Interplay between the Endogenous Opioid System and Proteasome Complex: Beyond Signaling


Abstract
:1. Introduction
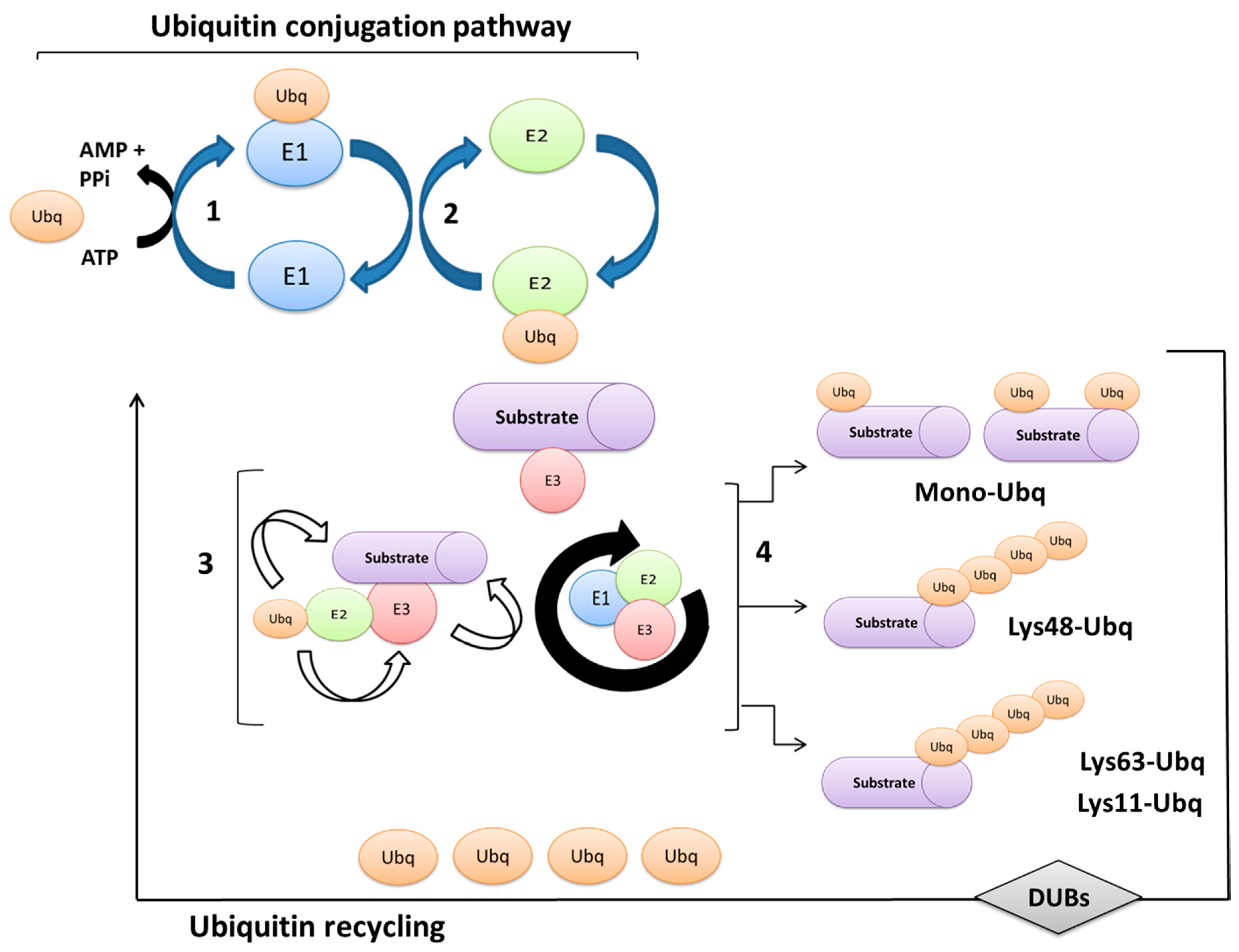
2. Ubiquitination Processes and Ubiquitin-Proteasome System
3. Involvement of UPS Machinery in the Opioid Receptor Signaling Associated with Analgesia, Neuropathic Pain, and Addictive Behavior
4. Modulation of Opioid Receptor Fate and Signaling
5. Conclusions
Funding
Conflicts of Interest
References
- Akil, H.; Watson, S.J.; Young, E.; Lewis, M.E.; Khachaturian, H.; Walker, J.M. Endogenous opioids: Biology and function. Annu. Rev. Neurosci. 1984, 7, 223–255. [Google Scholar] [CrossRef]
- Mansour, A.; Fox, C.A.; Akil, H.; Watson, S.J. Opioid-receptor mRNA expression in the rat CNS: Anatomical and functional implications. Trends Neurosci. 1995, 18, 22–29. [Google Scholar] [CrossRef]
- Toll, L.; Bruchas, M.R.; Calo’, G.; Cox, B.M.; Zaveri, N.T. Nociceptin/Orphanin FQ Receptor Structure, Signaling, Ligands, Functions, and Interactions with Opioid Systems. Pharmacol. Rev. 2016, 68, 419–457. [Google Scholar] [CrossRef] [PubMed]
- Kieffer, B.L. Recent advances in molecular recognition and signal transduction of active peptides: Receptors for opioid peptides. Cell Mol. Neurobiol. 1995, 15, 615–635. [Google Scholar] [CrossRef]
- Kieffer, B.L.; Evans, C.J. Opioid receptors: From binding sites to visible molecules in vivo. Neuropharmacology 2009, 56 (Suppl. 1), 205–212. [Google Scholar] [CrossRef] [PubMed]
- Drolet, G.; Dumont, E.C.; Gosselin, I.; Kinkead, R.; Laforest, S.; Trottier, J.F. Role of endogenous opioid system in the regulation of the stress response. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2001, 25, 729–741. [Google Scholar] [CrossRef]
- Le Merrer, J.; Becker, J.A.; Befort, K.; Kieffer, B.L. Reward processing by the opioid system in the brain. Physiol. Rev. 2009, 89, 1379–1412. [Google Scholar] [CrossRef] [PubMed]
- Bodnar, R.J. Endogenous Opiates and Behavior: 2015. Peptides 2017, 88, 126–188. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.J.; Terman, G.W.; Chavkin, C. Endogenous dynorphins inhibit excitatory neurotransmission and block LTP induction in the hippocampus. Nature 1993, 363, 451–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- North, R.A.; Williams, J.T.; Surprenant, A.; Christie, M.J. Mu and delta receptors belong to a family of receptors that are coupled to potassium channels. Proc. Natl. Acad. Sci. USA 1987, 84, 5487–5491. [Google Scholar] [CrossRef] [PubMed]
- Rusin, K.I.; Giovannucci, D.R.; Stuenkel, E.L.; Moises, H.C. Kappa-opioid receptor activation modulates Ca2+ currents and secretion in isolated neuroendocrine nerve terminals. J. Neurosci. 1997, 17, 6565–6574. [Google Scholar] [CrossRef] [PubMed]
- Torrecilla, M.; Marker, C.L.; Cintora, S.C.; Stoffel, M.; Williams, J.T.; Wickman, K. G-protein-gated potassium channels containing Kir3.2 and Kir3.3 subunits mediate the acute inhibitory effects of opioids on locus ceruleus neurons. J. Neurosci. 2002, 22, 4328–4334. [Google Scholar] [CrossRef] [PubMed]
- Waldhoer, M.; Bartlett, S.E.; Whistler, J.L. Opioid receptors. Annu. Rev. Biochem. 2004, 73, 953–990. [Google Scholar] [CrossRef]
- Blaesse, P.; Goedecke, L.; Bazelot, M.; Capogna, M.; Pape, H.C.; Jüngling, K. μ-Opioid Receptor-Mediated Inhibition of Intercalated Neurons and Effect on Synaptic Transmission to the Central Amygdala. J. Neurosci. 2015, 35, 7317–7325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winters, B.L.; Gregoriou, G.C.; Kissiwaa, S.A.; Wells, O.A.; Medagoda, D.I.; Hermes, S.M.; Burford, N.T.; Alt, A.; Aicher, S.A.; Bagley, E.E. Endogenous opioids regulate moment-to-moment neuronal communication and excitability. Nat. Commun. 2017, 8, 14611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McPherson, J.; Rivero, G.; Baptist, M.; Llorente, J.; Al-Sabah, S.; Krasel, C.; Dewey, W.L.; Bailey, C.P.; Rosethorne, E.M.; Charlton, S.J.; et al. μ-opioid receptors: Correlation of agonist efficacy for signalling with ability to activate internalization. Mol. Pharmacol. 2010, 78, 756–766. [Google Scholar] [CrossRef]
- Al-Hasani, R.; Bruchas, M.R. Molecular mechanisms of opioid receptor-dependent signaling and behavior. Anesthesiology 2011, 115, 1363–1381. [Google Scholar] [CrossRef] [PubMed]
- Raehal, K.M.; Schmid, C.L.; Groer, C.E.; Bohn, L.M. Functional selectivity at the μ-opioid receptor: Implications for understanding opioid analgesia and tolerance. Pharmacol. Rev. 2011, 63, 1001–1019. [Google Scholar] [CrossRef]
- Pradhan, A.A.; Smith, M.L.; Kieffer, B.L.; Evans, C.J. Ligand-directed signalling within the opioid receptor family. Br. J. Pharmacol. 2012, 167, 960–969. [Google Scholar] [CrossRef] [Green Version]
- Lamberts, J.T.; Traynor, J.R. Opioid receptor interacting proteins and the control of opioid signaling. Curr. Pharm. Des. 2013, 19, 7333–7347. [Google Scholar] [CrossRef]
- DiAntonio, A.; Haghighi, A.P.; Portman, S.L.; Lee, J.D.; Amaranto, A.M.; Goodman, C.S. Ubiquitination-dependent mechanisms regulate synaptic growth and function. Nature 2001, 412, 449–452. [Google Scholar] [CrossRef]
- Murphey, R.K.; Godenschwege, T.A. New roles for ubiquitin in the assembly and function of neuronal circuits. Neuron 2002, 36, 5–8. [Google Scholar] [CrossRef]
- Ehlers, M.D. Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat. Neurosci. 2003, 6, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Ryu, K.Y.; Garza, J.C.; Lu, X.Y.; Barsh, G.S.; Kopito, R.R. Hypothalamic neurodegeneration and adult-onset obesity in mice lacking the Ubb polyubiquitin gene. Proc. Natl. Acad. Sci. USA 2008, 105, 4016–4021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, P.C.; Dodart, J.C.; Aron, L.; Finley, L.W.; Bronson, R.T.; Haigis, M.C.; Yankner, B.A.; Harper, J.W. Altered social behavior and neuronal development in mice lacking the Uba6–Use1 ubiquitin transfer system. Mol. Cell 2013, 50, 172–184. [Google Scholar] [CrossRef]
- Rape, M. Ubiquitylation at the crossroads of development and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Marchese, A.; Chen, C.; Kim, Y.M.; Benovic, J.L. The ins and outs of G protein-coupled receptor trafficking. Trends Biochem. Sci. 2003, 28, 369–376. [Google Scholar] [CrossRef]
- Marchese, A.; Trejo, J. Ubiquitin-dependent regulation of G protein-coupled receptor trafficking and signaling. Cell Signal. 2013, 25, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, J.E.; Marchese, A. Regulation of GPCR Trafficking by Ubiquitin. Prog. Mol. Biol. Transl. Sci. 2015, 132, 15–38. [Google Scholar] [PubMed] [Green Version]
- Lefkowitz, R.J. G protein-coupled receptors. III. New roles for receptor kinases and beta-arrestins in receptor signaling and desensitization. J. Biol. Chem. 1998, 273, 18677–18680. [Google Scholar] [CrossRef]
- Chaturvedi, K.; Christoffers, K.H.; Singh, K.; Howells, R.D. Structure and regulation of opioid receptors. Biopolymers 2000, 55, 334–346. [Google Scholar] [CrossRef]
- Law, P.Y.; Wong, Y.H.; Loh, H.H. Molecular mechanisms and regulation of opioid receptor signaling. Annu. Rev. Pharmacol. Toxicol. 2000, 40, 389–430. [Google Scholar] [CrossRef] [PubMed]
- Appelqvist, H.; Wäster, P.; Kågedal, K.; Öllinger, K. The lysosome: From waste bag to potential therapeutic target. J. Mol. Cell Biol. 2013, 5, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Dikic, I. Proteasomal and Autophagic Degradation Systems. Annu. Rev. Biochem. 2017, 86, 193–224. [Google Scholar] [CrossRef] [PubMed]
- Peth, A.; Uchiki, T.; Goldberg, A.L. ATP-dependent steps in the binding of ubiquitin conjugates to the 26S proteasome that commit to degradation. Mol. Cell 2010, 40, 671–681. [Google Scholar] [CrossRef] [PubMed]
- Inobe, T.; Matouschek, A. Paradigms of protein degradation by the proteasome. Curr. Opin. Struct. Biol. 2014, 24, 156–164. [Google Scholar] [CrossRef] [Green Version]
- Collins, G.A.; Goldberg, A.L. The logic of the 26S proteasome. Cell 2017, 169, 792–806. [Google Scholar] [CrossRef]
- Saeki, Y. Ubiquitin recognition by the proteasome. J. Biochem. 2017, 161, 113–124. [Google Scholar] [CrossRef]
- Goldberg, A.L. Protein degradation and protection against misfolded or damaged proteins. Nature 2003, 426, 895–899. [Google Scholar] [CrossRef]
- Hochstrasser, M. Ubiquitin-dependent protein degradation. Annu. Rev. Genet. 1996, 30, 405–439. [Google Scholar] [CrossRef]
- Saeki, Y.; Tanaka, K. Assembly and Function of the Proteasome. Methods Mol. Biol. 2012, 832, 315–337. [Google Scholar] [PubMed]
- Schwartz, A.L.; Ciechanover, A. The ubiquitin-proteasome pathway and pathogenesis of human diseases. Annu. Rev. Med. 1999, 50, 57–74. [Google Scholar] [CrossRef]
- Wang, J.; Maldonado, M.A. The ubiquitin-proteasome system and its role in inflammatory and autoimmune diseases. Cell Mol. Immunol. 2006, 3, 255–261. [Google Scholar] [PubMed]
- Kammerl, I.E.; Meiners, S. Proteasome function shapes innate and adaptive immune responses. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 311, L328–L336. [Google Scholar] [CrossRef]
- Glickman, M.H.; Ciechanover, A. The ubiquitin-proteasome proteolytic pathway: Destruction for the sake of construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef] [PubMed]
- Gallestegui, N.; Groll, M. The 26S proteasome: Assembly and function of a destructive machine. Trends Biochem. Sci. 2010, 35, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Komander, D.; Rape, M. The ubiquitin code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef] [PubMed]
- Förster, F.; Sakata, E. 26S Proteasome: Structure and function. In Encyclopedia of Biological Chemistry, 2nd ed.; Lennarz, W.J., Lane, M.D., Eds.; Elsevier: Amsterdam, The Netherlands, 2013; pp. 595–600. [Google Scholar]
- Pickart, C.M. Mechanisms underlying ubiquitination. Annu. Rev. Biochem. 2001, 70, 503–533. [Google Scholar] [CrossRef]
- Pham, A.D.; Sauer, F. Ubiquitin-activating/conjugating activity of TAFII250, a mediator of activation of gene expression in Drosophila. Science 2000, 289, 2357–2360. [Google Scholar] [CrossRef]
- Robzyk, K.; Recht, J.; Osley, M.A. Rad6-dependent ubiquitination of histone H2B in yeast. Science 2000, 287, 501–504. [Google Scholar] [CrossRef]
- Strous, G.J.; van Kerkhof, P.; Govers, R.; Ciechanover, A.; Schwartz, A.L. The ubiquitin conjugation system is required for ligand-induced endocytosis and degradation of the growth hormone receptor. EMBO J. 1996, 15, 3806–3812. [Google Scholar] [CrossRef] [PubMed]
- Rotin, D.; Staub, O.; Haguenauer-Tsapis, R. Ubiquitination and endocytosis of plasma membrane proteins: Role of Nedd4/Rsp5p family of ubiquitin-protein ligases. J. Membr. Biol. 2000, 176, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef] [PubMed]
- Gadhave, K.; Bolshette, N.; Ahire, A.; Pardeshi, R.; Thakur, K.; Trandafir, C.; Istrate, A.; Ahmed, S.; Lahkar, M.; Muresanu, D.F.; et al. The ubiquitin proteasomal system: A potential target for the management of Alzheimer’s disease. J. Cell Mol. Med. 2016, 20, 1392–1407. [Google Scholar] [CrossRef] [PubMed]
- Lehman, N.L. The ubiquitin proteasome system in neuropathology. Acta Neuropathol. 2009, 118, 329–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, T.T.; D’Andrea, A.D. Regulation of DNA repair by ubiquitylation. Nat. Rev. Mol. Cell Biol. 2006, 7, 323–334. [Google Scholar] [CrossRef]
- Bremm, A.; Komander, D. Emerging roles for Lys11-linked polyubiquitin in cellular regulation. Trends Biochem. Sci. 2011, 36, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Kulathu, Y.; Komander, D. Atypical ubiquitylation—The unexplored world of polyubiquitin beyond Lys48 and Lys63 linkages. Nat. Rev. Mol. Cell Biol. 2012, 13, 508–523. [Google Scholar] [CrossRef]
- Morris, J.R.; Solomon, E. BRCA1: BARD1 induces the formation of conjugated ubiquitin structures, dependent on K6 of ubiquitin, in cells during DNA replication and repair. Hum. Mol. Genet. 2004, 13, 807–817. [Google Scholar] [CrossRef]
- Cunningham, C.N.; Baughman, J.M.; Phu, L.; Tea, J.S.; Yu, C.; Coons, M.; Kirkpatrick, D.S.; Bingol, B.; Corn, J.E. USP30 and parkin homeostatically regulate atypical ubiquitin chains on mitochondria. Nat. Cell Biol. 2015, 17, 160–169. [Google Scholar] [CrossRef]
- Swatek, K.N.; Komander, D. Ubiquitin modifications. Cell Res. 2016, 26, 399–422. [Google Scholar] [CrossRef]
- Komander, D.; Clague, M.J.; Urbé, S. Breaking the chains: Structure and function of the deubiquitinases. Nat. Rev. Mol. Cell Biol. 2009, 10, 550–563. [Google Scholar] [CrossRef] [PubMed]
- Clague, M.J.; Barsukov, I.; Coulson, J.M.; Liu, H.; Rigden, D.J.; Urbé, S. Deubiquitylases from genes to organism. Physiol. Rev. 2013, 93, 1289–1315. [Google Scholar] [CrossRef] [PubMed]
- Da Fonseca, P.C.; Morris, E.P. Structure of the human 26S proteasome: Subunit radial displacements open the gate into the proteolytic core. J. Biol. Chem. 2008, 283, 23305–23314. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.M.; Yu, Y.; Cheng, Y. Structure characterization of the 26S proteasome. Biochim. Biophys. Acta 2011, 1809, 67–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budenholzer, L.; Cheng, C.L.; Li, Y.; Hochstrasser, M. Proteasome Structure and Assembly. J. Mol. Biol. 2017, 429, 3500–3524. [Google Scholar] [CrossRef] [PubMed]
- Cascio, P.; Call, M.; Petre, B.M.; Walz, T.; Goldberg, A.L. Properties of the hybrid form of the 26S proteasome containing both 19S and PA28 complexes. EMBO J. 2002, 21, 2636–2645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rechsteiner, M.; Hill, C.P. Mobilizing the proteolytic machine: Cell biological roles of proteasome activators and inhibitors. Trends Cell Biol. 2005, 15, 27–33. [Google Scholar] [CrossRef]
- Tsvetkov, P.; Reuven, N.; Shaul, Y. The nanny model for IDPs. Nat. Chem. Biol. 2009, 5, 778–781. [Google Scholar] [CrossRef]
- Neefjes, J.; Jongsma, M.L.; Paul, P.; Bakke, O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat. Rev. Immunol. 2011, 11, 823–836. [Google Scholar] [CrossRef]
- Ebstein, F.; Kloetzel, P.M.; Kruger, E.; Seifert, U. Emerging roles of immunoproteasomes beyond MHC class I antigen processing. Cell Mol. Life Sci. 2012, 69, 2543–2558. [Google Scholar] [CrossRef]
- Nandi, D.; Jiang, H.; Monaco, J.J. Identification of MECL-1 (LMP-10) as the third IFN-gamma-inducible proteasome subunit. J. Immunol. 1996, 156, 2361–2364. [Google Scholar]
- Toes, R.E.; Nussbaum, A.K.; Degermann, S.; Schirle, M.; Emmerich, N.P.; Kraft, M.; Laplace, C.; Zwinderman, A.; Dick, T.P.; Müller, J.; et al. Discrete cleavage motifs of constitutive and immunoproteasomes revealed by quantitative analysis of cleavage products. J. Exp. Med. 2001, 194, 1–12. [Google Scholar] [CrossRef]
- Chaturvedi, K.; Bandari, P.; Chinen, N.; Howells, R.D. Proteasome involvement in agonist-induced down-regulation of mu and delta opioid receptors. J. Biol. Chem. 2001, 276, 12345–12355. [Google Scholar] [CrossRef]
- Moulédous, L.; Neasta, J.; Uttenweiler-Joseph, S.; Stella, A.; Matondo, M.; Corbani, M.; Monsarrat, B.; Meunier, J.C. Long-term morphine treatment enhances proteasome-dependent degradation of G beta in human neuroblastoma SH-SY5Y cells: Correlation with onset of adenylate cyclase sensitization. Mol. Pharmacol. 2005, 68, 467–476. [Google Scholar] [CrossRef]
- Caputi, F.F.; Candeletti, S.; Romualdi, P. Involvement of proteasome machinery in pain and addiction. In Proceedings of the 6th Mediterranean Neuroscience Society Conference (MNS), St. Julian’s, Malta, 12–15 June 2017; pp. 127–128. [Google Scholar]
- Yang, L.; Wang, S.; Sung, B.; Lim, G.; Mao, J. Morphine induces ubiquitin-proteasome activity and glutamate transporter degradation. J. Biol. Chem. 2008, 283, 21703–21713. [Google Scholar] [CrossRef]
- Zhou, J.; Li, Y.; Yan, G. Protective role of taurine against morphine-induced neurotoxicity in C6 cells via inhibition of oxidative stress. Neurotox. Res. 2011, 4, 334–342. [Google Scholar] [CrossRef]
- Guzmán, D.; Vázquez, I.; Brizuela, N.; Alvarez, R.; Mejía, G.; García, E.; Santamaría, D.; de Apreza, M.L.; Olguín, H.J. Assessment of oxidative damage induced by acute doses of morphine sulfate in postnatal and adult rat brain. Neurochem. Res. 2006, 31, 549–554. [Google Scholar] [CrossRef]
- Hutchinson, M.R.; Coats, B.D.; Lewis, S.S.; Zhang, Y.; Sprunger, D.B.; Rezvani, N.; Baker, E.M.; Jekich, B.M.; Wieseler, J.L.; Somogyi, A.A.; et al. Proinflammatory cytokines oppose opioid-induced acute and chronic analgesia. Brain Behav. Immun. 2008, 22, 1178–1189. [Google Scholar] [CrossRef] [Green Version]
- Ibi, M.; Matsuno, K.; Matsumoto, M.; Sasaki, M.; Nakagawa, T.; Katsuyama, M.; Iwata, K.; Zhang, J.; Kaneko, S.; Yabe-Nishimura, C. Involvement of NOX1/NADPH oxidase in morphine-induced analgesia and tolerance. J. Neurosci. 2011, 31, 18094–18103. [Google Scholar] [CrossRef]
- Ghavimi, H.; Charkhpour, M.; Ghasemi, S.; Mesgari, M.; Hamishehkar, H.; Hassanzadeh, K.; Arami, S.; Hassanzadeh, K. Pioglitazone prevents morphine antinociceptive tolerance via ameliorating neuroinflammation in rat cerebral cortex. Pharmacol. Rep. 2015, 67, 78–84. [Google Scholar] [CrossRef]
- Caputi, F.F.; Rullo, L.; Acquas, E.; Ciccocioppo, R.; Candeletti, S.; Romualdi, P. Evidence of a PPARγ-mediated mechanism in the ability of Withania somnifera to attenuate tolerance to the antinociceptive effects of morphine. Pharmacol. Res. 2018, 139, 422–430. [Google Scholar] [CrossRef]
- Jung, T.; Grune, T. The proteasome and its role in the degradation of oxidized proteins. IUBMB Life 2008, 60, 743–752. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Traynor, J.R. Opioid-induced down-regulation of RGS4: Role of ubiquitination and implications for receptor cross-talk. J. Biol. Chem. 2011, 286, 7854–7864. [Google Scholar] [CrossRef]
- Tsao, P.I.; von Zastrow, M. Type-specific sorting of G protein-coupled receptors after endocytosis. J. Biol. Chem. 2000, 275, 11130–11140. [Google Scholar] [CrossRef]
- Moss, A.; Blackburn-Munro, G.; Garry, E.M.; Blakemore, J.A.; Dickinson, T.; Rosie, R.; Mitchell, R.; Fleetwood-Walker, S.M. A role of the ubiquitin-proteasome system in neuropathic pain. J. Neurosci. 2002, 22, 1363–1372. [Google Scholar] [CrossRef]
- Ossipov, M.H.; Bazov, I.; Gardell, L.R.; Kowal, J.; Yakovleva, T.; Usynin, I.; Ekström, T.J.; Porreca, F.; Bakalkin, G. Control of chronic pain by the ubiquitin proteasome system in the spinal cord. J. Neurosci. 2007, 27, 8226–8237. [Google Scholar] [CrossRef]
- Yang, L.; Wang, S.; Lim, G.; Sung, B.; Zeng, Q.; Mao, J. Inhibition of the ubiquitin-proteasome activity prevents glutamate transporter degradation and morphine tolerance. Pain 2008, 140, 472–478. [Google Scholar] [CrossRef]
- Caputi, F.F.; Rullo, L.; Di Cesare Mannelli, L.; Ghelardini, C.; Candeletti, S.; A Romualdi, P. Protesome implication in oxaliplatin-induced neuropathy. In Proceedings of the Monothematic Conference of the Italian Society of Pharmacology (SIF): “The Pharmacological Basis of Novel Pain Therapeutics”, Florence, Italy, 4–5 May 2017. [Google Scholar]
- Zanardelli, M.; Micheli, L.; Cinci, L.; Failli, P.; Ghelardini, C.; Di Cesare Mannelli, L. Oxaliplatin neurotoxicity involves peroxisome alterations. PPARγ agonism as preventive pharmacological approach. PLoS ONE 2014, 9, e102758. [Google Scholar] [CrossRef]
- Massaly, N.; Francès, B.; Moulédous, L. Roles of the ubiquitin proteasome system in the effects of drugs of abuse. Front. Mol. Neurosci. 2015, 7, 99. [Google Scholar] [CrossRef]
- Caputi, F.F.; Carboni, L.; Mazza, D.; Candeletti, S.; Romualdi, P. Cocaine and ethanol target 26S proteasome activity and gene expression in neuroblastoma cells. Drug Alcohol Depend. 2016, 161, 265–275. [Google Scholar] [CrossRef]
- Caputi, F.F.; Carretta, D.; Lattanzio, F.; Palmisano, M.; Candeletti, S.; Romualdi, P. Proteasome subunit and opioid receptor gene expression down-regulation induced by paraquat and maneb in human neuroblastoma SH-SY5Y cells. Environ. Toxicol. Pharmacol. 2015, 40, 895–900. [Google Scholar] [CrossRef]
- Kumar, V.; Singh, D.; Singh, B.K.; Singh, S.; Mittra, N.; Jha, R.R.; Patel, D.K.; Singh, C. Alpha-synuclein aggregation, Ubiquitin proteasome system impairment, and L-Dopa response in zinc-induced Parkinsonism: Resemblance to sporadic Parkinson’s disease. Mol. Cell Biochem. 2018, 444, 149–160. [Google Scholar] [CrossRef]
- Massaly, N.; Dahan, L.; Baudonnat, M.; Hovnanian, C.; Rekik, K.; Solinas, M.; David, V.; Pech, S.; Zajac, J.M.; Roullet, P.; et al. Involvement of protein degradation by the ubiquitin proteasome system in opiate addictive behaviors. Neuropsychopharmacology 2013, 38, 596–604. [Google Scholar] [CrossRef]
- Trapaidze, N.; Keith, D.E.; Cvejic, S.; Evans, C.J.; Devi, L.A. Sequestration of the delta opioid receptor. Role of the C terminus in agonist-mediated internalization. J. Biol. Chem. 1996, 271, 29279–29285. [Google Scholar] [CrossRef]
- Cvejic, S.; Devi, L.A. Dimerization of the delta opioid receptor: Implication for a role in receptor internalization. J. Biol. Chem. 1997, 272, 26959–26964. [Google Scholar] [CrossRef]
- Chu, P.; Murray, S.; Lissin, D.; von Zastrow, M. Delta and kappa opioid receptors are differentially regulated by dynamin-dependent endocytosis when activated by the same alkaloid agonist. J. Biol. Chem. 1997, 272, 27124–27130. [Google Scholar] [CrossRef]
- Zhang, J.; Ferguson, S.S.; Barak, L.S.; Bodduluri, S.R.; Laporte, S.A.; Law, P.Y.; Caron, M.G. Role for G protein-coupled receptor kinase in agonist-specific regulation of mu-opioid receptor responsiveness. Proc. Natl. Acad. Sci. USA 1998, 95, 7157–7162. [Google Scholar] [CrossRef]
- Ko, J.L.; Arvidsson, U.; Williams, F.G.; Law, P.Y.; Elde, R.; Loh, H.H. Visualization of time-dependent redistribution of delta-opioid receptors in neuronal cells during prolonged agonist exposure. Brain Res. Mol. Brain Res. 1999, 69, 171–185. [Google Scholar] [CrossRef]
- Pei, G.; Kieffer, B.L.; Lefkowitz, R.J.; Freedman, N.J. Agonist-dependent phosphorylation of the mouse delta-opioid receptor: Involvement of G protein-coupled receptor kinases but not protein kinase C. Mol. Pharmacol. 1995, 48, 173–177. [Google Scholar]
- Caputi, F.F.; Lattanzio, F.; Carretta, D.; Mercatelli, D.; Candeletti, S.; Romualdi, P. Morphine and fentanyl differently affect MOP and NOP gene expression in human neuroblastoma SH-SY5Y cells. J. Mol. Neurosci. 2013, 51, 532–538. [Google Scholar] [CrossRef] [PubMed]
- Caputi, F.F.; Acquas, E.; Kasture, S.; Ruiu, S.; Candeletti, S.; Romualdi, P. The standardized Withania somnifera Dunal root extract alters basal and morphine-induced opioid receptor gene expression changes in neuroblastoma cells. BMC Complement. Altern. Med. 2018, 18, 9. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Z. The role of opioid receptor internalization and beta-arrestins in the development of opioid tolerance. Anesth. Analg. 2005, 101, 728–734. [Google Scholar] [CrossRef]
- Groer, C.E.; Tidgewell, K.; Moyer, R.A.; Harding, W.W.; Rothman, R.B.; Prisinzano, T.E.; Bohn, L.M. An opioid agonist that does not induce mu-opioid receptor—Arrestin interactions or receptor internalization. Mol. Pharmacol. 2007, 71, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Mores, K.L.; Cassell, R.J.; van Rijn, R.M. Arrestin recruitment and signaling by G protein-coupled receptor heteromers. Neuropharmacology 2018. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, S.K. Seven-transmembrane receptors and ubiquitination. Circ. Res. 2007, 100, 1142–1154. [Google Scholar] [CrossRef]
- Luttrell, L.M.; Lefkowitz, R.J. The role of beta-arrestins in the termination and transduction of G-protein-coupled receptor signals. J. Cell Sci. 2002, 115, 455–465. [Google Scholar] [PubMed]
- Oakley, R.H.; Laporte, S.A.; Holt, J.A.; Caron, M.G.; Barak, L.S. Differential affinities of visual arrestin, beta arrestin1, and beta arrestin2 for G protein-coupled receptors delineate two major classes of receptors. J. Biol. Chem. 2000, 275, 17201–17210. [Google Scholar] [CrossRef]
- Bohn, L.M.; Lefkowitz, R.J.; Caron, M.G. Differential mechanisms of morphine antinociceptive tolerance revealed in (beta)arrestin-2 knock-out mice. J. Neurosci. 2002, 22, 10494–10500. [Google Scholar] [CrossRef] [PubMed]
- Bohn, L.M.; Dykstra, L.A.; Lefkowitz, R.J.; Caron, M.G.; Barak, L.S. Relative opioid efficacy is determined by the complements of the G protein-coupled receptor desensitization machinery. Mol. Pharmacol. 2004, 66, 106–112. [Google Scholar] [CrossRef]
- Raehal, K.M.; Walker, J.K.; Bohn, L.M. Morphine side effects in beta-arrestin 2 knockout mice. J. Pharmacol. Exp. Ther. 2005, 314, 1195–1201. [Google Scholar] [CrossRef] [PubMed]
- Raehal, K.M.; Bohn, L.M. The role of beta-arrestin2 in the severity of antinociceptive tolerance and physical dependence induced by different opioid pain therapeutics. Neuropharmacology 2011, 60, 58–65. [Google Scholar] [CrossRef] [PubMed]
- DeWire, S.M.; Yamashita, D.S.; Rominger, D.H.; Liu, G.; Cowan, C.L.; Graczyk, T.M.; Chen, X.T.; Pitis, P.M.; Gotchev, D.; Yuan, C.; et al. A G protein-biased ligand at the μ-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. J. Pharmacol. Exp. Ther. 2013, 344, 708–717. [Google Scholar] [CrossRef] [PubMed]
- Soergel, D.G.; Subach, R.A.; Burnham, N.; Lark, M.W.; James, I.E.; Sadler, B.M.; Skobieranda, F.; Violin, J.D.; Webster, L.R. Biased agonism of the μ-opioid receptor by TRV130 increases analgesia and reduces on-target adverse effects versus morphine: A randomized, double-blind, placebo-controlled, crossover study in healthy volunteers. Pain 2014, 155, 1829–1835. [Google Scholar] [CrossRef] [PubMed]
- Rankovic, Z.; Brust, T.F.; Bohn, L.M. Biased agonism: An emerging paradigm in GPCR drug discovery. Bioorg. Med. Chem. Lett. 2016, 26, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Bohn, L.M.; Lefkowitz, R.J.; Gainetdinov, R.R.; Peppel, K.; Caron, M.G.; Lin, F.T. Enhanced morphine analgesia in mice lacking beta-arrestin 2. Science 1999, 286, 2495–2498. [Google Scholar] [CrossRef]
- Bohn, L.M.; Gainetdinov, R.R.; Lin, F.T.; Lefkowitz, R.J.; Caron, M.G. Mu-opioid receptor desensitization by beta-arrestin-2 determines morphine tolerance but not dependence. Nature 2000, 408, 720–723. [Google Scholar] [CrossRef]
- Dang, V.C.; Christie, M.J. Mechanisms of rapid opioid receptor desensitization, resensitization and tolerance in brain neurons. Br. J. Pharmacol. 2012, 165, 1704–1716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groer, C.E.; Schmid, C.L.; Jaeger, A.M.; Bohn, L.M. Agonist-directed interactions with specific beta-arrestins determine mu-opioid receptor trafficking, ubiquitination, and dephosphorylation. J. Biol. Chem. 2011, 286, 31731–31741. [Google Scholar] [CrossRef]
- Shenoy, S.K.; McDonald, P.H.; Kohout, T.A.; Lefkowitz, R.J. Regulation of receptor fate by ubiquitination of activated beta 2-adrenergic receptor and beta-arrestin. Science 2001, 294, 1307–1313. [Google Scholar] [CrossRef]
- Shenoy, S.K.; Xiao, K.; Venkataramanan, V.; Snyder, P.M.; Freedman, N.J.; Weissman, A.M. Nedd4 mediates agonist-dependent ubiquitination, lysosomal targeting, and degradation of the beta2-adrenergic receptor. J. Biol. Chem. 2008, 283, 22166–22176. [Google Scholar] [CrossRef] [PubMed]
- Martin, N.P.; Lefkowitz, R.J.; Shenoy, S.K. Regulation of V2 vasopressin receptor degradation by agonist-promoted ubiquitination. J. Biol. Chem. 2003, 278, 45954–45959. [Google Scholar] [CrossRef] [PubMed]
- Hicke, L. Protein regulation by monoubiquitin. Nat. Rev. Mol. Cell Biol. 2001, 2, 195–201. [Google Scholar] [CrossRef]
- Hicke, L.; Dunn, R. Regulation of membrane protein transport by ubiquitin and ubiquitin-binding proteins. Annu. Rev. Cell Dev. Biol. 2003, 19, 141–172. [Google Scholar] [CrossRef]
- Marchese, A.; Paing, M.M.; Temple, B.R.; Trejo, J. G protein-coupled receptor sorting to endosomes and lysosomes. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 601–629. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Drugs and Treatments | Experimental Paradigm | Key Finding | References |
|---|---|---|---|
| ZLLL and lactacystin (proteasome inhibitors) | Human embryonic kidney 293 cells transfected with murine μ or δ receptors | Attenuation of the agonist-induced μ (MOR) and δ (DOR) down-regulation | Chaturvedi et al. 2001 [75] |
| MG-115 or lactacystin (proteasome inhibitors) | Human neuroblastoma SH-SY5Y cells | Suppression of Gβ down-regulation induced by prolonged morphine exposure | Moulédous et al. 2005 [76] |
| Opioid agonists | Human neuroblastoma SH-SY5Y cells | Significant increase of the proteasomal proteolytic activity | Caputi et al. 2017 [77] |
| MG-132 and lactacystin (proteasome inhibitors) | Human neuroblastoma SH-SY5Y cells | Block of the regulator of G protein signaling protein 4 (RGS4) reduction induced by DAMGO or DPDPE opioid agonists | Wang and Traynor, 2011 [86] |
| Co-administration of MG-132 with morphine | Adult male Sprague Dawley rats | Prevention of morphine tolerance development | Yang et al. 2008 [90] |
| Epoxomicin and MG-132 (proteasome inhibitors) | Neuropathic pain model (spinal nerve ligation) | Decrease of painful signs and dynorphin level normalization | Ossipov et al. 2007 [89] |
| Oxaliplatin exposure | Neuropathic pain model | Activation of the proteasome degradation machinery | Caputi et al. 2017 [91] |
| Lactacystin or MG-132 (proteasome inhibitors) | Opiate addictive behavior | Obstruction of morphine-associated compartment preference | Massaly et al. 2013 [97] |
| Lactacystin (protesome inhibitor) | Opiate addictive behavior | Obstruction of the morphine behavioral sensitization development and decrease of heroin self-administration | Massaly et al. 2013 [97] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caputi, F.F.; Rullo, L.; Stamatakos, S.; Candeletti, S.; Romualdi, P. Interplay between the Endogenous Opioid System and Proteasome Complex: Beyond Signaling. Int. J. Mol. Sci. 2019, 20, 1441. https://doi.org/10.3390/ijms20061441
Caputi FF, Rullo L, Stamatakos S, Candeletti S, Romualdi P. Interplay between the Endogenous Opioid System and Proteasome Complex: Beyond Signaling. International Journal of Molecular Sciences. 2019; 20(6):1441. https://doi.org/10.3390/ijms20061441
Chicago/Turabian StyleCaputi, Francesca Felicia, Laura Rullo, Serena Stamatakos, Sanzio Candeletti, and Patrizia Romualdi. 2019. "Interplay between the Endogenous Opioid System and Proteasome Complex: Beyond Signaling" International Journal of Molecular Sciences 20, no. 6: 1441. https://doi.org/10.3390/ijms20061441