STAT3 Post-Translational Modifications Drive Cellular Signaling Pathways in Prostate Cancer Cells

 , , , , , , , , and
, , , , , , , , and
Abstract
:1. Introduction
- un-phosphorylated STAT3 (U-STAT3) can bind to the interferon γ-activated sequence (GAS), either as dimer or as monomer. The U-STAT3 regulates gene expression via its binding to AT-rich DNA sequences and drives chromatin structure remodeling [18];
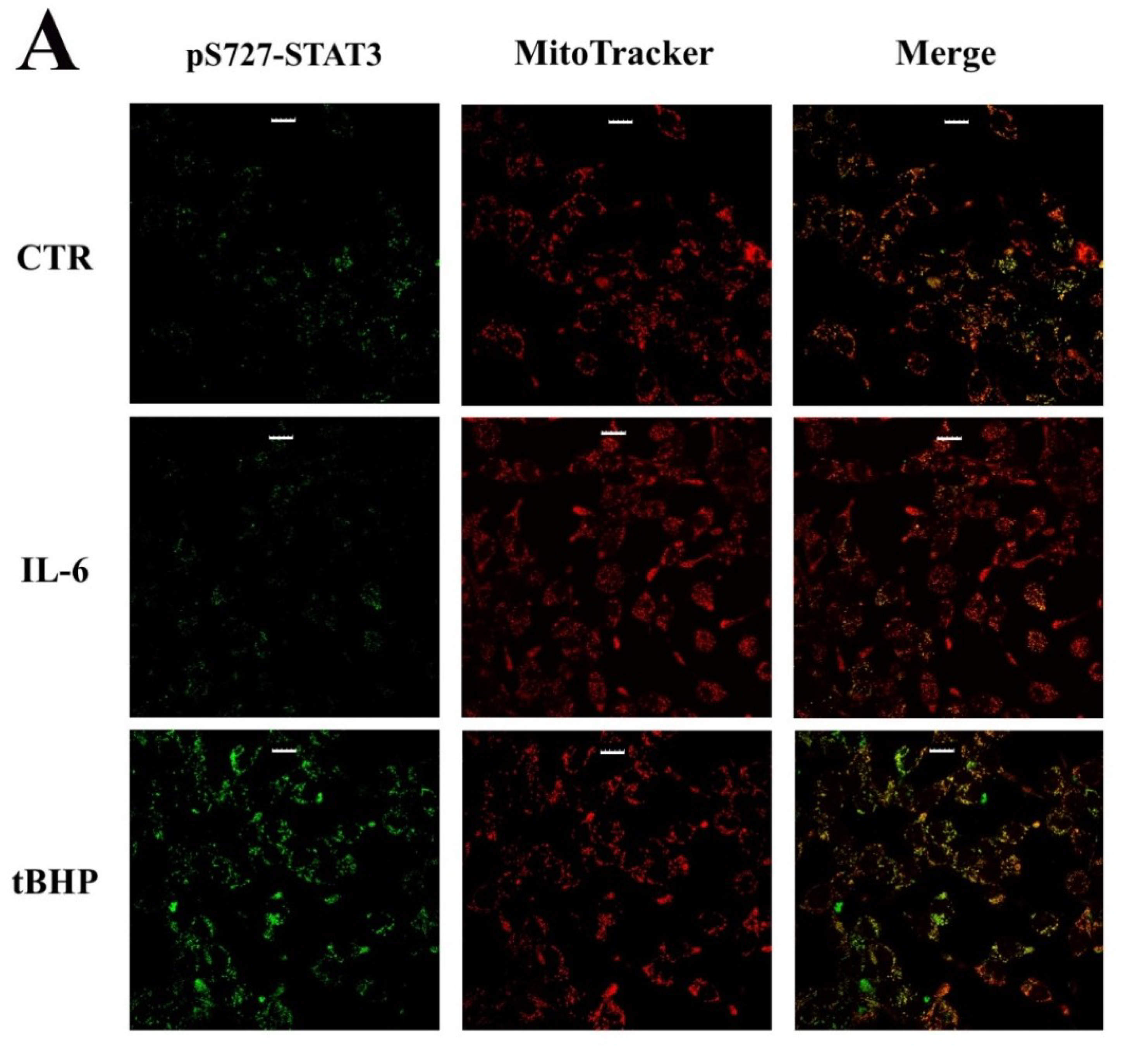
- STAT3 phosphorylation at S727, rather than Y705, is required for its mitochondrial activity [19]. In particular, has been reported that mitochondrial STAT3 controls cell respiration and metabolism by enhancing the activity of succinate oxidoreductase (complex II), ATP synthase (complex V) and lactate dehydrogenase. Affecting the balance between glycolytic and oxidative phosphorylation metabolisms, STAT3 provides the necessary requirements to support the metabolic shift toward aerobic glycolysis known as the Warburg effect [20];
- STAT3 was found associated with a variety of cytosolic structures, including focal adhesions, microtubules and mitotic spindles. Indeed, STAT3 plays an important role during the assembly of cytoskeleton networks, such as the actin filaments and microtubules, thus promoting cell migration and invasion [21].
2. Results
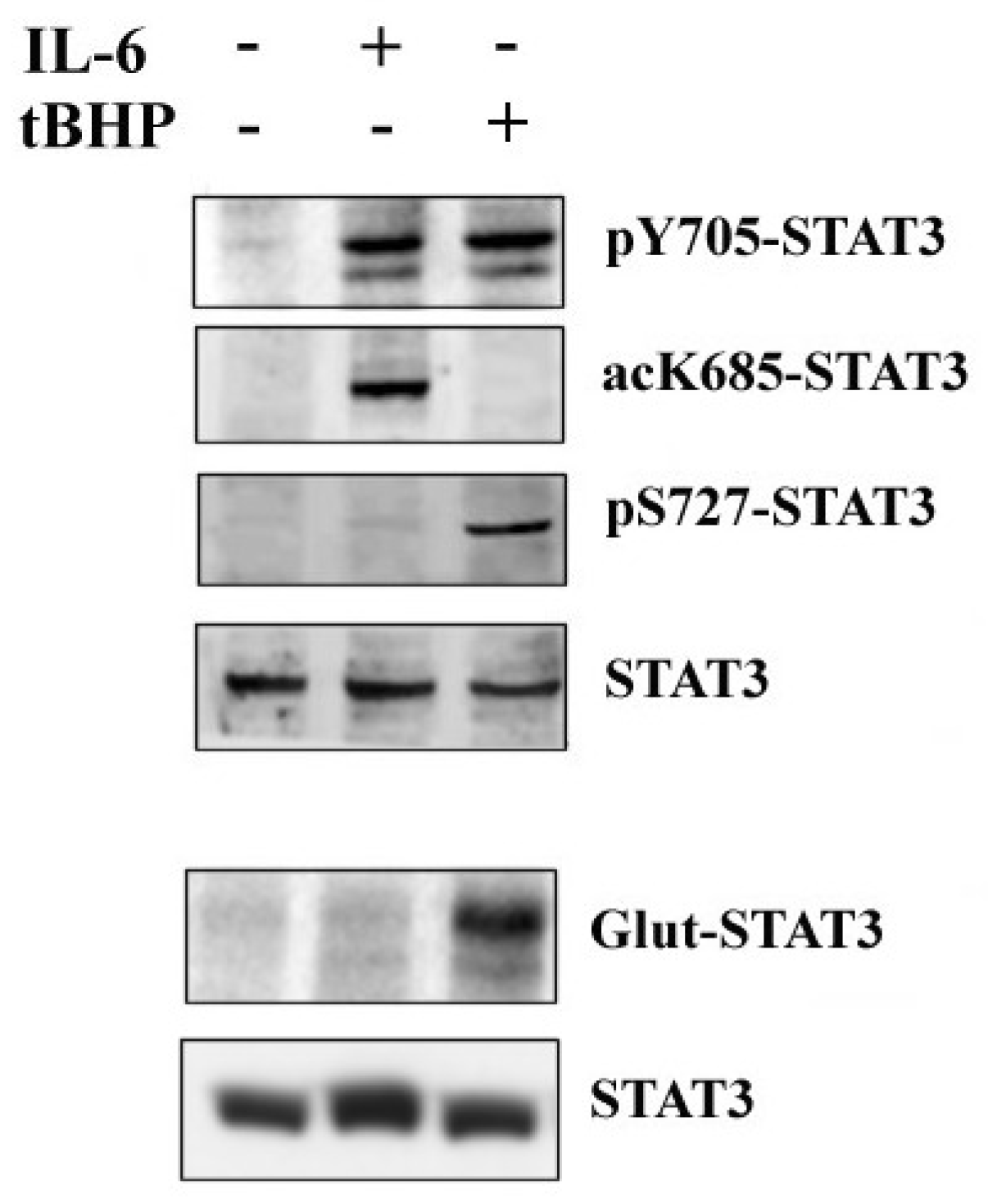
2.1. STAT3 Post-Translational Modifications (PTMs) Modulate Different Signal Transduction PATHWAYS in PCa
2.2. STAT3 PTMs: Cellular Condition Sensors and Trascriptional Program Drivers
2.3. STAT3 Partners: PTM Modulators
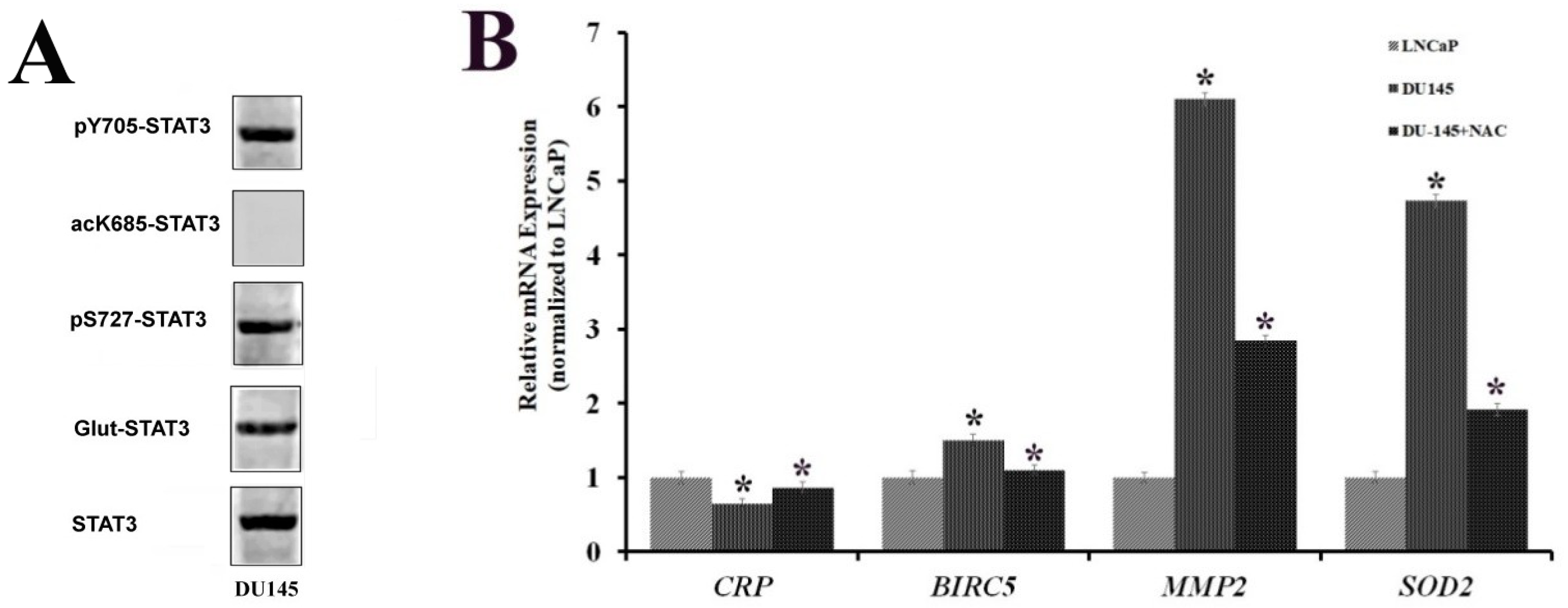
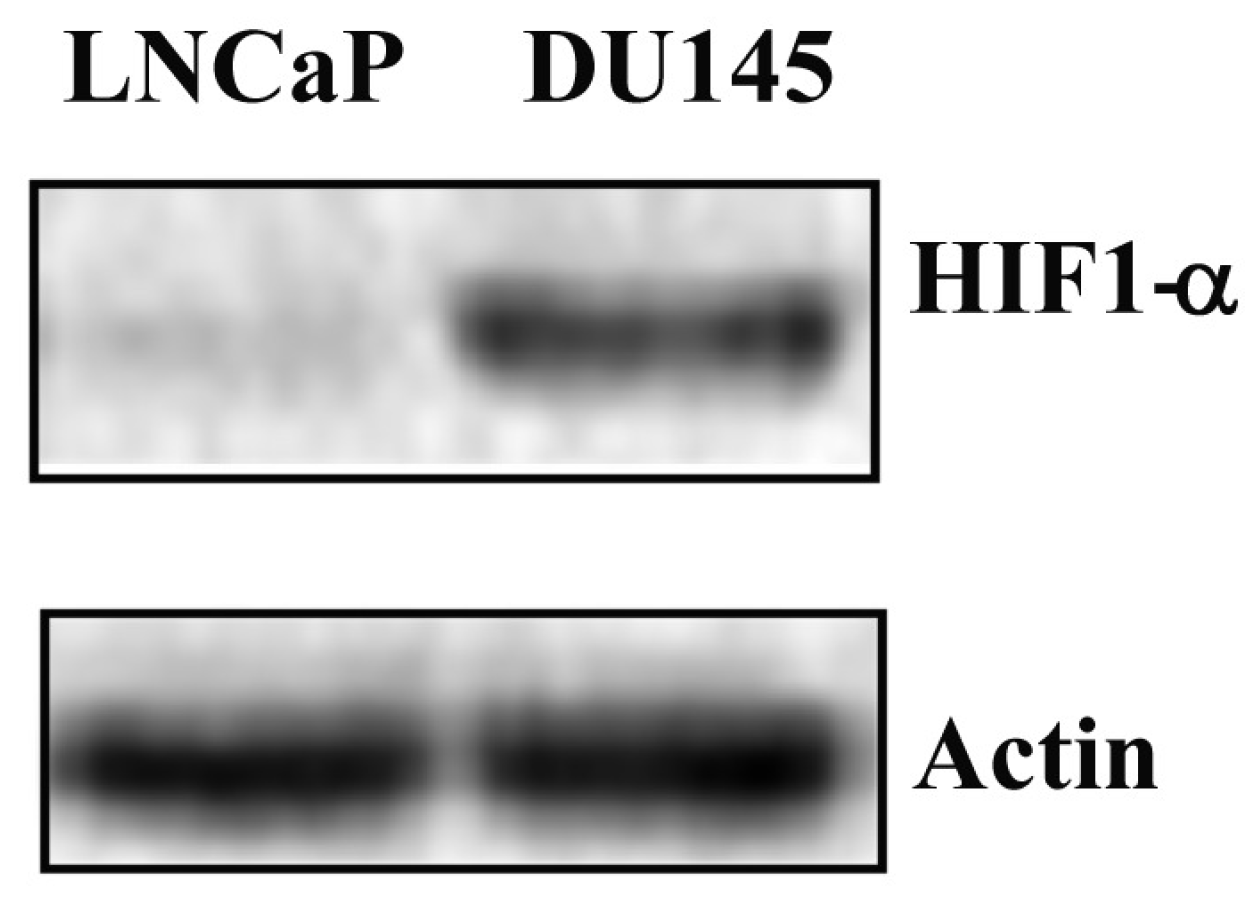
2.4. STAT3 PTMs in DU-145 Cells
3. Discussion and Conclusions
4. Material and Methods
4.1. Cell Culture
4.2. Proteins Extraction and Immunoblotting
4.3. Analysis of S-Glutathionylated STAT3
4.4. Reactive Oxygen Species (ROS) Detection
4.5. Extraction of RNA and RT-qPCR
4.6. Immunofluorescence
4.7. Mitochondria Staining
4.8. Chromatin Immunoprecipitation (ChIP) and Co-Immunoprecipitation Assays
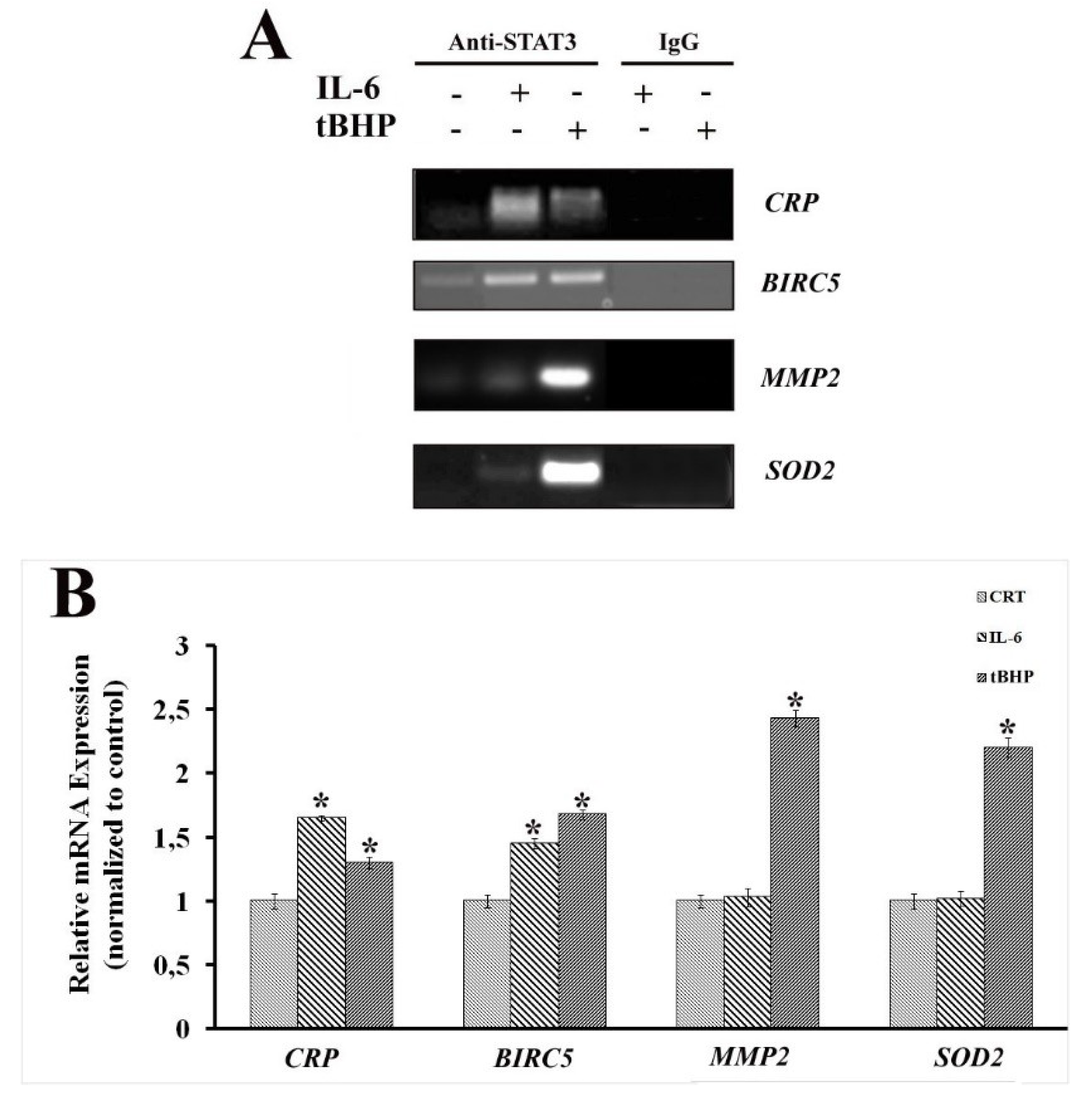
- Crp:
- For 5′-GAGTTTGTAATAAATAAC-3′ (TSS-172/-155)Rev 5′-CACTATGTAAATAATTTTC-3′ (TSS-85/-67),
- Birc5:
- For 5′-GTTGCAGTGAGCTGAGATC-3′ (TSS-1114/-1095)Rev 5′-GTGCCTCCACTGTCTTTTTC-3′ (TSS-917/937),
- Mmp2:
- For 5′-CAAGATGGAGTCGCTCTG-3′ (TSS -650/-633)Rev 5′-GTAAGCCTTAACTTGGCCTC-3′ (TSS-580/599),
- Sod2:
- For 5′-GTGGGTGTCCAAGAACTGT-3′ (TSS-748/730)Rev 5′-GTACTCTTGCGCCGTACC-3′ (TSS-643/-626).
4.9. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tolkach, Y.; Kristianse, G. The Heterogeneity of Prostate Cancer: A Practical Approach. Pathobiology 2018, 85, 108–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Zhao, D.; Spring, D.J.; DePinho, R.A. Genetics and biology of prostate cancer. Genes Dev. 2018, 32, 1105–1140. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Nonomura, N. Role of Androgen Receptor in Prostate Cancer: A Review. World J. Mens Health 2018, 36, e32. [Google Scholar] [CrossRef] [PubMed]
- Hoang, D.T.; Iczkowski, K.A.; Kilari, D.; See, W.; Nevalainen, M.T. Androgen receptor-dependent and -independent mechanisms driving prostate cancer progression: Opportunities for therapeutic targeting from multiple angles. Oncotarget 2017, 10, 3724–3745. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Chen, W.; Zhang, J.; Li, H. Androgen receptor: What we know and what we expect in castration-resistant prostate cancer. Int. Urol. Nephrol. 2018, 50, 1753–1764. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, S.; Ramamurthy, V.P.; Njar, V.C.O. Dissecting major signaling pathways in prostate cancer development and progression: Mechanisms and novel therapeutic targets. J. Steroid Biochem. Mol. Biol. 2017, 166, 16–27. [Google Scholar] [CrossRef]
- Culig, Z.; Puhr, M. Interleukin-6 and prostate cancer: Current developments and unsolved questions. Mol. Cell. Endocrinol. 2018, 15, 25–30. [Google Scholar] [CrossRef]
- Thaper, D.; Vahid, S.; Kaur, R.; Kumar, S.; Nouruzi, S.; Bishop, J.L.; Johansson, M.; Zoubeidi, A. Galiellalactone inhibits the STAT3/AR signaling axis and suppresses Enzalutamide-resistant Prostate Cancer. Sci. Rep. 2018, 8, 17307. [Google Scholar] [CrossRef]
- Bishop, J.L.; Thaper, D.; Zoubeidi, A. The Multifaceted Roles of STAT3 Signaling in the Progression of Prostate Cancer. Cancers 2014, 6, 829–859. [Google Scholar] [CrossRef]
- Don-Doncow, N.; Marginean, F.; Coleman, I.; Nelson, P.S.; Ehrnström, R.; Krzyzanowska, A.; Morrissey, C.; Hellsten, R.; Bjartell, A. Expression of STAT3 in Prostate Cancer Metastases. Eur. Urol. 2017, 71, 313–316. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.; Zhao, Y.; Yu, Y.; Fang, J.M.; Cui, R.; Liu, Z.Q.; Guo, X.L.; Xu, Q. Docetaxel-mediated autophagy promotes chemoresistance in castration-resistant prostate cancer cells by inhibiting STAT3. Cancer Lett. 2018, 462, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Abdulghani, J.; Gu, L.; Dagvadorj, A.; Lutz, J.; Leiby, B.; Bonuccelli, G.; Lisanti, M.P.; Zellweger, T.; Alanen, K.; Mirtti, T.; Visakorpi, T.; et al. Stat3 promotes metastatic progression of prostate cancer. Am. J. Pathol. 2008, 172, 1717–1728. [Google Scholar] [CrossRef]
- Balanis, N.; Carlin, C.R. Stress-induced EGF receptor signaling through STAT3 and tumor progression in triple-negative breast cancer. Mol. Cell. Endocrinol. 2017, 451, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, H.; Ohno, Y.; Toyoshima, Y.; Ohtake, J.; Homma, S.; Kawamura, H.; Takahashi, N.; Taketomi, A. Interleukin-6/STAT3 signaling as a promising target to improve the efficacy of cancer immunotherapy. Cancer Sci. 2017, 108, 1947–1952. [Google Scholar] [CrossRef]
- Yuan, J.; Zhang, F.; Niu, R. Multiple regulation pathways and pivotal biological functions of STAT3 in cancer. Sci Rep. 2015, 3, 17663. [Google Scholar] [CrossRef]
- Li, W.X. Canonical and non-canonical JAK-STAT signaling. Trends Cell Biol. 2008, 18, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, J.; DiGiovanni, J. Non-canonical Stat3 signaling in cancer. Mol. Carcinog. 2016, 55, 1889–1898. [Google Scholar] [CrossRef]
- Yan, S.J.; Lim, S.J.; Shi, S.; Dutta, P.; Li, W.X. Unphosphorylated STAT and heterochromatin protect genome stability. FASEB J. 2011, 25, 232–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Raje, V.; Yakovlev, V.A.; Yacoub, A.; Szczepanek, K.; Meier, J.; Derecka, M.; Chen, Q.; Hu, Y.; Sisler, J.; et al. Mitochondrial localized Stat3 promotes breast cancer growth via phosphorylation of serine 727. J. Biol. Chem. 2013, 25, 31280–31288. [Google Scholar] [CrossRef]
- Camporeale, A.; Demaria, M.; Monteleone, E.; Giorgi, C.; Wieckowski, M.R.; Pinton, P.; Poli, V. STAT3 Activities and Energy Metabolism: Dangerous Liaisons. Cancers 2014, 31, 1579–1596. [Google Scholar] [CrossRef]
- Verma, N.K.; Dourlat, J.; Davies, A.M.; Long, A.; Liu, W.Q.; Garbay, C.; Kelleher, D.; Volkov, Y. STAT3-stathmin interactions control microtubule dynamics in migrating T-cells. J. Biol. Chem. 2009, 284, 12349–12362. [Google Scholar] [CrossRef]
- Avalle, L.; Camporeale, A.; Camperi, A.; Poli, V. STAT3 in cancer: A double edged sword. Cytokine 2017, 98, 42–50. [Google Scholar] [CrossRef]
- Meier, J.A.; Hyun, M.; Cantwell, M.; Raza, A.; Mertens, C.; Raje, V.; Sisler, J.; Tracy, E.; Torres-Odio, S.; Gispert, S.; et al. Stress-induced dynamic regulation of mitochondrial STAT3 and its association with cyclophilin D reduce mitochondrial ROS production. Sci. Signal. 2017, 28, 10. [Google Scholar] [CrossRef]
- Butturini, E.; de Carcereri Prati, A.; Chiavegato, G.; Rigo, A.; Cavalieri, E.; Darra, E.; Mariotto, S. Mild oxidative stress induces S-glutathionylation of STAT3 and enhances chemosensitivity of tumoural cells to chemotherapeutic drugs. Free Radic. Biol. Med. 2013, 65, 1322–1330. [Google Scholar] [CrossRef]
- Ohbayashi, N.; Ikeda, O.; Taira, N.; Yamamoto, Y.; Muromoto, R.; Sekine, Y.; Sugiyama, K.; Honjoh, T.; Matsuda, T. LIF- and IL-6-induced acetylation of STAT3 at Lys-685 through PI3K/Akt activation. Biol. Pharm. Bull. 2007, 30, 1860–1864. [Google Scholar] [CrossRef]
- Benayoun, B.A.; Veitia, R.A. A post-translational modification code for transcription factors: Sorting through a sea of signals. Trends Cell Biol. 2009, 19, 189–197. [Google Scholar] [CrossRef]
- Guanizo, A.C.; Fernando, C.D.; Garama, D.J.; Gough, D.J. STAT3: A multifaceted oncoprotein. Growth Factors 2018, 36, 1–14. [Google Scholar] [CrossRef]
- Cocchiola, R.; Romaniello, D.; Grillo, C.; Altieri, F.; Liberti, M.; Magliocca, F.M.; Chichiarelli, S.; Marrocco, I.; Borgoni, G.; Perugia, G.; Eufemi, M. Analysis of STAT3 post-translational modifications (PTMs) in human prostate cancer with different Gleason Score. Oncotarget 2017, 27, 42560–42570. [Google Scholar] [CrossRef]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef] [Green Version]
- Karan, D.; Dubey, S. From Inflammation to Prostate Cancer: The Role of Inflammasomes. Adv. Urol. 2016, 2016, 3140372. [Google Scholar] [CrossRef]
- Chaiswing, L.; St Clair, W.H.; St. Clair, D.K. Redox Paradox: A Novel Approach to Therapeutics-Resistant Cancer. Antioxid. Redox Signal. 2018, 29, 1237–1272. [Google Scholar] [CrossRef]
- Li, S.P.; Goldman, N.D. Regulation of human C-reactive protein gene expression by two synergistic IL-6 responsive elements. Biochemistry 1996, 35, 9060–9068. [Google Scholar] [CrossRef]
- Wu, Y.; Potempa, L.A.; El Kebir, D. Filep JGC-reactive protein and inflammation: Conformational changes affect function. Biol. Chem. 2015, 396, 1181–1197. [Google Scholar] [CrossRef]
- Sun, J.; Ren, X.; Simpkins, J.W. Sequential Upregulation of Superoxide Dismutase 2 and Heme Oxygenase 1 by tert-Butylhydroquinone Protects Mitochondria during Oxidative Stress. Mol. Pharmacol. 2015, 88, 437–449. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Wang, Y.; Liang, Y.; Zhang, M.; Wei, J.; Zheng, X.; Li, F.; Meng, Y.; Zhu, N.W.; Li, J.; et al. Loss of p27 upregulates MnSOD in a STAT3-dependent manner, disrupts intracellular redox activity and enhances cell migration. J. Cell Sci. 2014, 127, 2920–2933. [Google Scholar] [CrossRef] [Green Version]
- Xuan, X.; Li, S.; Lou, X.; Zheng, X.; Li, Y.; Wang, F.; Gao, Y.; Zhang, H.; He, H.; Zeng, Q. STAT3 promotes invasion of esophageal squamous cell carcinoma through up-regulation of MMP2. Mol. Biol. Rep. 2015, 42, 907–915. [Google Scholar] [CrossRef]
- Gopcevic, K.; Rovcanin, B.; Kekic, D.; Krivokapic, Z.; Dragutinovic, V. Matrix Metalloproteinase-2 and -9, Lactate, and Malate Dehydrogenase and Lipid Peroxides in Sera of Patients with Colorectal Carcinoma. Folia Biol. 2017, 63, 190–196. [Google Scholar]
- Kim, K.W.; Mutter, R.W.; Cao, C.; Albert, J.M.; Shinohara, E.T.; Sekhar, K.R.; Lu, B. Inhibition of signal transducer and activator of transcription 3 activity results in down-regulation of Survivin following irradiation. Mol. Cancer Ther. 2006, 5, 2659–2665. [Google Scholar] [CrossRef] [Green Version]
- Laudisi, F.; Cherubini, F.; Monteleone, G.; Stolfi, C. STAT3 Interactors as Potential Therapeutic Targets for Cancer Treatment. Int. J. Mol. Sci. 2018, 19, 1787. [Google Scholar] [CrossRef]
- Hou, T.; Ray, S.; Lee, C.; Brasier, A.R. The STAT3 NH2-terminal domain stabilizes enhanceosome assembly by interacting with the p300 bromodomain. J. Biol. Chem. 2008, 283, 30725–30734. [Google Scholar] [CrossRef]
- Cardoso, A.A.; Jiang, Y.; Luo, M.; Reed, A.M.; Shahda, S.; He, Y.; Maitra, A.; Kelley, M.R.; Fishel, M.L. APE1/Ref-1 regulates STAT3 transcriptional activity and APE1/Ref-1-STAT3 dual-targeting effectively inhibits pancreatic cancer cell survival. PLoS ONE 2012, 7, e47462. [Google Scholar] [CrossRef]
- Liu, X.; Wang, L.; Zhao, K.; Thompson, P.R.; Hwang, Y.; Marmorstein, R.; Cole, P.A. The structural basis of protein acetylation by the p300/CBP transcriptional coactivator. Nature 2008, 451, 846–850. [Google Scholar] [CrossRef]
- Bhakat, K.K.; Mantha, A.K.; Mitra, S. Transcriptional regulatory functions of mammalian AP-endonuclease (APE1/Ref-1), an essential multifunctional protein. Antioxid. Redox Signal. 2009, 11, 621–638. [Google Scholar] [CrossRef]
- Wang, Y.M.; Gu, M.L.; Meng, F.S.; Jiao, W.R.; Zhou, X.X.; Yao, H.P.; Ji, F. Histone acetyltransferase p300/CBP inhibitor C646 blocks the survival and invasion pathways of gastric cancer cell lines. Int. J. Oncol. 2017, 51, 1860–1868. [Google Scholar] [CrossRef]
- Kim, Y.J.; Kim, D.; Illuzzi, J.L.; Delaplane, S.; Su, D.; Bernier, M.; Gross, M.L.; Georgiadis, M.M.; Wilson, D.M., 3rd. S-glutathionylation of cysteine 99 in the APE1 protein impairs abasic endonuclease activity. J. Mol. Biol. 2011, 414, 313–326. [Google Scholar] [CrossRef]
- Butturini, E.; Darra, E.; Chiavegato, G.; Cellini, B.; Cozzolino, F.; Monti, M.; Pucci, P.; Dell’Orco, D.; Mariotto, S. S-Glutathionylation at Cys328 and Cys542 impairs STAT3 phosphorylation. ACS Chem. Biol. 2014, 9, 1885–1893. [Google Scholar] [CrossRef]
- Sakaguchi, M.; Oka, M.; Iwasaki, T.; Fukami, Y.; Nishigori, C. Role and regulation of STAT3 phosphorylation at Ser727 in melanocytes and melanoma cells. J. Investig. Dermatol. 2012, 132, 1877–1885. [Google Scholar] [CrossRef]
- Bak, Y.; Kang, J.W.; Kim, M.S.; Park, Y.S.; Kwon, T.; Kim, S.; Hong, J.; Yoon, D.Y. IL-32θ downregulates CCL5 expression through its interaction with PKCδ and STAT3. Cell. Signal. 2014, 26, 3007–3015. [Google Scholar] [CrossRef]
- Hsu, F.N.; Chen, M.C.; Lin, K.C.; Peng, Y.T.; Li, P.C.; Lin, E.; Chiang, M.C.; Hsieh, J.T.; Lin, H. Cyclin-dependent kinase 5 modulates STAT3 and androgen receptor activation through phosphorylation of Ser727 on STAT3 in prostate cancer cells. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E975–E986. [Google Scholar] [CrossRef]
- Masliantsev, K.; Pinel, B.; Balbous, A.; Guichet, P.O.; Tachon, G.; Milin, S.; Godet, J.; Duchesne, M.; Berger, A.; Petropoulos, C.; et al. Impact of STAT3 phosphorylation in glioblastoma stem cells radiosensitization and patient outcome. Oncotarget 2017, 9, 3968–3979. [Google Scholar] [CrossRef] [Green Version]
- Poli, V.; Camporeale, A. STAT3-Mediated Metabolic Reprograming in Cellular Transformation and Implications for Drug Resistance. Front. Oncol. 2015, 5, 121. [Google Scholar] [CrossRef]
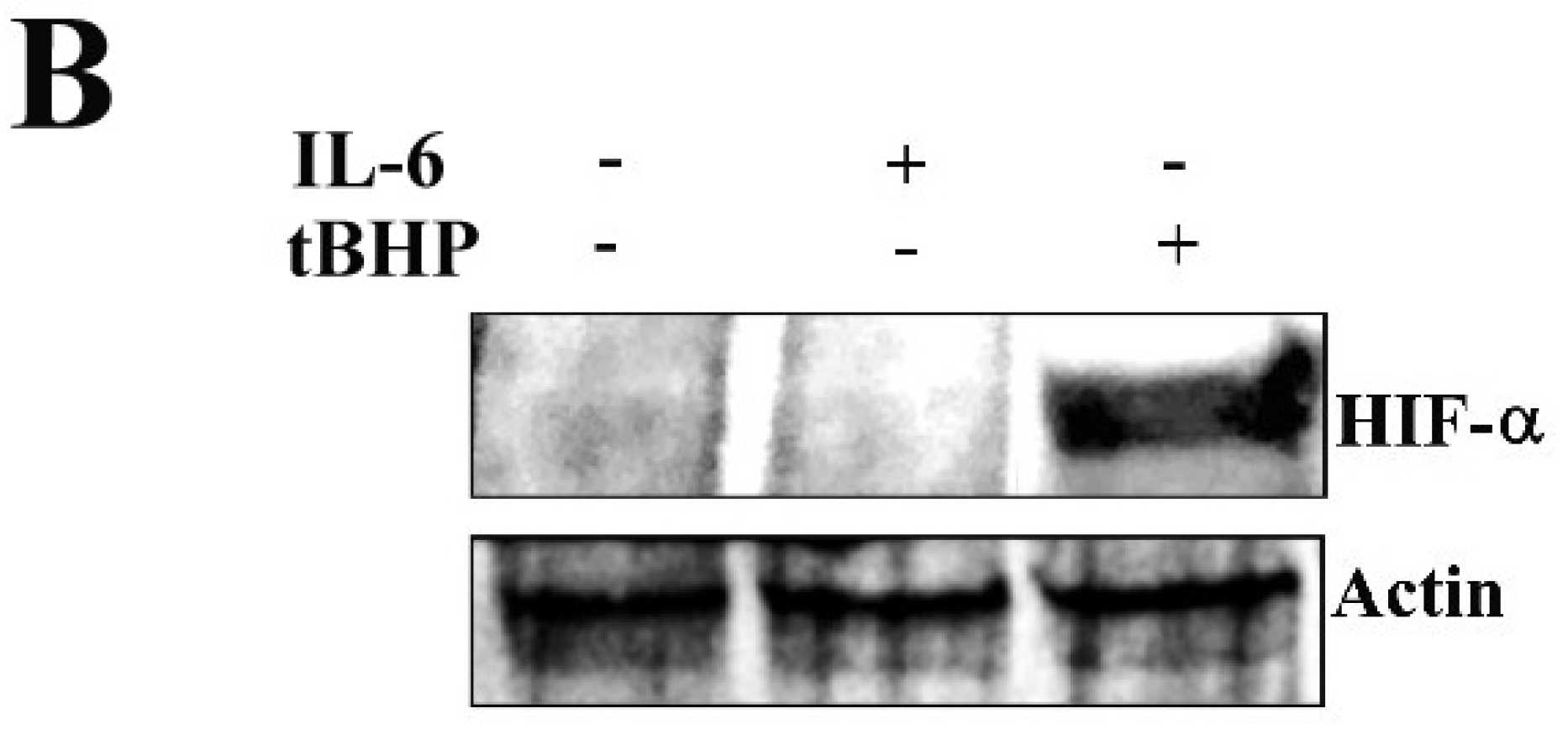
- Darnell, J.E., Jr. STAT3, HIF-1, glucose addiction and Warburg effect. Aging 2010, 2, 890–891. [Google Scholar] [CrossRef] [Green Version]
- Zepeda, A.B.; Pessoa, A., Jr.; Castillo, R.L.; Figueroa, C.A.; Pulgar, V.M.; Farías, J.G. Cellular and molecular mechanisms in the hypoxic tissue: Role of HIF-1 and ROS. Cell Biochem. Funct. 2013, 31, 451–459. [Google Scholar] [CrossRef]
- Demaria, M.; Poli, V. PKM2, STAT3 and HIF-1α: The Warburg’s vicious circle. JAK-Stat 2012, 1, 194–196. [Google Scholar] [CrossRef]
- Jiang, C.; Masood, M.; Rasul, A.; Wei, W.; Wang, Y.; Ali, M.; Mustaqeem, M.; Li, J.; Li, X. Altholactone Inhibits NF-κB and STAT3 Activation and Induces Reactive Oxygen Species-Mediated Apoptosis in Prostate Cancer DU145 Cells. Molecules 2017, 22, 240. [Google Scholar] [CrossRef]
- Maryam, A.; Mehmood, T.; Zhang, H.; Li, Y.; Khan, M.; Ma, T. Alantolactone induces apoptosis, promotes STAT3 glutathionylation and enhanceschemosensitivity of A549 lung adenocarcinoma cells to doxorubicin via oxidativestress. Sci. Rep. 2017, 24, 6242. [Google Scholar] [CrossRef]
- Eufemi, M.; Coppari, S.; Altieri, F.; Grillo, C.; Ferraro, A.; Turano, C. ERp57 is present in STAT3-DNA complexes. Biochem. Biophys. Res. Commun. 2004, 29, 1306–1312. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Cell ROS (Mean Fluorescence) |
|---|---|
| LNCaP | 32,435.25 |
| LNCaP + tBHP | 45,390.62 |
| DU-145 | 41,480.41 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cocchiola, R.; Rubini, E.; Altieri, F.; Chichiarelli, S.; Paglia, G.; Romaniello, D.; Carissimi, S.; Giorgi, A.; Giamogante, F.; Macone, A.; et al. STAT3 Post-Translational Modifications Drive Cellular Signaling Pathways in Prostate Cancer Cells. Int. J. Mol. Sci. 2019, 20, 1815. https://doi.org/10.3390/ijms20081815
Cocchiola R, Rubini E, Altieri F, Chichiarelli S, Paglia G, Romaniello D, Carissimi S, Giorgi A, Giamogante F, Macone A, et al. STAT3 Post-Translational Modifications Drive Cellular Signaling Pathways in Prostate Cancer Cells. International Journal of Molecular Sciences. 2019; 20(8):1815. https://doi.org/10.3390/ijms20081815
Chicago/Turabian StyleCocchiola, Rossana, Elisabetta Rubini, Fabio Altieri, Silvia Chichiarelli, Giuliano Paglia, Donatella Romaniello, Stefania Carissimi, Alessandra Giorgi, Flavia Giamogante, Alberto Macone, and et al. 2019. "STAT3 Post-Translational Modifications Drive Cellular Signaling Pathways in Prostate Cancer Cells" International Journal of Molecular Sciences 20, no. 8: 1815. https://doi.org/10.3390/ijms20081815