Role of SIRT-3, p-mTOR and HIF-1α in Hepatocellular Carcinoma Patients Affected by Metabolic Dysfunctions and in Chronic Treatment with Metformin

 , ,
, , 
 ,
, 

Abstract
1. Introduction
2. Results
2.1. Patient Characteristics
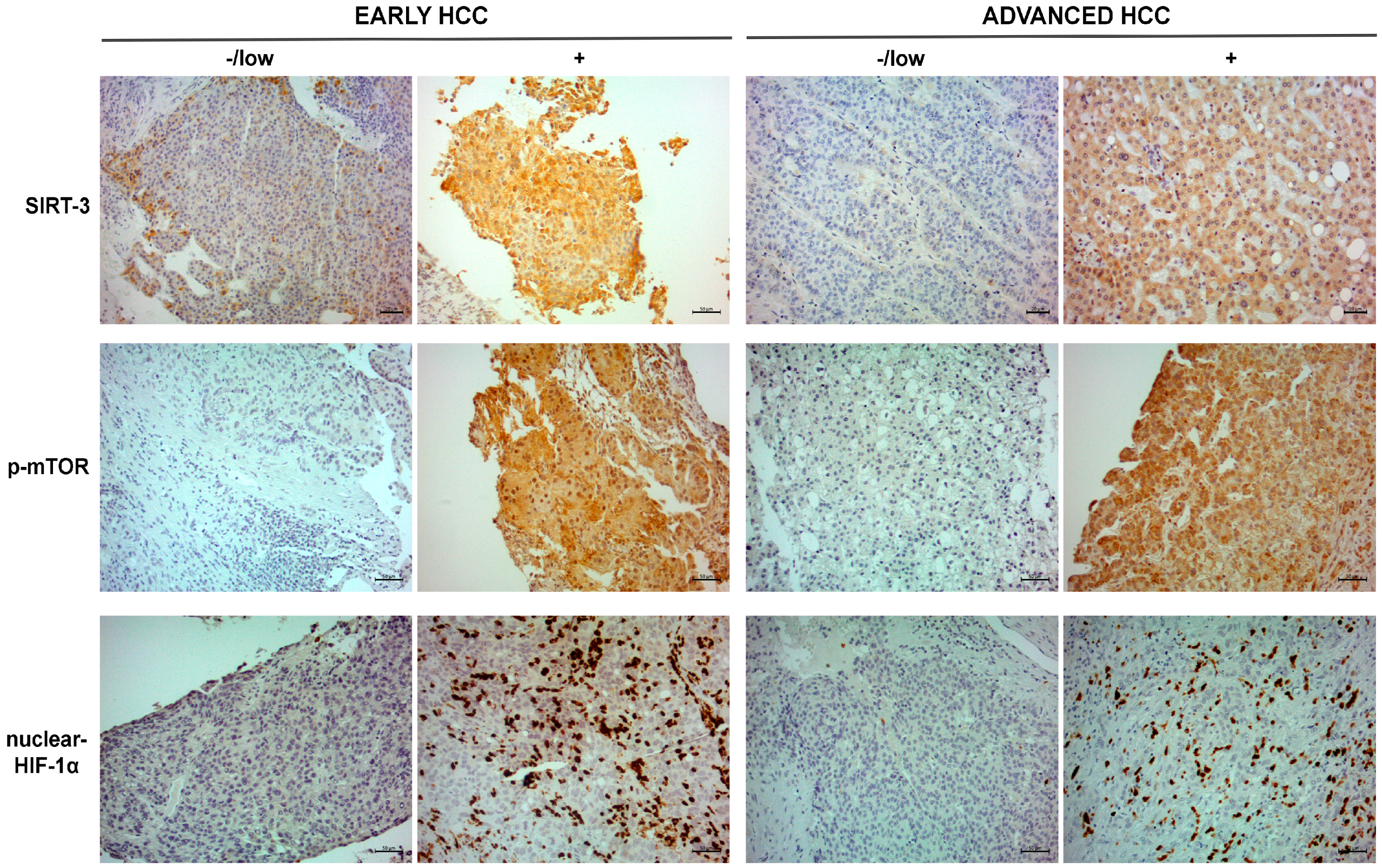
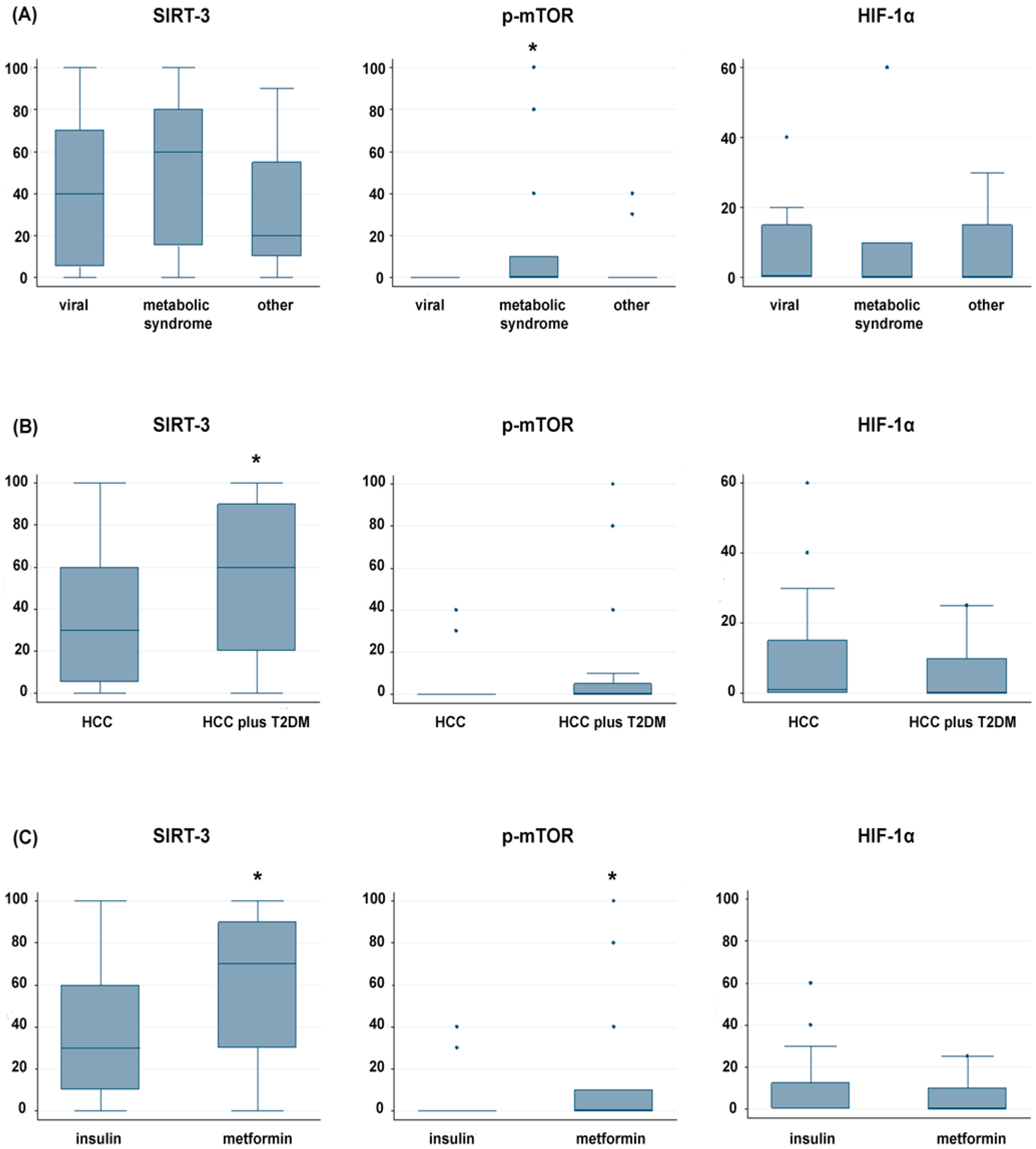
2.2. Immunohistochemical Expression Pattern of SIRT-3, p-mTOR and Nuclear HIF-1α in Tumor Tissues
2.3. In Vitro Effect of Metformin and Sorafenib on HCC Cell Lines
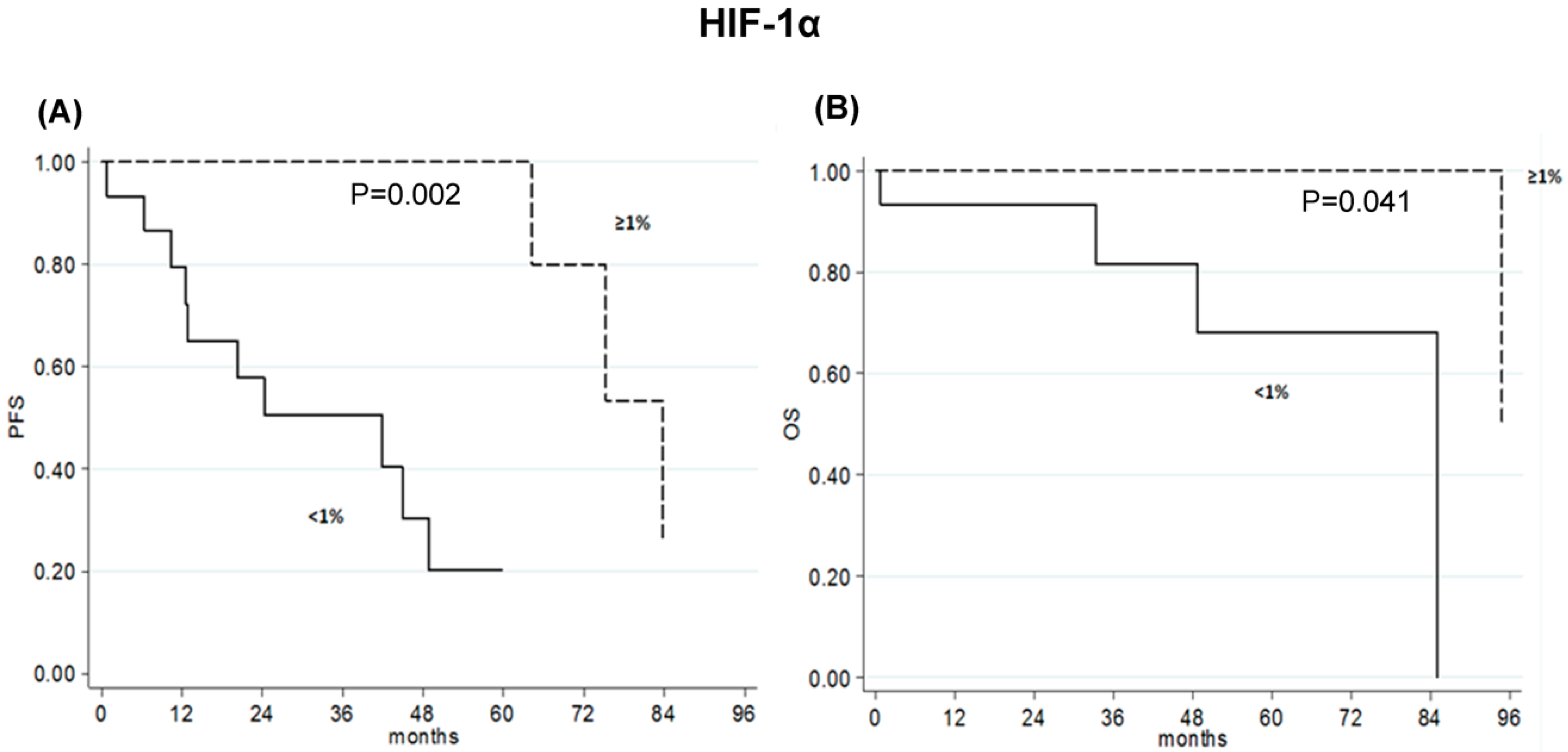
2.4. Association between SIRT-3, p-mTOR and Nuclear HIF-1α Expression and Clinical Outcome of Early-Stage HCC Patients
2.5. High p-mTOR Levels Correlated with Poor Prognosis in Advanced-Stage HCC Patients
3. Discussion
4. Materials and Methods
4.1. Patient Population
4.2. Immunohistochemistry Analysis
4.3. HCC Cell Lines and Drugs
4.4. Cell Viability Assay
4.5. Annexin V Assay
4.6. Western Blot Analysis
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| HCC | hepatocellular carcinoma |
| NASH | non-alcoholic steatohepatitis |
| SIRT-3 | sirtuin-3 |
| T2DM | type 2 diabetes mellitus |
| mTOR | mammalian target of rapamycin |
| AMPK | AMP-activated protein kinase |
| HIF-1α | hypoxia-induced factor-1α |
| AASLD | American Association for the Study of Liver Diseases |
| IQR | interquartile range |
| DFS | disease-free survival |
| PFS | progression-free survival |
| OS | overall survival |
| FBS | fetal bovine serum |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| DMSO | dimethyl sulfoxide |
| IR | insulin resistance |
References
- Seeff, L.B.; Hoofnagle, J.H. Epidemiology of hepatocellular carcinoma in areas of low hepatitis B and hepatitis C endemicity. Oncogene 2006, 25, 3771–3777. [Google Scholar] [CrossRef]
- Souza, M.R.; Diniz Mde, F.; Medeiros-Filho, J.E.; Araújo, M.S. Metabolic syndrome and risk factors for non-alcoholic fatty liver disease. Arq. Gastroenterol. 2012, 49, 89–96. [Google Scholar] [CrossRef]
- De Matteis, S.; Granato, A.M.; Napolitano, R.; Molinari, C.; Valgiusti, M.; Santini, D.; Foschi, F.G.; Ercolani, G.; Vespasiani Gentilucci, U.; Faloppi, L.; et al. Interplay Between SIRT-3, Metabolism and Its Tumor Suppressor Role in Hepatocellular Carcinoma. Dig. Dis. Sci. 2017, 62, 1872–1880. [Google Scholar] [CrossRef]
- Casadei Gardini, A.; Faloppi, L.; De Matteis, S.; Foschi, F.G.; Silvestris, N.; Tovoli, F.; Palmieri, V.; Marisi, G.; Brunetti, O.; Vespasiani-Gentilucci, U.; et al. Metformin and insulin impact on clinical outcome in patients with advanced hepatocellular carcinoma receiving sorafenib: Validation study and biological rationale. Eur. J. Cancer 2017, 86, 106–114. [Google Scholar] [CrossRef]
- Casadei Gardini, A.; Faloppi, L.; De Matteis, S.; Foschi, F.G.; Silvestris, N.; Tovoli, F.; Palmieri, V.; Marisi, G.; Brunetti, O.; Vespasiani-Gentilucci, U.; et al. Effects of metformin on clinical outcome in diabetic patients with advanced HCC receiving sorafenib. Expert Opin. Pharmacother. 2015, 16, 2719–2725. [Google Scholar] [CrossRef]
- Kimura, N.; Tokunaga, C.; Dalal, S.; Richardson, C.; Yoshino, K.; Hara, K.; Kemp, B.E.; Witters, L.A.; Mimura, O.; Yonezawa, K. A possible linkage between AMP-activated protein kinase (AMPK) and mammalian target of rapamycin (mTOR) signalling pathway. Genes Cells 2003, 8, 65e79. [Google Scholar] [CrossRef]
- Zhou, X.; Chen, J.; Yi, G.; Deng, M.; Liu, H.; Liang, M.; Shi, B.; Fu, X.; Chen, Y.; Chen, L.; et al. Metformin suppresses hypoxiainduced stabilization of HIF-1a through reprogramming of oxygen metabolism in hepatocellular carcinoma. Oncotarget 2016, 7, 873e84. [Google Scholar]
- Bell, E.L.; Emerling, B.M.; Ricoult, S.J.; Guarente, L. SirT3 suppresses hypoxia inducible factor 1alpha and tumor growth by inhibiting mitochondrial ROS production. Oncogene 2011, 30, 2986e96T. [Google Scholar] [CrossRef]
- Alhazzazi, T.Y.; Kamarajan, P.; Joo, N.; Huang, J.Y.; Verdin, E.; D’Silva, N.J.; Kapila, Y.L. Sirtuin-3 (SIRT3), a novel potential therapeutic target for oral cancer. Cancer 2011, 117, 1670–1678. [Google Scholar] [CrossRef]
- Alhazzazi, T.Y.; Kamarajan, P.; Verdin, E.; Kapila, Y.L. SIRT3 and cancer: Tumor promoter or suppressor. Biochim. Biophys. Acta 2011, 1816, 80–88. [Google Scholar] [CrossRef]
- Desouki, M.M.; Doubinskaia, I.; Gius, D.; Abdulkadir, S.A. Decreased mitochondrial SIRT3 expression is a potential molecular biomarker associated with poor outcome in breast cancer. Hum. Pathol. 2014, 45, 1071–1077. [Google Scholar] [CrossRef]
- Zhang, Y.Y.; Zhou, L.M. Sirt3 inhibits hepatocellular carcinoma cell growth through reducing Mdm2-mediated p53 degradation. Biochem. Biophys. Res. Commun. 2012, 423, 26–31. [Google Scholar] [CrossRef]
- Song, C.L.; Tang, H.; Ran, L.K.; Ko, B.C.; Zhang, Z.Z.; Chen, X.; Ren, J.H.; Tao, N.N.; Li, W.Y.; Huang, A.L.; Chen, J. Sirtuin 3 inhibits hepatocellular carcinoma growth through the glycogen synthase kinase-3β/BCL2-associated X protein-dependent apoptotic pathway. Oncogene 2016, 35, 631–641. [Google Scholar] [CrossRef]
- Yang, S.L.; Liu, L.P.; Jiang, J.X.; Xiong, Z.F.; He, Q.J.; Wu, C. The correlation of expression levels of HIF-1alpha and HIF-2alpha in hepatocellular carcinoma with capsular invasion, portal vein tumor thrombi and patients’ clinical outcome. J. Clin. Oncol. 2014, 44, 159–167. [Google Scholar]
- Villanueva, A.; Chiang, D.Y.; Newell, P.; Peix, J.; Thung, S.; Alsinet, C.; Tovar, V.; Roayaie, S.; Minguez, B.; Sole, M.; et al. Pivotal role of mTOR signaling in hepatocellular carcinoma. Gastroenterol 2008, 135, 1972–1983. [Google Scholar] [CrossRef]
- Hirsch, F.R.; Varella-Garcia, M.; Bunn, P.A., Jr.; Di Maria, M.V.; Veve, R.; Bremmes, R.M.; Barón, A.E.; Zeng, C.; Franklin, W.A. Epidermal growth factor receptor in non-small-cell lung carcinomas: Correlation between gene copy number and protein expression and impact on prognosis. J. Clin. Oncol. 2003, 21, 3798–3807. [Google Scholar] [CrossRef]
- Al-Bahrani, R.; Tuertcher, D.; Zailaie, S.; Abuetabh, Y.; Nagamori, S.; Zetouni, N.; Bahitham, W.; Sergi, C. Differential SIRT1 expression in hepatocellular carcinomas and cholangiocarcinoma of the liver. Ann. Clin. Lab. Sci. 2015, 45, 3–9. [Google Scholar]
- Teodoro, J.S.; Duarte, F.V.; Gomes, A.P.; Varela, A.T.; Peixoto, F.M.; Rolo, A.P.; Palmeira, C.M. Berberine reverts hepatic mitochondrial dysfunction in high-fat fed rats: A possible role for SirT3 activation. Mitochondrion 2013, 13, 637–646. [Google Scholar] [CrossRef]
- Weir, H.J.; Lane, J.D.; Balthasar, N. SIRT3: A Central Regulator of Mitochondrial Adaptation in Health and Disease. Genes Cancer 2013, 4, 118–124. [Google Scholar] [CrossRef]
- Hirschey, M.D.; Shimazu, T.; Goetzman, E.; Jing, E.; Schwer, B.; Lombard, D.B.; Grueter, C.A.; Harris, C.; Biddinger, S.; Ilkayeva, O.R.; et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 2010, 464, 121–125. [Google Scholar] [CrossRef]
- Hallows, W.C.; Yu, W.; Smith, B.C.; Devries, M.K.; Ellinger, J.J.; Someya, S.; Shortreed, M.R.; Prolla, T.; Markley, J.L.; Smith, L.M.; et al. Sirt3 promotes the urea cycle and fatty acid oxidation during dietary restriction. Mol. Cell 2011, 41, 139–149. [Google Scholar] [CrossRef]
- Shimazu, T.; Hirschey, M.D.; Hua, L.; Dittenhafer-Reed, K.E.; Schwer, B.; Lombard, D.B.; Li, Y.; Bunkenborg, J.; Alt, F.W.; Denu, J.M.; et al. SIRT3 deacetylates mitochondrial 3- hydroxy-3-methylglutaryl CoA synthase 2 and regulates ketone body production. Cell Metab. 2010, 12, 654–661. [Google Scholar] [CrossRef]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef]
- Kim, Y.C.; Guan, K.L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef]
- Feng, Y.M.; Zhao, D.; Zhang, N.; Yu, C.G.; Zhang, Q.; Thijs, L.; Staessen, J.A. Insulin resistance in relation to lipids and inflammation in type-2 diabetic patients and non-diabetic people. PLoS ONE 2016, 11, e0153171. [Google Scholar] [CrossRef]
- Liu, H.Y.; Han, J.; Cao, S.Y.; Hong, T.; Zhuo, D.; Shi, J.; Liu, Z.; Cao, W. Hepatic autophagy is suppressed in the presence of insulin resistance and hyperinsulinemia: Inhibition of FoxO1- dependent expression of key autophagy genes by insulin. J. Biol. Chem. 2009, 284, 31484–31492. [Google Scholar] [CrossRef]
- Zhang, N.; Cao, M.M.; Liu, H.; Xie, G.Y.; Li, Y.B. Autophagy regulates insulin resistance following endoplasmic reticulum stress in diabetes. J. Physiol. Biochem. 2015, 71, 319–327. [Google Scholar] [CrossRef]
- Qian, Q.; Zhang, Z.; Orwig, A.; Chen, S.; Ding, W.X.; Xu, Y.; Kunz, R.C.; Lind, N.R.L.; Stamler, J.S.; Yang, L. S-nitrosoglutathione reductase dysfunction contributes to obesity-associated hepatic insulin resistance via regulating autophagy. Diabetes 2018, 67, 193–207. [Google Scholar] [CrossRef]
- Zhou, W.; Ye, S. Rapamycin improves insulin resistance and hepatic steatosis in type 2 diabetes rats through activation of autophagy. Cell Biol. Int. 2018. [Google Scholar] [CrossRef]
- Zheng, S.S.; Chen, X.H.; Yin, X.; Zhang, B.H. Prognostic significance of HIF-1α expression in hepatocellular carcinoma: A meta-analysis. PLoS ONE 2013, 8, e65753. [Google Scholar] [CrossRef]
- Dai, X.; Pi, G.; Yang, S.L.; Chen, G.G.; Liu, L.P.; Dong, H.H. Association of PD-L1 and HIF-1α Coexpression with Poor Prognosis in Hepatocellular Carcinoma. Transl. Oncol. 2018, 1, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Ponziani, F.; Ojetti, V.; Tortora, A.; Di Maurizio, L.; Purchiaroni, F.; Gasbarrini, A. The metabolic and toxicological considerations for mTOR inhibitors in the treatment of hepatocarcinoma. Exp. Opin. Drug Metab. Toxicol. 2011, 7, 1535–1546. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Huang, Y.; Li, J.; Wang, Z. The mTOR pathway is associated with the poor prognosis of human hepatocellular carcinoma. Med. Oncol. 2010, 27, 255–261. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patients | N. (%) |
|---|---|
| Median age (range) | 70 (28–89) |
| Gender | |
| Male | 60 (85.7) |
| Female | 10 (14.3) |
| Etiology | |
| Viral infection | 31 (44.3) |
| Metabolic syndrome | 23 (32.9) |
| Alcoholic | 7 (10.0) |
| Other | 9 (8.6) |
| Diabetes | |
| No | 38 (55.1) |
| Yes | 31 (44.9) |
| Metformin | |
| No | 46 (66.7) |
| Yes | 23 (33.3) |
| Performance Status (ECOG) | |
| 0 | 56 (80) |
| 1 | 14 (20) |
| Child–Pugh | |
| A | 67 (89.7) |
| B | 3 (10.3) |
| BCLC staging | |
| A | 41 (58.6) |
| B | 9 (12.9) |
| C | 20 (28.5) |
| Alfafetoprotein levels | |
| <400 | 17 (63.0) |
| ≥400 | 10 (37.0) |
| Unknown | 43 |
| MELD score | |
| ≤10 | 18 (25.7) |
| >10 | 52 (74.3) |
| Metastasis | |
| No | 56 (80) |
| Yes | 14 (20) |
| Markers | No. Patients | Five-Year Percent DFS (months) (95% CI) | p | Eight-Year Percent OS (months) (95% CI) | p |
|---|---|---|---|---|---|
| SIRT-3 (%) | |||||
| <35 | 23 | 23 (4–41) | 20 (0–49) | ||
| ≥35 | 23 | 51 (28–73) | 0.308 | 32 (0–78) | 0.117 |
| p-mTor (%) | |||||
| 0 | 23 | 47 (22–73) | 39 (0–95) | ||
| >0 | 4 | 50 (1–99) | 0.952 | 0 | 0.577 |
| HIF-1α (%) | |||||
| <1 | 15 | 20 (0–44) | 0 | ||
| ≥1 | 10 | 100 | 0.002 | 50 (0-100) | 0.041 |
| Response | No. Patients | SIRT-3 (%) Median Value (range) | p | No. Patients | p-mTOR (%) Median value (range) | p | No. Patients | HIF-1α (%) Median Value (range) | p |
|---|---|---|---|---|---|---|---|---|---|
| CR | 4 | 80 (60–100) | 2 | 0 (0–0) | 2 | 20 (0–40) | |||
| SD | 10 | 45 (0–100) | 7 | 0 (0–0) | 7 | 0 (0–8) | |||
| PD | 10 | 55 (0–90) | 0.276 | 5 | 0 (0–80) | 0.407 | 4 | 0 (0–60) | 0.752 |
| Markers | No. Patients | Median PFS (months) (95% CI) | p | Median OS (months) (95% CI) | p |
|---|---|---|---|---|---|
| SIRT-3 (%) | |||||
| <60 | 11 | 3.7 (1.6–6.0) | 12.0 (5.2–nr) | ||
| ≥60 | 10 | 5.3 (1.7–12.9) | 0.542 | 14.3 (2.0–15.8) | 0.624 |
| p-mTor (%) | |||||
| 0 | 13 | 5.3 (2.3–10.7) | 13.9 (6.7–15.8) | ||
| >0 | 3 | 1.8 (1.6–4.0) | 0.055 | 6.1 (2.6–nr) | 0.098 |
| HIF-1α (%) | |||||
| 0 | 10 | 3.5 (1.2–6.0) | 13.9 (2.2–15.8) | ||
| >0 | 6 | 4.7 (1.6–22.8) | 0.382 | 12.0 (2.6–22.8) | 0.682 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Matteis, S.; Scarpi, E.; Granato, A.M.; Vespasiani-Gentilucci, U.; La Barba, G.; Foschi, F.G.; Bandini, E.; Ghetti, M.; Marisi, G.; Cravero, P.; et al. Role of SIRT-3, p-mTOR and HIF-1α in Hepatocellular Carcinoma Patients Affected by Metabolic Dysfunctions and in Chronic Treatment with Metformin. Int. J. Mol. Sci. 2019, 20, 1503. https://doi.org/10.3390/ijms20061503
De Matteis S, Scarpi E, Granato AM, Vespasiani-Gentilucci U, La Barba G, Foschi FG, Bandini E, Ghetti M, Marisi G, Cravero P, et al. Role of SIRT-3, p-mTOR and HIF-1α in Hepatocellular Carcinoma Patients Affected by Metabolic Dysfunctions and in Chronic Treatment with Metformin. International Journal of Molecular Sciences. 2019; 20(6):1503. https://doi.org/10.3390/ijms20061503
Chicago/Turabian StyleDe Matteis, Serena, Emanuela Scarpi, Anna Maria Granato, Umberto Vespasiani-Gentilucci, Giuliano La Barba, Francesco Giuseppe Foschi, Erika Bandini, Martina Ghetti, Giorgia Marisi, Paola Cravero, and et al. 2019. "Role of SIRT-3, p-mTOR and HIF-1α in Hepatocellular Carcinoma Patients Affected by Metabolic Dysfunctions and in Chronic Treatment with Metformin" International Journal of Molecular Sciences 20, no. 6: 1503. https://doi.org/10.3390/ijms20061503
APA StyleDe Matteis, S., Scarpi, E., Granato, A. M., Vespasiani-Gentilucci, U., La Barba, G., Foschi, F. G., Bandini, E., Ghetti, M., Marisi, G., Cravero, P., Gramantieri, L., Cucchetti, A., Ercolani, G., Santini, D., Frassineti, G. L., Faloppi, L., Scartozzi, M., Cascinu, S., & Casadei-Gardini, A. (2019). Role of SIRT-3, p-mTOR and HIF-1α in Hepatocellular Carcinoma Patients Affected by Metabolic Dysfunctions and in Chronic Treatment with Metformin. International Journal of Molecular Sciences, 20(6), 1503. https://doi.org/10.3390/ijms20061503