Y.; Lees-Miller, S.P.; Alessi, D.R. Ionizing radiation induces ataxia telangiectasia mutated kinase (ATM)-mediated phosphorylation of LKB1/STK11 at Thr-366. Biochem. J. 2002, 368, 507–516. [Google Scholar] [CrossRef]
- Feng, F.Y.; de Bono, J.S.; Rubin, M.A.; Knudsen, K.E. Chromatin to Clinic: The Molecular Rationale for PARP1 Inhibitor Function. Mol. Cell 2015, 58, 925–934. [Google Scholar] [CrossRef]
- Yap, T.A.; Lau, A.; O’Connor, M.J.; Schellens, J.H.M.; Tutt, A.; Boss, D.S.; Carmichael, J.; Mortimer, P.; de Bono, J.S.; Swaisland, H.; et al. Inhibition of Poly(ADP-Ribose) Polymerase in Tumors from BRCA Mutation Carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar]
- Zulato, E.; Ciccarese, F.; Agnusdei, V.; Pinazza, M.; Nardo, G.; Iorio, E.; Curtarello, M.; Silic-Benussi, M.; Rossi, E.; Venturoli, C.; et al. LKB1 loss is associated with glutathione deficiency under oxidative stress and sensitivity of cancer cells to cytotoxic drugs and γ-irradiation. Biochem. Pharmacol. 2018, 156, 479–490. [Google Scholar] [CrossRef]
- Bonanno, L.; De Paoli, A.; Zulato, E.; Esposito, G.; Calabrese, F.; Favaretto, A.; Santo, A.; Del Conte, A.; Chilosi, M.; Oniga, F.; et al. LKB1 Expression Correlates with Increased Survival in Patients with Advanced Non–Small Cell Lung Cancer Treated with Chemotherapy and Bevacizumab. Clin. Cancer Res. 2017, 23, 3316–3324. [Google Scholar] [CrossRef]
- Leinonen, H.M.; Kansanen, E.; Pölönen, P.; Heinäniemi, M.; Levonen, A.-L. Role of the Keap1–Nrf2 Pathway in Cancer. Adv. Cancer Res. 2014, 281–320. [Google Scholar]
- Seo, H.; Jung, D.K.; Kang, H.-G.; Jeong, J.Y.; Lee, S.Y.; Choi, J.E.; Hong, M.J.; Do, S.K.; Lee, J.H.; Lee, W.K.; et al. An expression quantitative trait locus variant for LKB1 gene predicts the clinical outcomes of chemotherapy in patients with non-small cell lung cancer. Cancer Genet. 2018, 228–229, 73–82. [Google Scholar] [CrossRef]
- Zulato, E.; Curtarello, M.; Nardo, G.; Indraccolo, S. Metabolic effects of anti-angiogenic therapy in tumors. Biochimie 2012, 94, 925–931. [Google Scholar] [CrossRef]
- Curtarello, M.; Zulato, E.; Nardo, G.; Valtorta, S.; Guzzo, G.; Rossi, E.; Esposito, G.; Msaki, A.; Pasto, A.; Rasola, A.; et al. VEGF-Targeted Therapy Stably Modulates the Glycolytic Phenotype of Tumor Cells. Cancer Res. 2015, 75, 120–133. [Google Scholar] [CrossRef]
- Noy, R.; Pollard, J.W. Tumor-Associated Macrophages: From Mechanisms to Therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef]
- Wong, K.-K.; Liu, Y.; Li, Y.; Wang, X.; Liu, F.; Gao, P.; Quinn, M.M.; Li, F.; Merlino, A.A.; Benes, C.H.; et al. Gemcitabine and Chk1 inhibitor AZD7762 synergistically suppress the growth of Lkb1-deficient lung adenocarcinoma. Cancer Res. 2017, 5068–5076. [Google Scholar] [CrossRef]
- Thompson, R.; Eastman, A. The cancer therapeutic potential of Chk1 inhibitors: How mechanistic studies impact on clinical trial design. Br. J. Clin. Pharmacol. 2013, 76, 358–369. [Google Scholar] [CrossRef]
- Richer, A.L.; Cala, J.M.; O’Brien, K.; Carson, V.M.; Inge, L.J.; Whitsett, T.G. WEE1 Kinase Inhibitor AZD1775 Has Preclinical Efficacy in LKB1-Deficient Non–Small Cell Lung Cancer. Cancer Res. 2017, 77, 4663–4672. [Google Scholar] [CrossRef]
- Parker, L.; Piwnica-Worms, H. Inactivation of the p34cdc2-cyclin B complex by the human WEE1 tyrosine kinase. Science 1992, 257, 1955–1957. [Google Scholar] [CrossRef]
- Russell, P.; Nurse, P. Negative regulation of mitosis by wee1+, a gene encoding a protein kinase homolog. Cell 1987, 49, 559–567. [Google Scholar] [CrossRef]
- Matheson, C.J.; Backos, D.S.; Reigan, P. Targeting WEE1 Kinase in Cancer. Trends Pharmacol. Sci. 2016, 37, 872–881. [Google Scholar] [CrossRef]
- Koivunen, J.P.; Kim, J.; Lee, J.; Rogers, A.M.; Park, J.O.; Zhao, X.; Naoki, K.; Okamoto, I.; Nakagawa, K.; Yeap, B.Y.; et al. Mutations in the LKB1 tumour suppressor are frequently detected in tumours from Caucasian but not Asian lung cancer patients. Br. J. Cancer 2008, 99, 245–252. [Google Scholar] [CrossRef]
- Memmott, R.M.; Dennis, P.A. LKB1 and Mammalian Target of Rapamycin As Predictive Factors for the Anticancer Efficacy of Metformin. J. Clin. Oncol. 2009, 27, e226. [Google Scholar] [CrossRef]
- Daaka, Y.; Luttrell, L.M.; Lefkowitz, R.J. Switching of the coupling of the β2-adrenergic receptor to different G proteins by protein kinase A. Nature 1997, 390, 88–91. [Google Scholar] [CrossRef]
- Nilsson, M.B.; Sun, H.; Diao, L.; Tong, P.; Liu, D.; Li, L.; Fan, Y.; Poteete, A.; Lim, S.-O.; Howells, K.; et al. Stress hormones promote EGFR inhibitor resistance in NSCLC: Implications for combinations with β-blockers. Sci. Transl. Med. 2017, 9, eaao4307. [Google Scholar] [CrossRef]
- Nardo, G.; Favaro, E.; Curtarello, M.; Moserle, L.; Zulato, E.; Persano, L.; Rossi, E.; Esposito, G.; Crescenzi, M.; Casanovas, O.; et al. Glycolytic Phenotype and AMP Kinase Modify the Pathologic Response of Tumor Xenografts to VEGF Neutralization. Cancer Res. 2011, 71, 4214–4225. [Google Scholar] [CrossRef] [PubMed]
- Moro, M.; Caiola, E.; Ganzinelli, M.; Zulato, E.; Rulli, E.; Marabese, M.; Centonze, G.; Busico, A.; Pastorino, U.; de Braud, F.G.; et al. Metformin Enhances Cisplatin-Induced Apoptosis and Prevents Resistance to Cisplatin in Co-mutated KRAS/LKB1 NSCLC. J. Thorac. Oncol. 2018, 13, 1692–1704. [Google Scholar] [CrossRef]
- El-Mir, M.Y.; Nogueira, V.; Fontaine, E.; Avéret, N.; Rigoulet, M.; Leverve, X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J. Biol. Chem. 2000, 275, 223–228. [Google Scholar] [CrossRef]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef]
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crinò, L.; Eberhardt, W.E.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1–Positive Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Baas, P.; Kim, D.-W.; Felip, E.; Pérez-Gracia, J.L.; Han, J.-Y.; Molina, J.; Kim, J.-H.; Arvis, C.D.; Ahn, M.-J.; et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): A randomised controlled trial. Lancet 2016, 387, 1540–1550. [Google Scholar] [CrossRef]
- Rittmeyer, A.; Barlesi, F.; Waterkamp, D.; Park, K.; Ciardiello, F.; von Pawel, J.; Gadgeel, S.M.; Hida, T.; Kowalski, D.M.; Dols, M.C.; et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): A phase 3, open-label, multicentre randomised controlled trial. Lancet 2017, 389, 255–265. [Google Scholar] [CrossRef]
- Wu, D.; Luo, Y.; Guo, W.; Niu, Q.; Xue, T.; Yang, F.; Sun, X.; Chen, S.; Liu, Y.; Liu, J.; et al. Lkb1 maintains Treg cell lineage identity. Nat. Commun. 2017, 8, 15876. [Google Scholar] [CrossRef]
- MacIver, N.J.; Blagih, J.; Saucillo, D.C.; Tonelli, L.; Griss, T.; Rathmell, J.C.; Jones, R.G. The Liver Kinase B1 Is a Central Regulator of T Cell Development, Activation, and Metabolism. J. Immunol. 2011, 187, 4187–4198. [Google Scholar] [CrossRef]
- Windt, G.J.W.; Pearce, E.L. Metabolic switching and fuel choice during T-cell differentiation and memory development. Immunol. Rev. 2012, 249, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhang, W.; Zhang, M.; Zhu, H.; Moriasi, C.; Zou, M.-H. Liver Kinase B1 Suppresses Lipopolysaccharide-induced Nuclear Factor κB (NF-κB) Activation in Macrophages. J. Biol. Chem. 2015, 290, 2312–2320. [Google Scholar] [CrossRef]
- Chen, S.; Fang, L.; Guo, W.; Zhou, Y.; Yu, G.; Li, W.; Dong, K.; Liu, J.; Luo, Y.; Wang, B.; et al. Control of Treg cell homeostasis and immune equilibrium by Lkb1 in dendritic cells. Nat. Commun. 2018, 9, 5298. [Google Scholar] [CrossRef]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Aref, A.R.; Skoulidis, F.; Herter-Sprie, G.S.; Buczkowski, K.A.; Liu, Y.; Awad, M.M.; Denning, W.L.; et al. STK11/LKB1 Deficiency Promotes Neutrophil Recruitment and Proinflammatory Cytokine Production to Suppress T-cell Activity in the Lung Tumor Microenvironment. Cancer Res. 2016, 76, 999–1008. [Google Scholar] [CrossRef]
- Attili, I.; Karachaliou, N.; Bonanno, L.; Berenguer, J.; Bracht, J.; Codony-Servat, J.; Codony-Servat, C.; Ito, M.; Rosell, R. STAT3 as a potential immunotherapy biomarker in oncogene-addicted non-small cell lung cancer. Ther. Adv. Med. Oncol. 2018, 10. [Google Scholar] [CrossRef]
- Barber, G.N. STING: Infection, inflammation and cancer. Nat. Rev. Immunol. 2015, 15, 760–770. [Google Scholar] [CrossRef]
- Rizvi, H.; Sanchez-Vega, F.; La, K.; Chatila, W.; Jonsson, P.; Halpenny, D.; Plodkowski, A.; Long, N.; Sauter, J.L.; Rekhtman, N.; et al. Molecular Determinants of Response to Anti–Programmed Cell Death (PD)-1 and Anti–Programmed Death-Ligand 1 (PD-L1) Blockade in Patients With Non–Small-Cell Lung Cancer Profiled With Targeted Next-Generation Sequencing. J. Clin. Oncol. 2018, 36, 633–641. [Google Scholar] [CrossRef]
- Hato, T.; Zhu, A.X.; Duda, D.G. Rationally combining anti-VEGF therapy with checkpoint inhibitors in hepatocellular carcinoma. Immunotherapy 2016, 8, 299–313. [Google Scholar] [CrossRef]
- Ohm, J.E. VEGF inhibits T-cell development and may contribute to tumor-induced immune suppression. Blood 2003, 101, 4878–4886. [Google Scholar] [CrossRef]
- Colussi, O.; Taieb, J.; Pernot, S.; Sandoval, F.; Tartour, E.; Benhamouda, N.; Terme, M.; Marcheteau, E.; Dubreuil, O.; Carpentier, A.F. VEGFA-VEGFR Pathway Blockade Inhibits Tumor-Induced Regulatory T-cell Proliferation in Colorectal Cancer. Cancer Res. 2012, 73, 539–549. [Google Scholar]
- Koinis, F.; Vetsika, E.K.; Aggouraki, D.; Skalidaki, E.; Koutoulaki, A.; Gkioulmpasani, M.; Georgoulias, V.; Kotsakis, A. Effect of First-Line Treatment on Myeloid-Derived Suppressor Cells’ Subpopulations in the Peripheral Blood of Patients with Non–Small Cell Lung Cancer. J. Thorac. Oncol. 2016, 11, 1263–1272. [Google Scholar] [CrossRef]
- Borden, E.C. Interferons α and β in cancer: Therapeutic opportunities from new insights. Nat. Rev. Drug Discov. 2019, 18, 219–234. [Google Scholar] [CrossRef]
- Huang, S.; Bucana, C.D.; Van Arsdall, M.; Fidler, I.J. Stat1 negatively regulates angiogenesis, tumorigenicity and metastasis of tumor cells. Oncogene 2002, 21, 2504–2512. [Google Scholar] [CrossRef]
- Eubank, T.D.; Roberts, R.D.; Khan, M.; Curry, J.M.; Nuovo, G.J.; Kuppusamy, P.; Marsh, C.B. Granulocyte Macrophage Colony-Stimulating Factor Inhibits Breast Cancer Growth and Metastasis by Invoking an Anti-Angiogenic Program in Tumor-Educated Macrophages. Cancer Res. 2009, 69, 2133–2140. [Google Scholar] [CrossRef]
- Yates-Binder, C.C.; Rodgers, M.; Jaynes, J.; Wells, A.; Bodnar, R.J.; Turner, T. An IP-10 (CXCL10)-derived peptide inhibits angiogenesis. PLoS ONE 2012, 7, e40812. [Google Scholar] [CrossRef]
- Bouzin, C.; Brouet, A.; De Vriese, J.; DeWever, J.; Feron, O. Effects of Vascular Endothelial Growth Factor on the Lymphocyte-Endothelium Interactions: Identification of Caveolin-1 and Nitric Oxide as Control Points of Endothelial Cell Anergy. J. Immunol. 2007, 178, 1505–1511. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, Y.; Iwamoto, H.; Hosaka, K.; Seki, T.; Andersson, P.; Lim, S.; Fischer, C.; Nakamura, M.; Abe, M.; et al. Discontinuation of anti-VEGF cancer therapy promotes metastasis through a liver revascularization mechanism. Nat. Commun. 2016, 7, 12680. [Google Scholar] [CrossRef]
- Tagliamonte, M.; Petrizzo, A.; Tornesello, M.L.; Ciliberto, G.; Buonaguro, F.M.; Buonaguro, L. Combinatorial immunotherapy strategies for hepatocellular carcinoma. Curr. Opin. Immunol. 2016, 39, 103–113. [Google Scholar] [CrossRef]
- Attili, I.; Passaro, A.; Pavan, A.; Conte, P.; De Marinis, F.; Bonanno, L. Combination immunotherapy strategies in advanced non-small cell lung cancer (NSCLC): Does biological rationale meet clinical needs? Crit. Rev. Oncol. Hematol. 2017, 119, 30–39. [Google Scholar] [CrossRef]
- Rusek, A.; Abba, M.; Eljaszewicz, A.; Moniuszko, M.; Niklinski, J.; Allgayer, H. MicroRNA modulators of epigenetic regulation, the tumor microenvironment and the immune system in lung cancer. Mol. Cancer 2015, 14, 34. [Google Scholar] [CrossRef]
- Wu, X.; Li, J.; Connolly, E.M.; Liao, X.; Ouyang, J.; Giobbie-Hurder, A.; Lawrence, D.; McDermott, D.; Murphy, G.; Zhou, J.; et al. Combined Anti-VEGF and Anti–CTLA-4 Therapy Elicits Humoral Immunity to Galectin-1 Which Is Associated with Favorable Clinical Outcomes. Cancer Immunol. Res. 2017, 5, 446–454. [Google Scholar] [CrossRef]
- Reck, M.; Nogami, N.; Rodríguez-Abreu, D.; Stroyakovskiy, D.; Lopez-Chavez, A.; Sandler, A.; Socinski, M.A.; Lee, A.; Moro-Sibilot, D.; Barlesi, F.; et al. Atezolizumab for First-Line Treatment of Metastatic Nonsquamous NSCLC. N. Engl. J. Med. 2018, 378, 2288–2301. [Google Scholar]
- Rhodes, L.V.; Tate, C.R.; Hoang, V.T.; Burks, H.E.; Gilliam, D.; Martin, E.C.; Elliott, S.; Miller, D.B.; Buechlein, A.; Rusch, D.; et al. Regulation of triple-negative breast cancer cell metastasis by the tumor-suppressor liver kinase B1. Oncogenesis 2015, 4, e168. [Google Scholar] [CrossRef]
- Zeng, Q.; Chen, J.; Li, Y.; Werle, K.D.; Zhao, R.-X.; Quan, C.-S.; Wang, Y.-S.; Zhai, Y.-X.; Wang, J.-W.; Youssef, M.; et al. LKB1 inhibits HPV-associated cancer progression by targeting cellular metabolism. Oncogene 2017, 36, 1245–1255. [Google Scholar] [CrossRef]



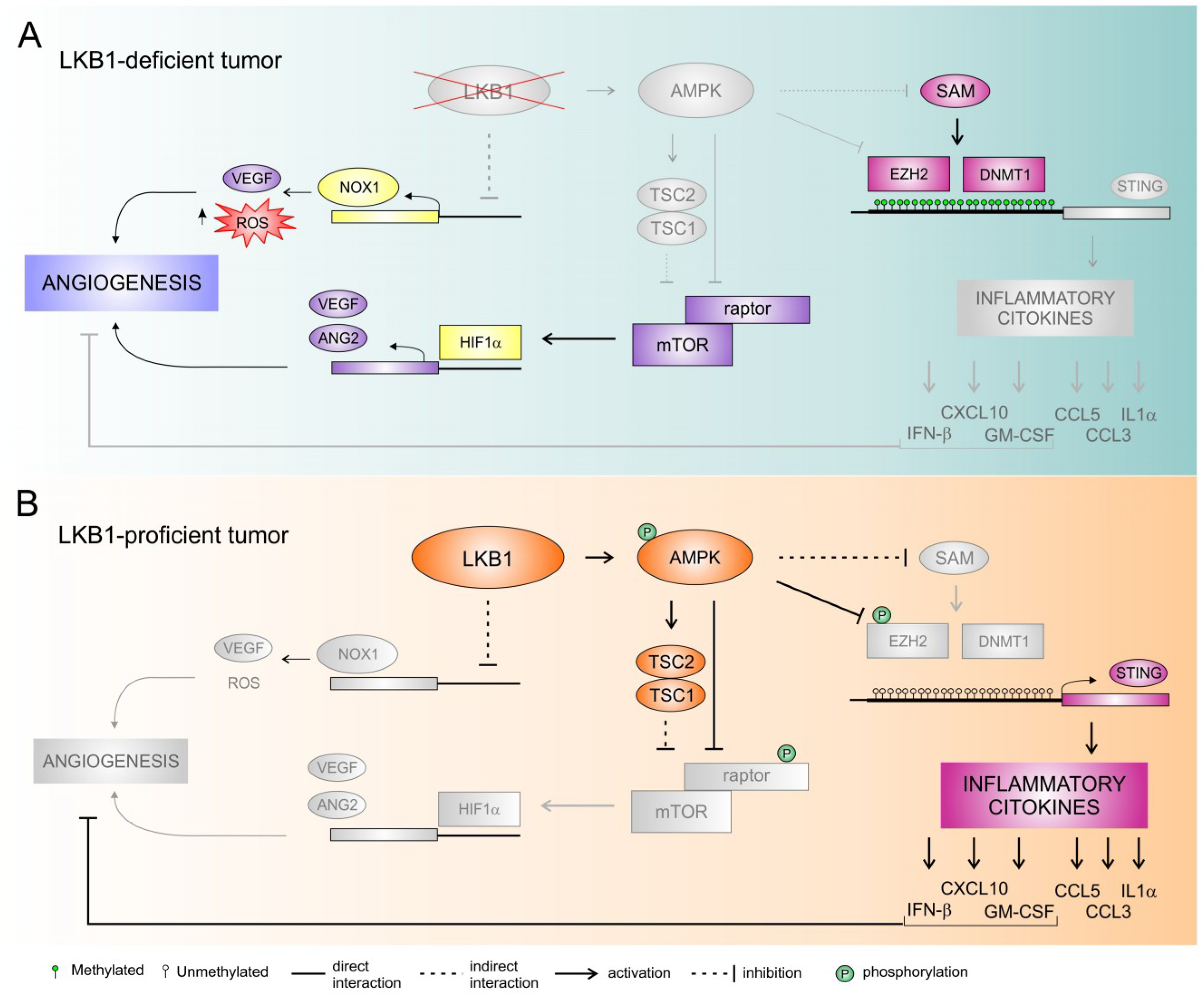{kind=link}
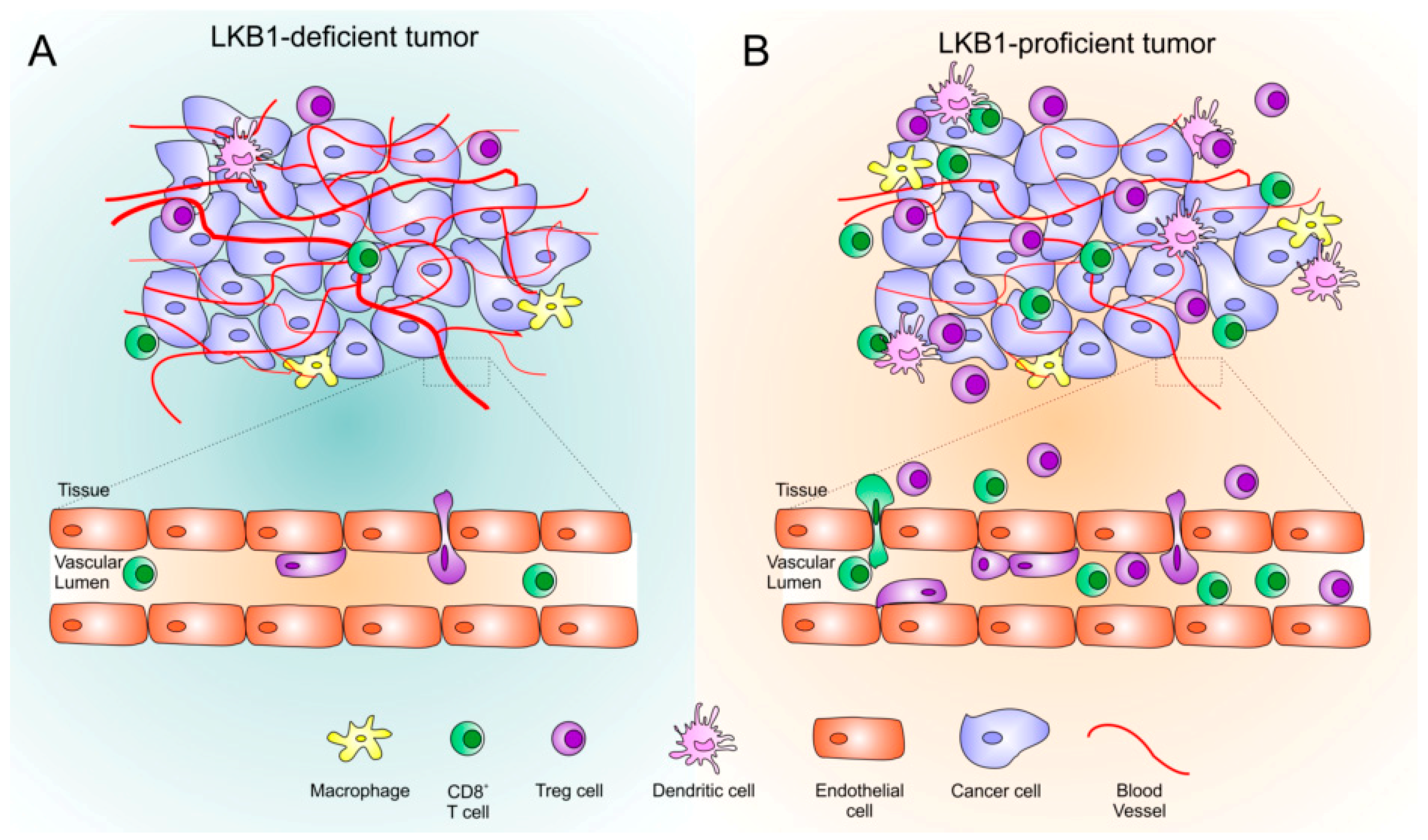{kind=link}
| A. Clinical Trials Requiring STK11/LKB1 Determination as Inclusion Criteria | ||||
|---|---|---|---|---|
| Clinical Trial ID | Phase | Brief Description | Study Population | Primary Endpoint |
| NCT03709147 (FAME trial) | II | Exploiting metformin plus/minus cyclic fasting mimicking diet (FMD) to improve the efficacy of platinum-pemetrexed chemotherapy in advanced LKB1-inactive lung adenocarcinoma | LKB1 inactive advanced NSCLC | PFS |
| NCT03872427 (BeGIN trial) | II | A phase II Basket trial of Glutaminase Inhibitor (BeGIN) CB-839 HCl in patients with NF1 aberrations, NF1 Mutant Malignant Peripheral Nerve Sheath Tumors (MPNST), KEAP1/NRF2 and LKB1 aberrant tumors. | NF1 aberrations, NF1 Mutant Malignant Peripheral Nerve Sheath Tumors (MPNST), KEAP1/NRF2 and LKB1 aberrant tumors | ORR |
| NCT03375307 | II | A phase II study of Olaparib (AZD2281) in patients with metastatic/advanced urothelial carcinoma with DNA-repair defects. | Metastatic/advanced urothelial carcinoma with DNA-repair defects (among these: STK11 gene mutations). | ORR |
| NCT02352844 | II | A Phase II Study of Everolimus in Patients With Advanced Solid Malignancies With TSC1, TSC2, NF1, NF2, or STK11 mutations. | Advanced solid malignancies with TSC1, TSC2, NF1, NF2, or STK11 mutations. | ORR |
| NCT02645149 | IV | Molecular profiling and matched targeted therapy for patients with metastatic melanoma. Once standard therapies have been exhausted, patients receive a targeted therapy matched for their genetic result, if applicable. If STK11 mutated, they receive everolimus. | BRAF and NRAS wild-type unresectable Stage III or Stage IV metastatic melanoma. | Type and frequency of genetic aberrations in BRAF/NRAS wild-type metastatic melanoma and proportion of patients with BRAS/NRAS wild-type melanoma receiving targeted therapy |
| B. Clinical Trials Investigating STK11/LKB1 Status Among Secondary Objectives | ||||
|---|---|---|---|---|
| Clinical Trial ID | Phase | Brief Description | Study Population | Primary Endpoint |
| NCT01470209 | I | A phase I study assessing the safety of the combination of everolimus and BKM120 for the treatment of advanced solid tumors cancer in patients who are no longer benefiting from or unable to withstand standard treatment. | Solid tumors (including lung cancer). Alterations in PIK3CA, NF1, TSC1/TSC2, mTOR, KRAS, LKB1, PTEN will be accessed. | DLT |
| NCT02642042 | II | A phase II Trial of Trametinib with docetaxel in Non-Small Cell Lung Cancer (NSCLC) KRAS mutated patients after one or two prior systemic therapies. | Advanced NSCLC carrying KRAS mutation Tertiary objective: To evaluate the response rate in the presence of co-mutations TP53 and LKB1 | ORR |
| NCT01310231 | II | A randomized phase II, double blind trial of standard chemotherapy with metformin (versus placebo) in women with metastatic breast cancer receiving first, second, third or fourth line chemotherapy with anthracycline, taxane, platinum, capecitabine or vinorelbine based regimens. | Metastatic breast cancer in first, second, third or fourth line chemotherapy treatment. Immunohistochemistry analysis of different markers (IR, LKB1, phosphorylated AKT, S6K, ribosomal protein S6, 4E-BP1, and stathmin) performed. | PFS |
| NCT02285855 | II | Tumor mutation status and metabolic response to metformin in non-small cell lung cancer (NSCLC). | NSCLC undergoing Stereotactic body Radiotherapy (SBRT). Genotype comparisons of JKRAS, STK11, and TP53 mutations assessed. | ORR [Closed for poor accrual] |
| NCT03495544 | Observational | Comparative multicenter study estimating association between germline DNA-repair genes mutations and PD-L1 expression level in breast cancer. | Breast cancer. Association between germline mutations (TP53 MLH1 MSH2 MSH6 PMS2 EPCAM APC MUTYH CDKN2A CDK4 ATM KIT PDGFRA CDH1 CTNNA1 PRSS1 SPINK1 BRCA1 BRCA2 FANCI FANCL PALB2 RAD51B RAD51C RAD54L RAD51D CHEK1 CHEK2 CDK12 BRIP1 PPP2R2A BARD1 PARP1 STK11 XRCC3) and PD-L1 expression. | Diagnostic performance of PD-L1 expression in breast cancer |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bonanno, L.; Zulato, E.; Pavan, A.; Attili, I.; Pasello, G.; Conte, P.; Indraccolo, S. LKB1 and Tumor Metabolism: The Interplay of Immune and Angiogenic Microenvironment in Lung Cancer. Int. J. Mol. Sci. 2019, 20, 1874. https://doi.org/10.3390/ijms20081874
Bonanno L, Zulato E, Pavan A, Attili I, Pasello G, Conte P, Indraccolo S. LKB1 and Tumor Metabolism: The Interplay of Immune and Angiogenic Microenvironment in Lung Cancer. International Journal of Molecular Sciences. 2019; 20(8):1874. https://doi.org/10.3390/ijms20081874
Chicago/Turabian StyleBonanno, Laura, Elisabetta Zulato, Alberto Pavan, Ilaria Attili, Giulia Pasello, PierFranco Conte, and Stefano Indraccolo. 2019. "LKB1 and Tumor Metabolism: The Interplay of Immune and Angiogenic Microenvironment in Lung Cancer" International Journal of Molecular Sciences 20, no. 8: 1874. https://doi.org/10.3390/ijms20081874