Nifedipine Modulates Renal Lipogenesis via the AMPK-SREBP Transcriptional Pathway

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Effect of Nifedipine on Cell Viability
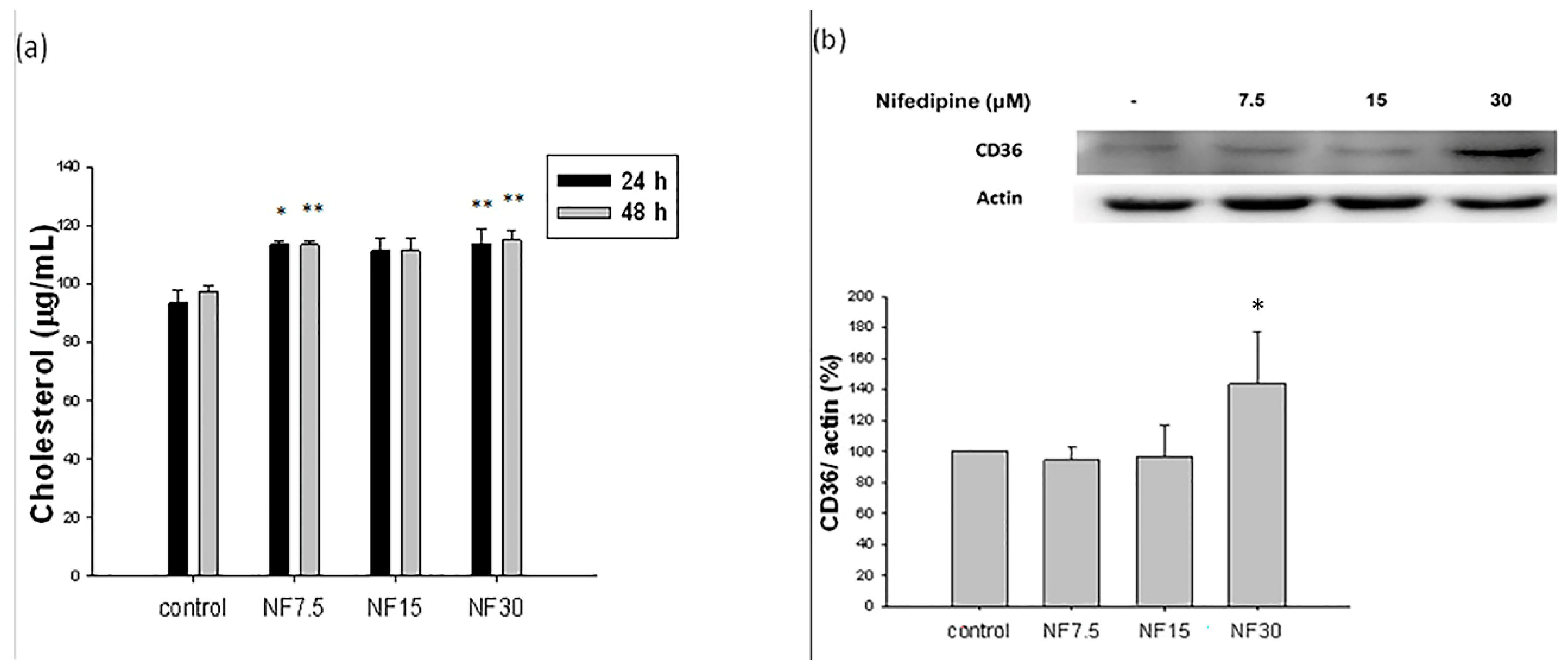
2.2. Effect of Nifedipine on Intracellular Lipid Accumulation
2.3. Nifedipine Stimulated Production of Reactive Oxygen Species (ROS)
2.4. Nifedipine Promoted the Expression of Proteins Related to Kidney Injury
2.5. Nifedipine Stimulated Cholesterol De Novo Biosynthesis
2.6. Nifedipine Upregulated the Expression of CD36 Proteins Related to Lipid Translocation
2.7. Nifedipine Stimulated the Production of Acetyl CoA Synthetase (ACS), Phosphorylated Acetyl-CoA Carboxylase (p-ACC) over Total Acetyl-CoA Carboxylase Acetyl (ACC), and Long Chain Fatty Acyl Elongase (ACSL1), but Not Fatty Acid Synthase (FAS) in 48 h
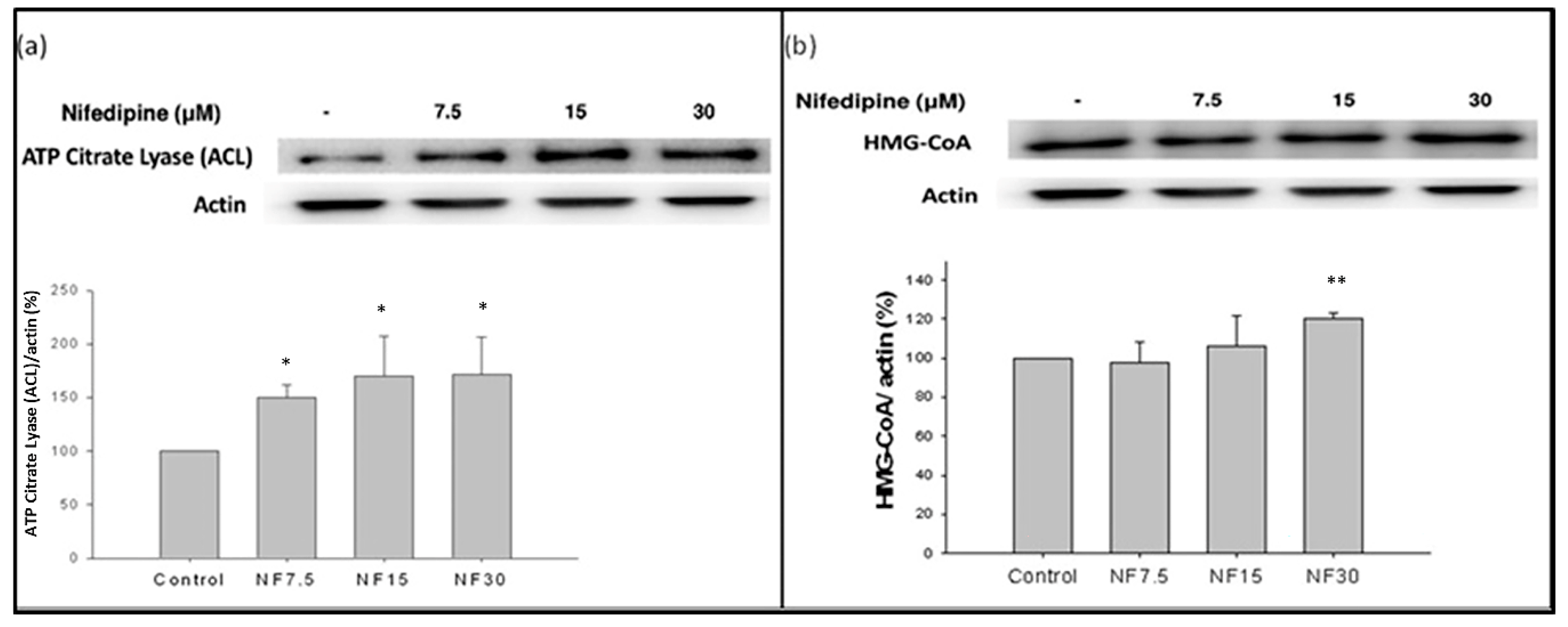
2.8. Nifedipine Stimulated the Production of ATP Citrate Lyase (ACL) and HMG-CoA Reductase (HMGCR)
2.9. Nifedipine Downregulated the Expression of Phosphorylated AMPK
2.10. Nifedipine Upregulated the Expression of SREBP-1 and SREBP-2
2.11. Nifedipine Upregulated the Expression of PPAR-α
2.12. Nifedipine Upregulated the Expression of lipin-1
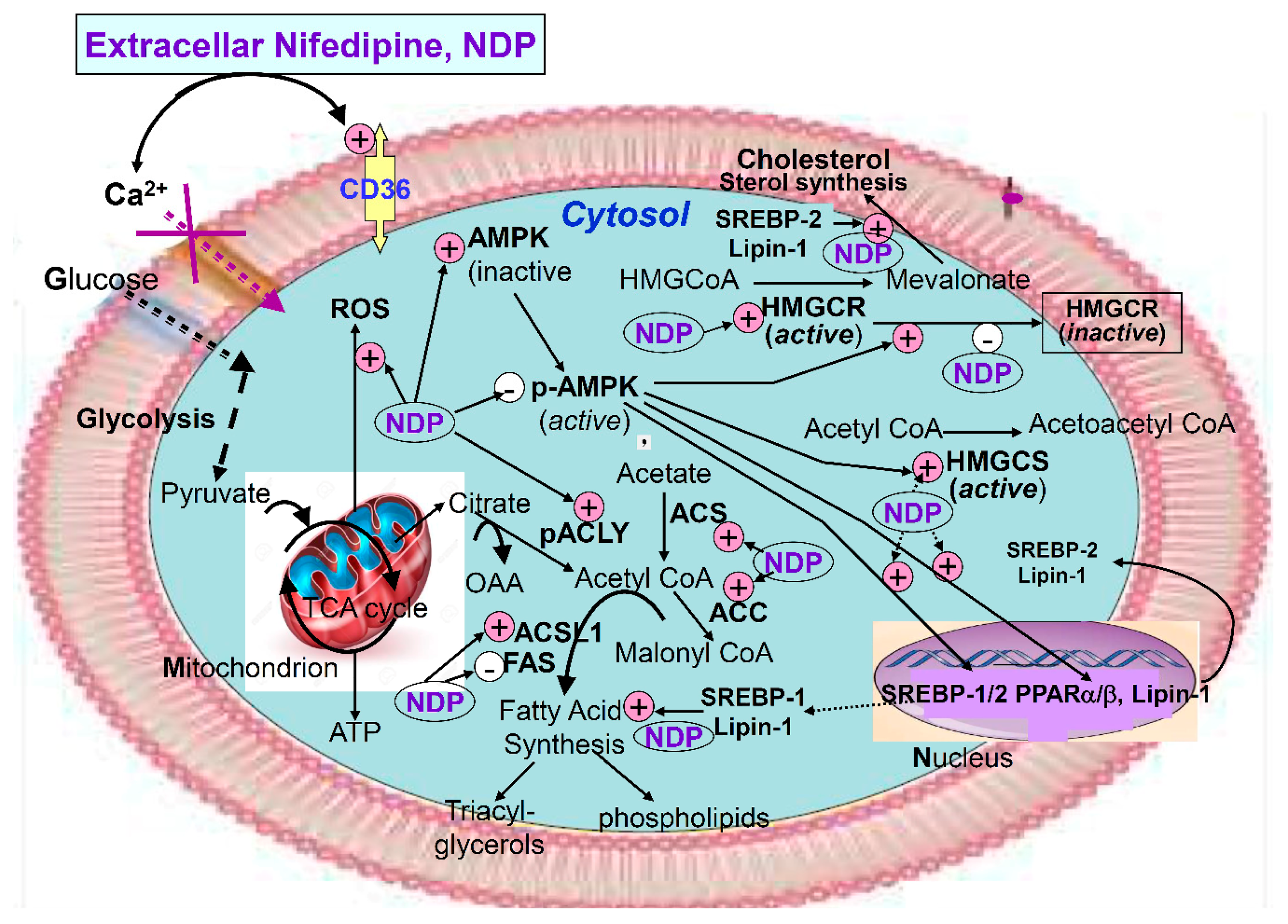
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Cell Survival Assay
4.3. Oil Red O Staining
4.4. Lipid Assay
4.5. Lipid Peroxidation
4.6. Measurement of ROS Production
4.7. Western Blotting Procedure
4.8. Statistical Analysis
5. Limitation
6. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- De Vries, A.P.; Ruggenenti, P.; Ruan, X.Z.; Praga, M.; Cruzado, J.M.; Bajema, I.M.; D’Agati, V.D.; Lamb, H.J.; Pongrac Barlovic, D.; Hojs, R.; et al. Fatty kidney: Emerging role of ectopic lipid in obesity-related renal disease. Lancet Diabetes Endocrinol. 2014, 2, 417–426. [Google Scholar] [CrossRef]
- Engin, A.B. What is lipotoxicity? Adv. Exp. Med. Biol. 2017, 960, 197–220. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.M.; Ahn, S.H.; Choi, P.; Ko, Y.A.; Han, S.H.; Chinga, F.; Park, A.S.; Tao, J.; Sharma, K.; Pullman, J.; et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 2015, 21, 37–46. [Google Scholar] [CrossRef]
- Tang, C.; Cai, J.; Dong, Z. Mitochondrial dysfunction in obesity-related kidney disease: A novel therapeutic target. Kidney Int. 2016, 90, 930–933. [Google Scholar] [CrossRef]
- Pepino, M.Y.; Kuda, O.; Samovski, D.; Abumrad, N.A. Structure-function of CD36 and importance of fatty acid signal transduction in fat metabolism. Annu. Rev. Nutr. 2014, 34, 281–303. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Okamura, D.M.; Lu, X.; Chen, Y.; Moorhead, J.; Varghese, Z.; Ruan, X.Z. CD36 in chronic kidney disease: Novel insights and therapeutic opportunities. Nat. Rev. Nephrol. 2017, 13, 769–781. [Google Scholar] [CrossRef]
- Marchal, L.; Laviolette, M.; Rodrigue-Way, A.; Sow, B.; Brochu, M.; Caron, V.; Tremblay, A. The CD36-PPARγ Pathway in Metabolic Disorders. Int. J. Mol. Sci. 2018, 19, 1529. [Google Scholar] [CrossRef]
- Jung, E.J.; Kwon, S.W.; Jung, B.H.; Oh, S.H.; Lee, B.H. Role of the AMPK/SREBP-1 pathway in the development of orotic acid-induced fatty liver. J. Lipid Res. 2011, 52, 1617–1625. [Google Scholar] [CrossRef] [PubMed]
- Mahali, S.K.; Verma, N.; Manna, S.K. Advanced glycation end products induce lipogenesis: Regulation by natural xanthone through inhibition of ERK and NF-κB. J. Cell Physiol. 2014, 229, 1972–1980. [Google Scholar] [CrossRef]
- Feng, L.; Gu, C.; Li, Y.; Huang, J. High Glucose Promotes CD36 Expression by Upregulating Peroxisome Proliferator-Activated Receptor γ Levels to Exacerbate Lipid Deposition in Renal Tubular Cells. BioMed Res. Int. 2017, 2017, 1414070; [Google Scholar] [CrossRef]
- Kohjima, M.; Higuchi, N.; Kato, M.; Kotoh, K.; Yoshimoto, T.; Fujino, T.; Yada, M.; Yada, R.; Harada, N.; Enjoji, M.; et al. SREBP-1c, regulated by the insulin and AMPK signaling pathways, plays a role in nonalcoholic fatty liver disease. Int. J. Mol. Med. 2008, 21, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.P.; Tsai, C.W.; Shen, C.Y.; Day, C.H.; Yeh, Y.L.; Chen, R.J.; Ho, T.J.; Padma, V.V.; Kuo, W.W.; Huang, C.Y. Palmitic acid interferes with energy metabolism balance by adversely switching the SIRT1-CD36-fatty acid pathway to the PKC zeta-GLUT4-glucose pathway in cardiomyoblasts. J. Nutr. Biochem. 2016, 31, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Peterson, T.R.; Sengupta, S.S.; Harris, T.E.; Carmack, A.E.; Kang, S.A.; Balderas, E.; Guertin, D.A.; Madden, K.L.; Carpenter, A.E.; Finck, B.N.; et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell 2011, 146, 408–420. [Google Scholar] [CrossRef] [PubMed]
- Ahaneku, J.E.; Sakata, K.; Urano, T.; Takada, Y.; Takada, A. Influence of baseline values on lipids, lipoproteins and fibrinolytic parameters during treatment of hypertension with cilnidipine. Pharmacol. Res. 2000, 41, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Ahaneku, J.E.; Sakata, K.; Uranol, T.; Takada, Y.; Takada, A. Effects of cilnidipine on lipids, lipoproteins and fibrinolytic system in hypertensive patients. Drugs Exp. Clin. Res. 2000, 26, 119–123. [Google Scholar]
- Christensen, P.K.; Akram, K.; K?nig, K.B.; Parving, H.H. Autoregulation of glomerular filtration rate in patients with type 2 diabetes during isradipine therapy. Diabetes Care 2003, 26, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Griffin, K.A.; Bidani, A.K. Potential risks of calcium channel blockers in chronic kidney disease. Curr. Cardiol. Rep. 2008, 10, 448–455. [Google Scholar] [CrossRef]
- Kloke, H.J.; Branten, A.J.; Huysmans, F.T.; Wetzels, J.F. Antihypertensive treatment of patients with proteinuric renal diseases: Risks or benefits of calcium channel blockers? Kidney Int. 1998, 53, 1559–1573. [Google Scholar] [CrossRef]
- Donkor, J.; Zhang, P.; Wong, S.; O’Loughlin, L.; Dewald, J.; Kok, B.P.; Brindley, D.N.; Reue, K. A conserved serine residue is required for the phosphatidate phosphatase activity but not the transcriptional coactivator functions of lipin-1 and lipin-2. J. Biol. Chem. 2009, 284, 29968–29978. [Google Scholar] [CrossRef]
- Ichimura, T.; Asseldonk, E.J.; Humphreys, B.D.; Gunaratnam, L.; Duffield, J.S.; Bonventre, J.V. Kidney injury molecule-1 is a phosphatidylserine receptor that confers a phagocytic phenotype on epithelial cells. J. Clin. Investig. 2008, 118, 1657–1668. [Google Scholar] [CrossRef]
- Gou, R.; Chen, J.; Sheng, S.; Wang, R.; Fang, Y.; Yang, Z.; Wang, L.; Tang, L. KIM-1 Mediates High Glucose-Induced Autophagy and Apoptosis in Renal Tubular Epithelial Cells. Cell Physiol. Biochem. 2016, 38, 2479–2488. [Google Scholar] [CrossRef] [PubMed]
- Myers, B.D.; Chui, F.; Hilberman, M.; Michaels, A.S. Transtubular leakage of glomerular filtrate in human acute renal failure. Am. J. Physiol. 1979, 237, F319–F325. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, B.D.; Xu, F.; Sabbisetti, V.; Grgic, I.; Movahedi Naini, S.; Wang, N.; et al. Chronic epithelial kidney injury molecule-1 expression causes murine kidney fibrosis. J. Clin. Investig. 2013, 123, 4023–4035. [Google Scholar] [CrossRef]
- Bland, S.K.; Schmiedt, C.W.; Clark, M.E.; DeLay, J.; Bienzle, D. Expression of Kidney Injury Molecule-1 in Healthy and Diseased Feline Kidney Tissue. Vet. Pathol. 2017, 54, 490–510. [Google Scholar] [CrossRef]
- Baines, R.J.; Chana, R.S.; Hall, M.; Febbraio, M.; Kennedy, D.; Brunskill, N.J. CD36 mediates proximal tubular binding and uptake of albumin and is upregulated in proteinuric nephropathies. Am. J. Physiol. Ren. Physiol. 2012, 303, F1006–F1014. [Google Scholar] [CrossRef]
- Grone, E.F.; Walli, A.K.; Grone, H.J.; Miller, B.; Seidel, D. The role of lipids in nephrosclerosis and glomerulosclerosis. Atherosclerosis 1994, 107, 1–13. [Google Scholar] [CrossRef]
- Takemura, T.; Yoshioka, K.; Aya, N.; Murakami, K.; Matumoto, A.; Itakura, H.; Kodama, T.; Suzuki, H.; Maki, S. Apolipoproteins and lipoprotein receptors in glomeruli in human kidney diseases. Kidney Int. 1993, 43, 918–927. [Google Scholar] [CrossRef]
- Chade, A.R.; Mushin, O.P.; Zhu, X.; Rodriguez-Porcel, M.; Grande, J.P.; Textor, S.C.; Lerman, A.; Lerman, L.O. Pathways of renal fibrosis and modulation of matrix turnover in experimental hypercholesterolemia. Hypertension 2005, 46, 772–779. [Google Scholar] [CrossRef]
- Drover, V.A.; Abumrad, N.A. CD36-dependent fatty acid uptake regulates expression of peroxisome proliferator activated receptors. Biochem. Soc. Trans. 2005, 33, 311–315. [Google Scholar] [CrossRef]
- Yasu, T.; Kobayashi, M.; Mutoh, A.; Yamakawa, K.; Momomura, S.; Ueda, S. Dihydropyridine calcium channel blockers inhibit non-esterified-fatty-acid-induced endothelial and rheological dysfunction. Clin. Sci. 2013, 125, 247–255. [Google Scholar] [CrossRef]
- Decleves, A.E.; Zolkipli, Z.; Satriano, J.; Wang, L.; Nakayama, T.; Rogac, M.; Le, T.P.; Nortier, J.L.; Farquhar, M.G.; Naviaux, R.K.; et al. Regulation of lipid accumulation by AMP-activated kinase in high fat diet-induced kidney injury. Kidney Int. 2014, 85, 611–623. [Google Scholar] [CrossRef]
- Sung, J.Y.; Choi, H.C. Nifedipine inhibits vascular smooth muscle cell proliferation and reactive oxygen species production through AMP-activated protein kinase signaling pathway. Vascul. Pharmacol. 2012, 56, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ricoult, S.J.H.; Manning, B.D. The multifaceted role of mTORC1 in the control of lipid metabolism. EMBO Rep. 2013, 14, 242–251. [Google Scholar] [CrossRef]
- Lage, R.; Dieguez, C.; Vidal-Puig, A.; Lopez, M. AMPK: A metabolic gauge regulating whole-body energy homeostasis. Trends Mol. Med. 2008, 14, 539–549. [Google Scholar] [CrossRef]
- Decleves, A.E.; Mathew, A.V.; Cunard, R. AMPK mediates the initiation of kidney disease induced by a high-fat diet. J. Am. Soc. Nephrol. 2011, 22, 1846–1855. [Google Scholar] [CrossRef]
- Eberlé, D.; Hegarty, B.; Bossard, P.; Ferré, P.; Foufelle, F. SREBP transcription factors: Master regulators of lipid homeostasis. Biochimie 2004, 86, 839–848. [Google Scholar] [CrossRef] [PubMed]
- Kok, B.P.C.; Brindley, D.N. “The Role of Phosphatidate Phosphatase in Glycerolipid Biosynthesis” in “Regulation of Lipins and Their Role in Lipid Metabolism”; AOCS Lipid Library: Urbana, IL, USA, 2010. [Google Scholar]
- Chen, Y.; Rui, B.B.; Tang, L.Y.; Hu, C.M. Lipin Family Proteins - Key Regulators in lipid metabolism. Ann. Nutr. Metab. 2015, 66, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, S.; Gupta, P.; Saini, A.S.; Kaushal, C.; Sharma, S. The peroxisome proliferator-activated receptor: A family of nuclear receptors role in various diseases. J. Adv. Pharm. Technol. Res. 2011, 2, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Reue, K.; Brindley, D.N. Thematic Review Series: Glycerolipids. Multiple roles for lipins/phosphatidate phosphatase enzymes in lipid metabolism. J. Lipid Res. 2008, 49, 2493–2503. [Google Scholar] [CrossRef]
- Favreau, F.; Thuillier, R.; Cau, J.; Milin, S.; Manguy, E.; Mauco, G.; Zhu, X.; Lerman, L.O.; Hauet, T. Anti-thrombin therapy during warm ischemia and cold preservation prevents chronic kidney graft fibrosis in a DCD model. Am. J. Transplant. 2007, 10, 30–39. [Google Scholar] [CrossRef]
- Favreau, F.; Zhu, X.Y.; Krier, J.D.; Lin, J.; Warner, L.; Textor, S.C.; Lerman, L.O. Revascularization of swine renal artery stenosis improves renal function but not the changes in vascular structure. Kidney Int. 2010, 78, 1110–1118. [Google Scholar] [CrossRef] [PubMed]
- Hauck, A.K.; Bernlohr, D.A. Oxidative stress and lipotoxicity. J. Lipid Res. 2016, 57, 1976–1986. [Google Scholar] [CrossRef] [PubMed]
- Qin, G.; Ma, J.; Huang, Q.; Yin, H.; Han, J.; Li, M.; Deng, Y.; Wang, B.; Hassan, W.; Shang, J. Isoquercetin Improves Hepatic Lipid Accumulation by Activating AMPK Pathway and Suppressing TGF-β Signaling on an HFD-Induced Nonalcoholic Fatty Liver Disease Rat Model. Int. J. Mol. Sci. 2018, 19, 4126. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, Y.-C.; Wu, M.-S.; Lin, Y.-F.; Chen, C.-R.; Chen, C.-Y.; Chen, C.-J.; Shen, C.-C.; Chen, K.-C.; Peng, C.-C. Nifedipine Modulates Renal Lipogenesis via the AMPK-SREBP Transcriptional Pathway. Int. J. Mol. Sci. 2019, 20, 1570. https://doi.org/10.3390/ijms20071570
Lin Y-C, Wu M-S, Lin Y-F, Chen C-R, Chen C-Y, Chen C-J, Shen C-C, Chen K-C, Peng C-C. Nifedipine Modulates Renal Lipogenesis via the AMPK-SREBP Transcriptional Pathway. International Journal of Molecular Sciences. 2019; 20(7):1570. https://doi.org/10.3390/ijms20071570
Chicago/Turabian StyleLin, Yen-Chung, Mai-Szu Wu, Yuh-Feng Lin, Chang-Rong Chen, Chang-Yu Chen, Chang-Jui Chen, Che-Chou Shen, Kuan-Chou Chen, and Chiung-Chi Peng. 2019. "Nifedipine Modulates Renal Lipogenesis via the AMPK-SREBP Transcriptional Pathway" International Journal of Molecular Sciences 20, no. 7: 1570. https://doi.org/10.3390/ijms20071570
APA StyleLin, Y.-C., Wu, M.-S., Lin, Y.-F., Chen, C.-R., Chen, C.-Y., Chen, C.-J., Shen, C.-C., Chen, K.-C., & Peng, C.-C. (2019). Nifedipine Modulates Renal Lipogenesis via the AMPK-SREBP Transcriptional Pathway. International Journal of Molecular Sciences, 20(7), 1570. https://doi.org/10.3390/ijms20071570