Synthesis of New GABAA Receptor Modulator with Pyrazolo[1,5-a]quinazoline (PQ) Scaffold

 , , , and
, , , and 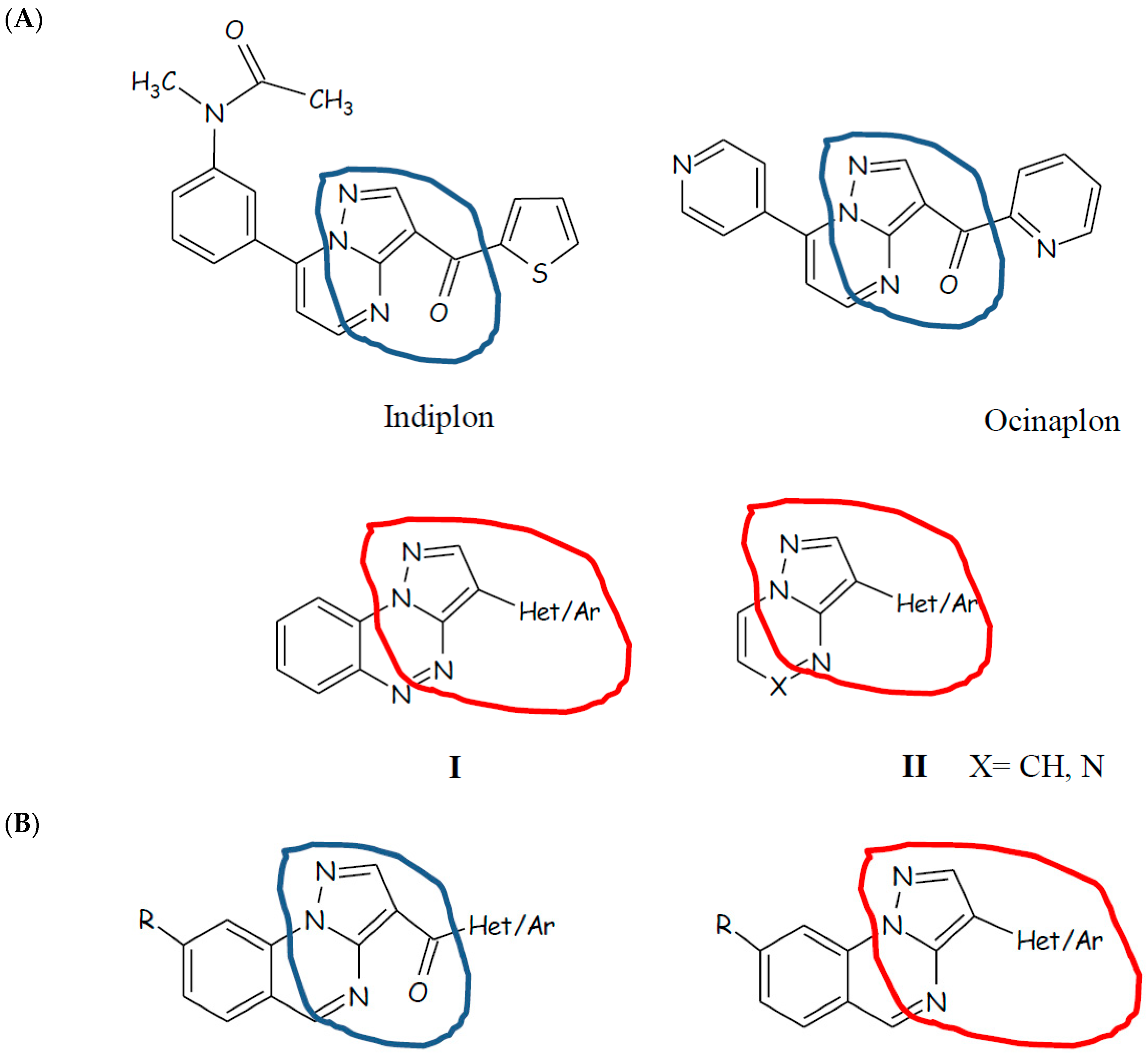{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
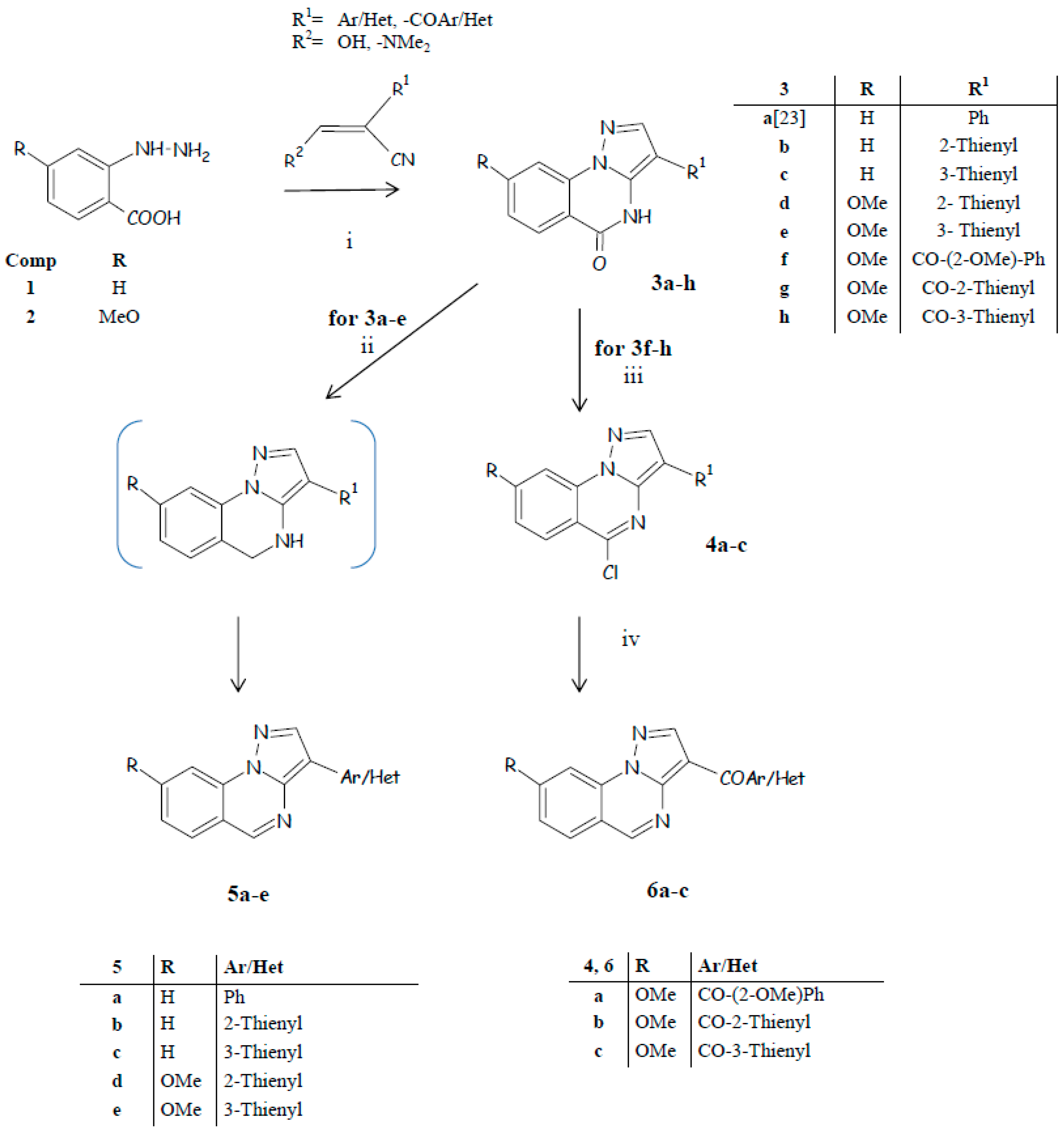
2.1. Chemistry
- 1-
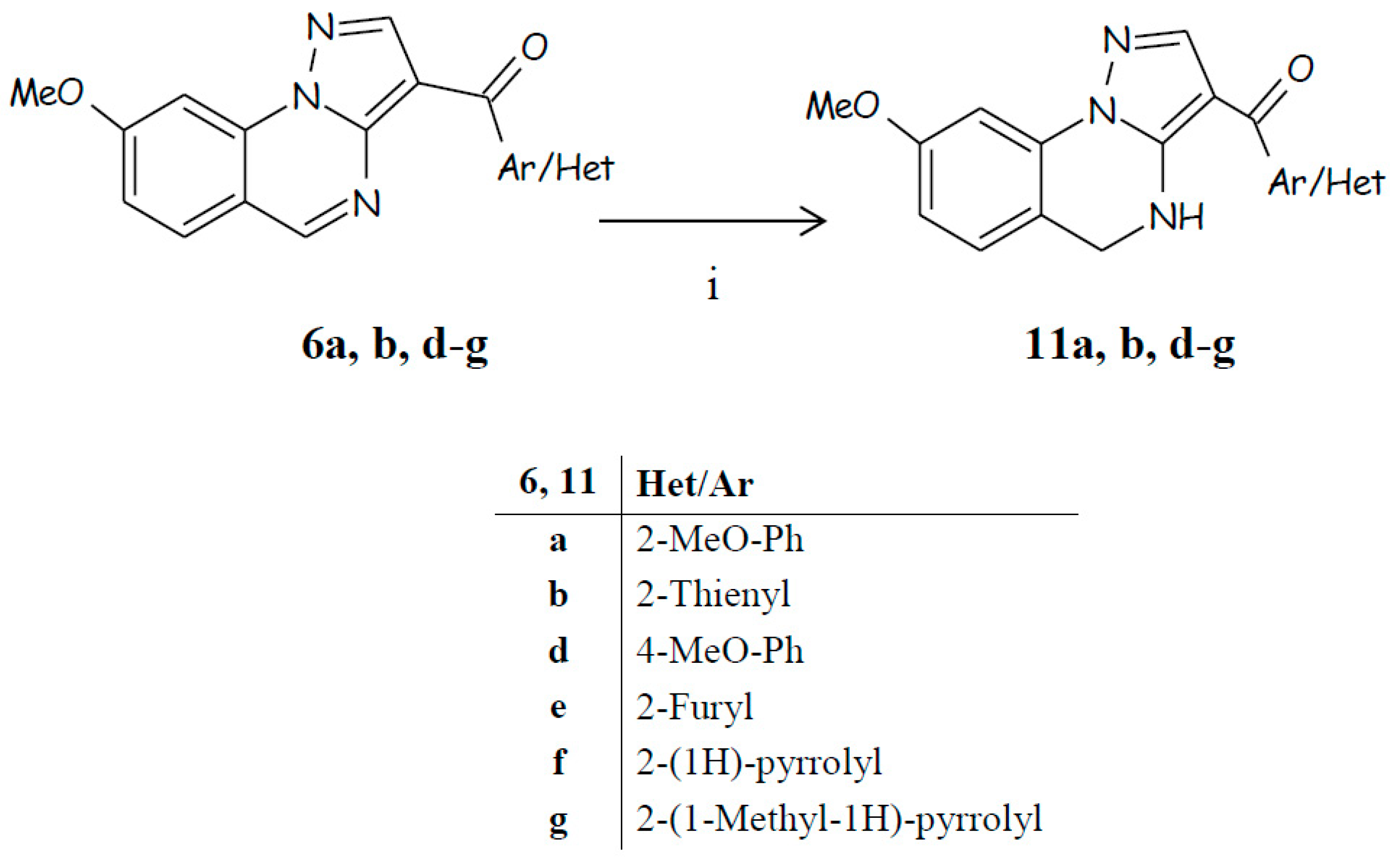
- condensation and next cyclization of the hydrazinobenzoic acid with suitable propanenitrile to directly obtain the pyrazolo[1,5-a]quinazoline scaffold bearing at position 3 the proper (hetero)aryl or (hetero)aroyl group (Scheme 1);
- 2-
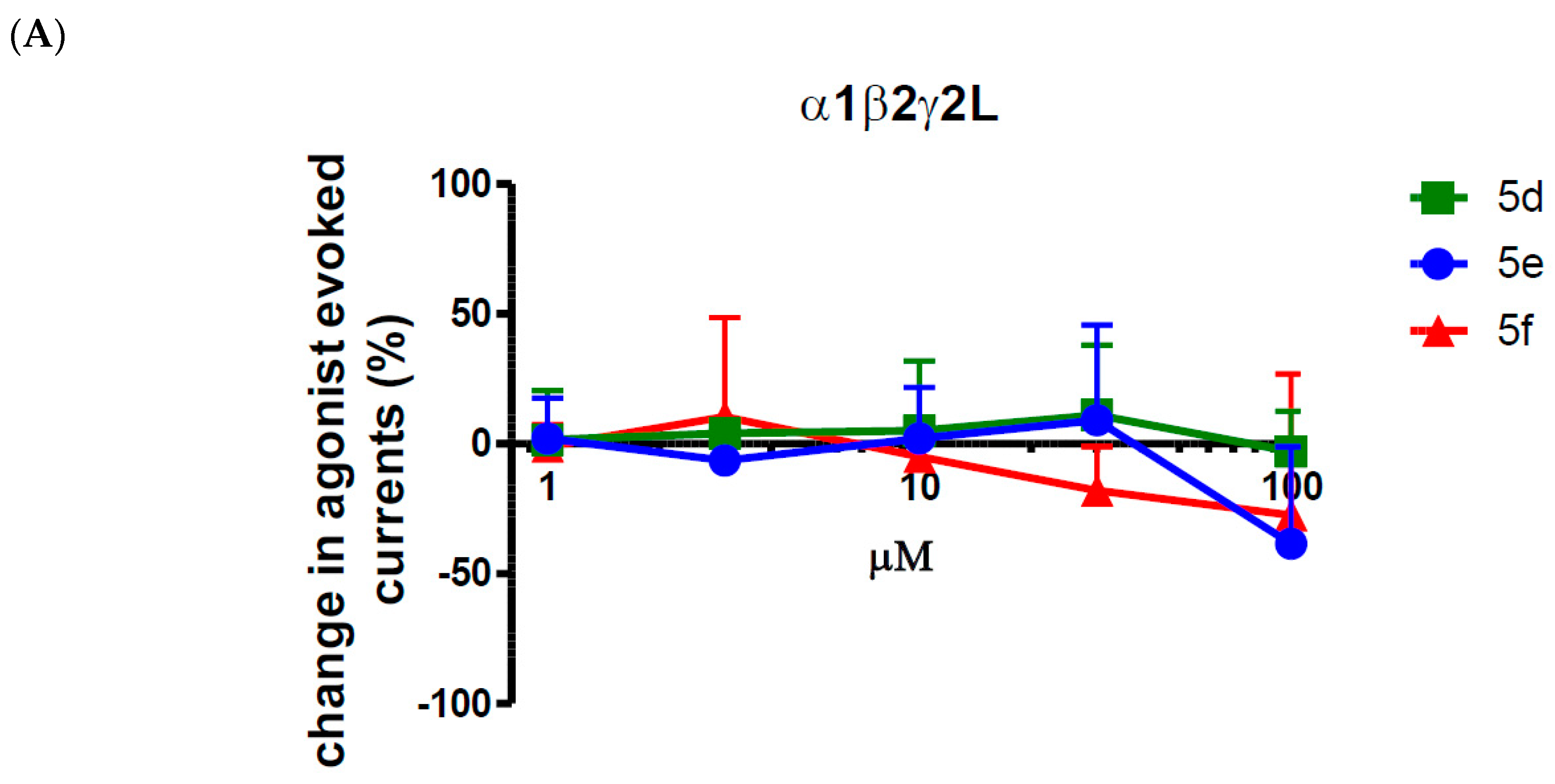
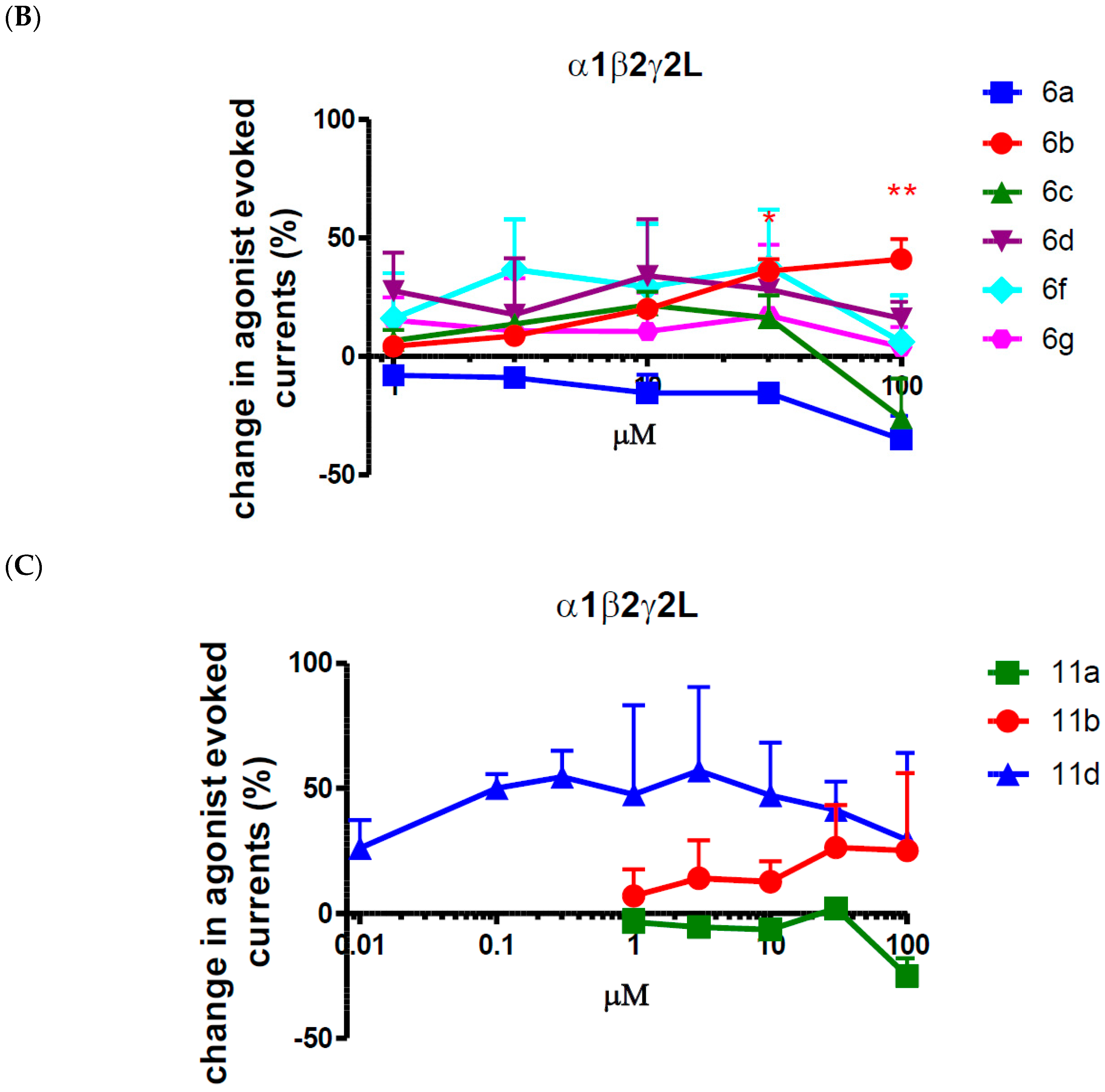
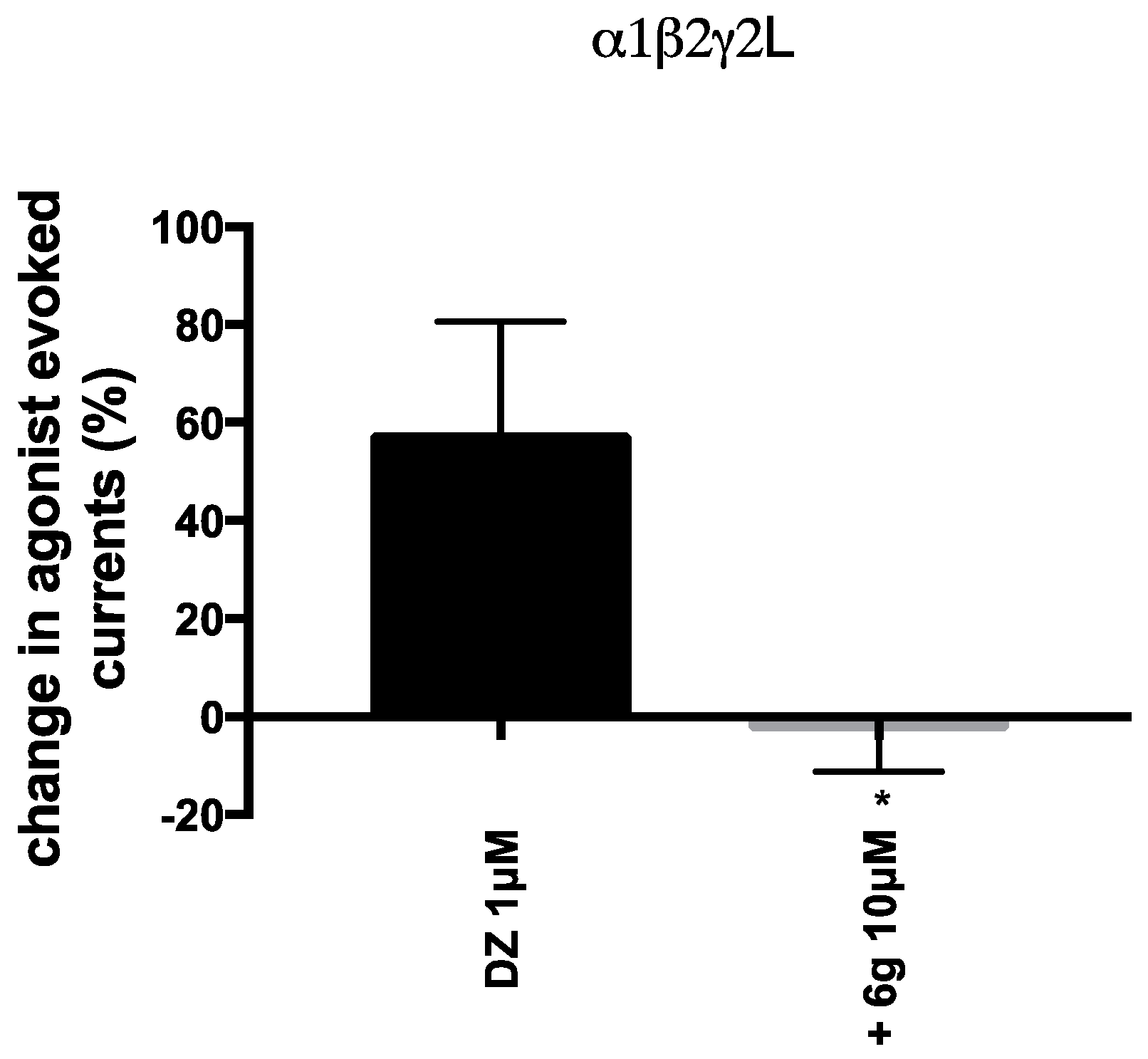
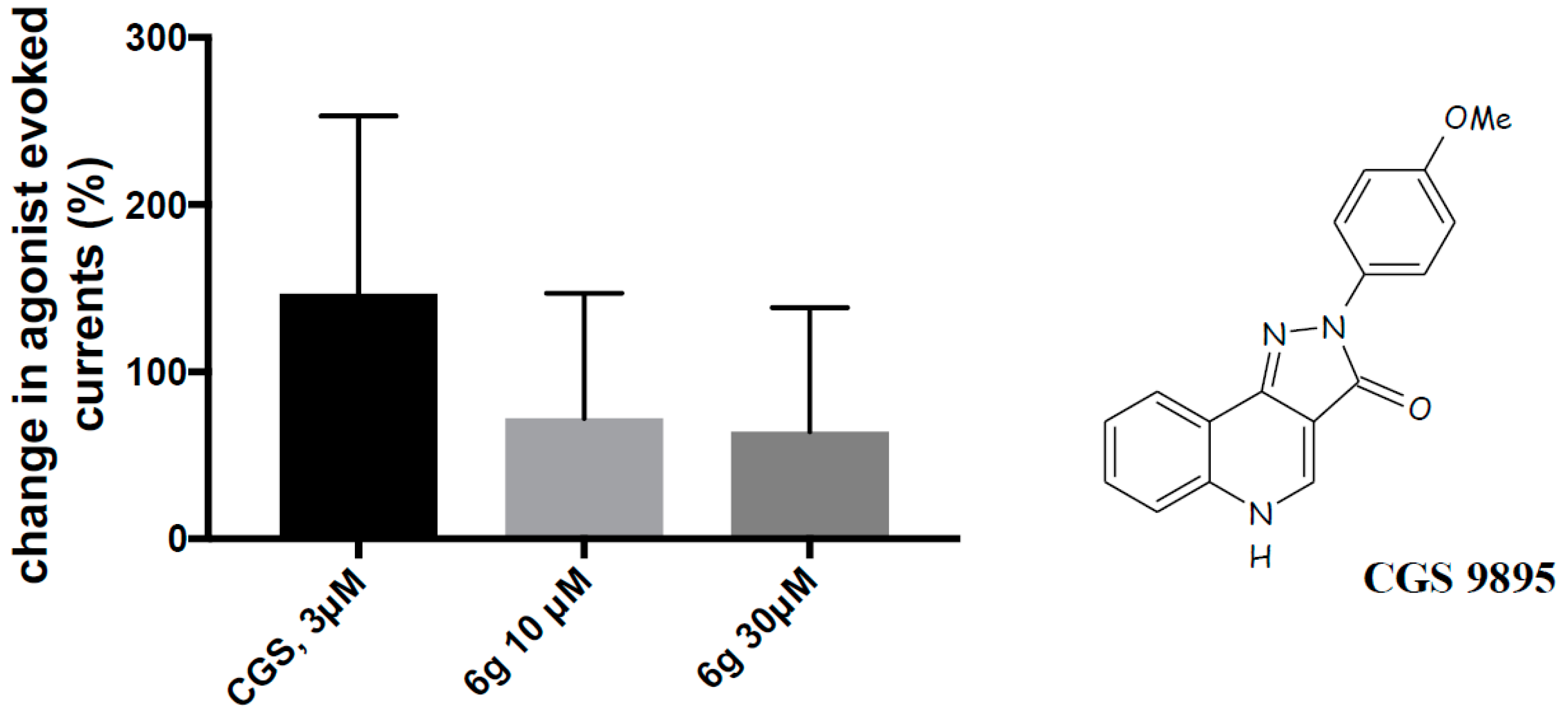
2.2. Biological Evaluation
3. Material and Methods
3.1. General Procedure for the Synthesis of 3b–e
3.1.1. 3-(Thien-2-yl)pyrazolo[1,5-a]quinazolin-5(4H)-one (3b)
3.1.2. 3-(Thien-3-yl)pyrazolo[1,5-a]quinazolin-5(4H)-one (3c)
3.1.3. 8-Methoxy-3-(thien-2-yl)pyrazolo[1,5-a]quinazolin-5(4H)-one (3d)
3.1.4. 8-Methoxy-3-(thien-3-yl)pyrazolo[1,5-a]quinazolin-5(4H)-one (3e)
3.2. General Procedure for the Synthesis of 3f–h
3.2.1. 8-Methoxy-3-(2-methoxybenzoyl)pyrazolo[1,5-a]quinazolin-5(4H)-one (3f)
3.2.2. 8-Methoxy-3-(thien-2-ylcarbonyl)pyrazolo[1,5-a]quinazolin-5(4H)-one (3g)
3.2.3. 8-Methoxy-3-(thien-3-ylcarbonyl)pyrazolo[1,5-a]quinazolin-5(4H)-one (3h)
3.3. General Procedure for the Synthesis of 4a–c
3.3.1. 3-(2-Methoxyphenylcarbonyl)-5-chloro-8-methoxypyrazolo[1,5-a]quinazoline (4a)
3.3.2. 3-(Thien-2-ylcarbonyl)-5-chloro-8-methoxypyrazolo[1,5-a]quinazoline (4b)
3.3.3. 3-(Thien-3-ylcarbonyl)-5-chloro-8-methoxypyrazolo[1,5-a]quinazoline (4c)
3.4. General Procedure for the Synthesis of 5a–e
3.4.1. 3-Phenylpyrazolo[1,5-a]quinazoline (5a)
3.4.2. 3-(Thien-2-yl)pyrazolo[1,5-a]quinazoline (5b)
3.4.3. 3-(Thien-3-yl)pyrazolo[1,5-a]quinazoline (5c)
3.4.4. 3-(Thien-2-yl)-8-methoxypyrazolo[1,5-a]quinazoline (5d)
3.4.5. 3-(Thien-3-yl)-8-methoxypyrazolo[1,5-a]quinazoline (5e)
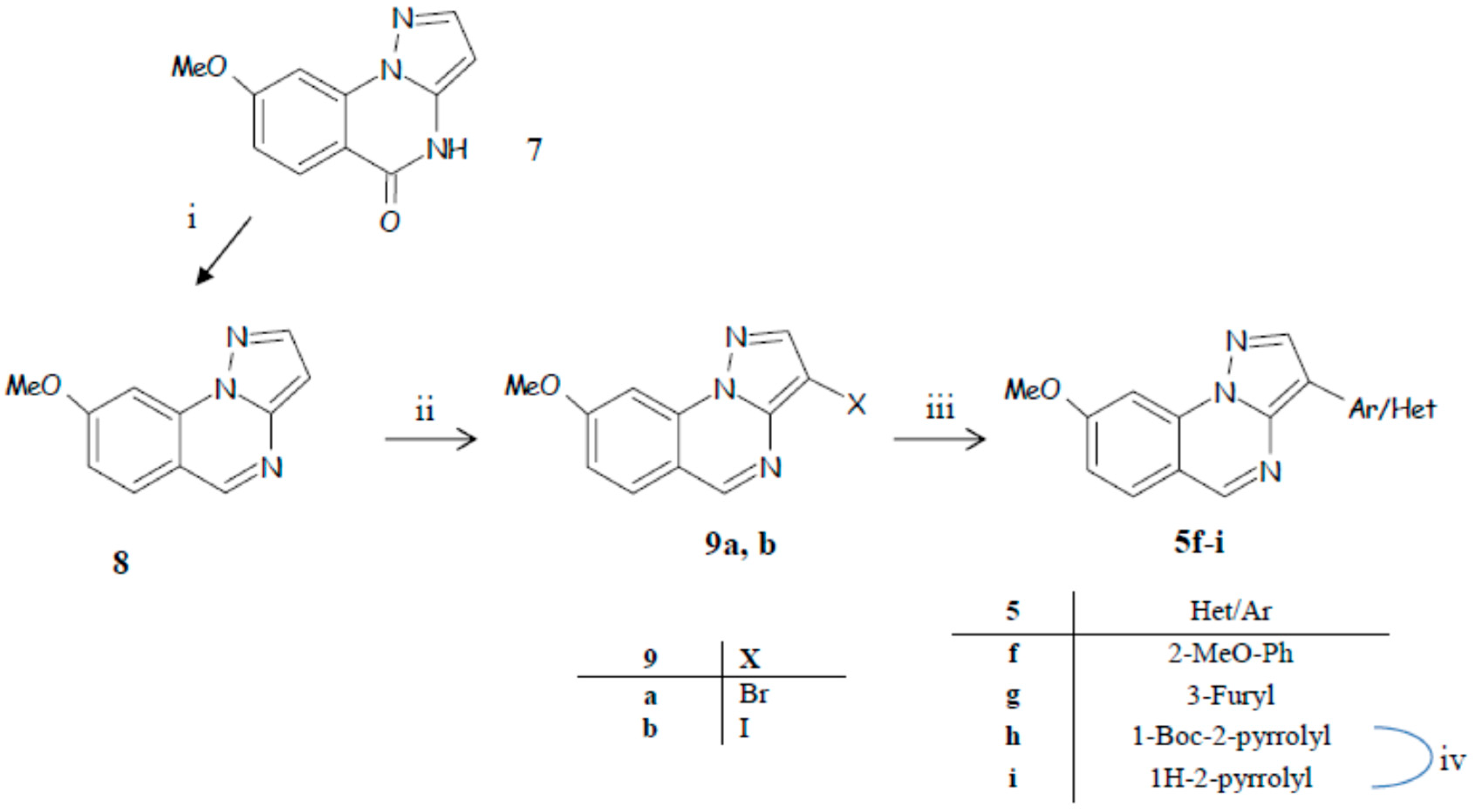
3.5. General Procedure for the Synthesis of 5f–h
3.5.1. 3-(2-Methoxyphenyl)-8-methoxypyrazolo[1,5-a]quinazoline (5f)
3.5.2. 3-(Fur-3-yl)-8-methoxypyrazolo[1,5-a]quinazoline (5g)
3.5.3. Tert-Butyl 2-(8-methoxypyrazolo[1,5-a]quinazolin-3-yl)-1H-pyrrole-1-carboxylate (5h)
3.6. 3-(1H-Pyrrol-2-yl)-8-methoxypyrazolo[1,5-a]quinazoline (5i)
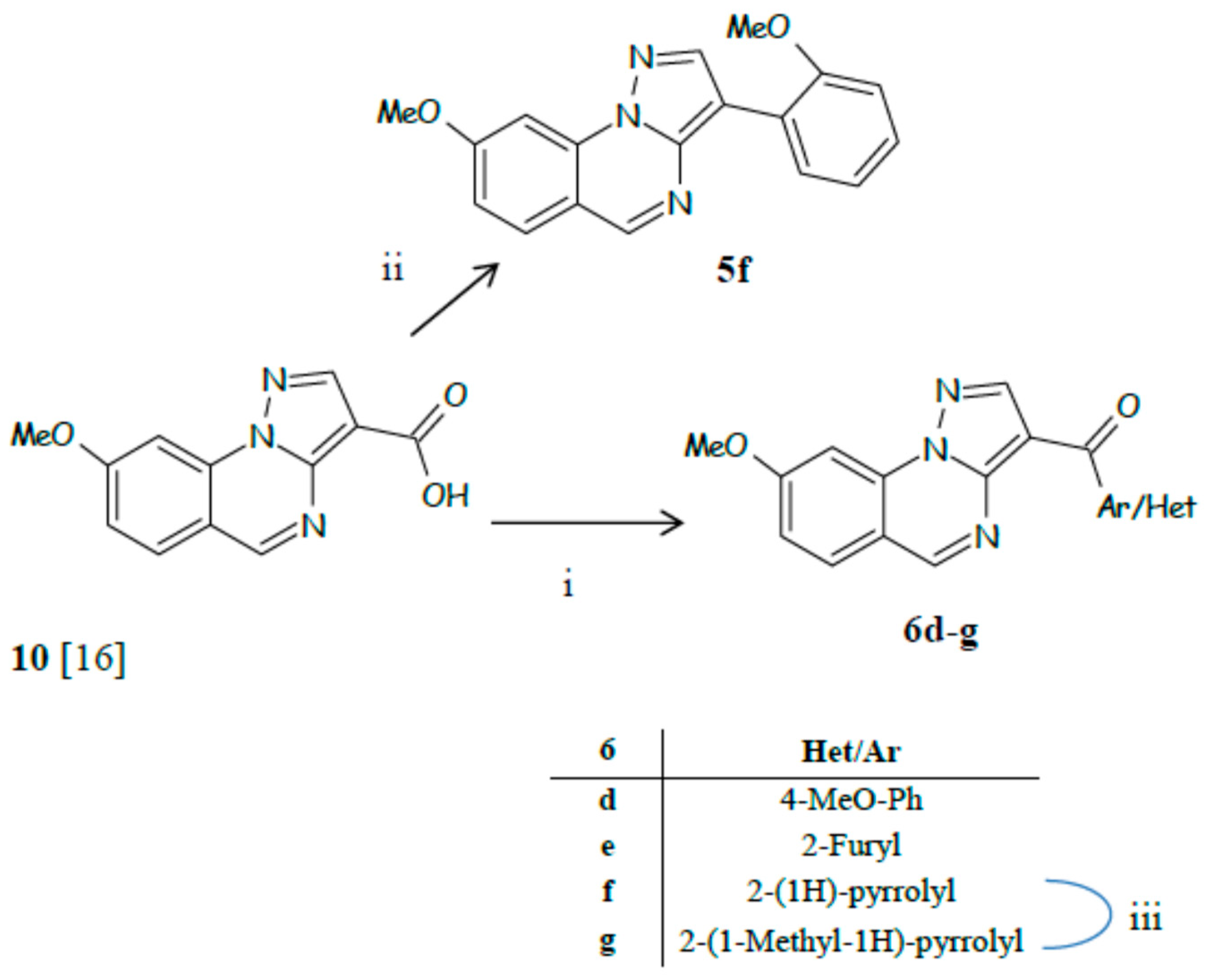
3.7. General Procedure for the Synthesis of 6a–c
3.7.1. 3-(2-Methoxyphenylcarbonyl)-8-methoxypyrazolo[1,5-a]quinazoline (6a)
3.7.2. 3-(Thien-2-ylcarbonyl)-8-methoxypyrazolo[1,5-a]quinazoline (6b)
3.7.3. 3-(Thien-3-ylcarbonyl)-8-methoxypyrazolo[1,5-a]quinazoline (6c)
3.8. 3-(4-Methoxyphenylcarbonyl)-8-methoxypyrazolo[1,5-a]quinazoline (6d)
3.9. General Procedure for The Synthesis of 6e–g
3.9.1. 3-(Fur-2-ylcarbonyl)-8-methoxypyrazolo[1,5-a]quinazoline (6e)
3.9.2. 3-(1H-pyrrol-2-ylcarbonyl)-8-methoxypyrazolo[1,5-a]quinazoline (6f)
3.9.3. 3-(1-Methyl-1H-pyrrol-2-ylcarbonyl)-8-methoxypyrazolo[1,5-a]quinazoline (6g)
3.10. 8-Methoxypyrazolo[1,5-a]quinazolin-5(4H)-one (7)
3.11. 8-Methoxypyrazolo[1,5-a]quinazoline (8)
3.12. General procedure for the synthesis of 9a,b
3.12.1. 3-Bromo-8-methoxypyrazolo[1,5-a]quinazoline (9a)
3.12.2. 3-Iodo-8-methoxypyrazolo[1,5-a]quinazoline (9b)
3.13. General procedure for the synthesis of 11a,b,d–g
3.13.1. 3-(2-Methoxyphenylcarbonyl)-8-methoxy-4,5-dihydropyrazolo[1,5-a]quinazoline (11a)
3.13.2. 3-(Thien-2-ylcarbonyl)-8-methoxy-4,5-dihydropyrazolo[1,5-a]quinazoline (11b)
3.13.3. 3-(4-Methoxyphenylcarbonyl)-8-methoxy-4,5-dihydropyrazolo[1,5-a]quinazoline (11d)
3.13.4. 3-(Fur-2-ylcarbonyl)-8-methoxy-4,5-dihydropyrazolo[1,5-a]quinazoline (11e)
3.13.5. 3-(1H-pyrrol-2-ylcarbonyl)-8-methoxy-4,5-dihydropyrazolo[1,5-a]quinazoline (11f)
3.13.6. 3-(1-Methyl-1H-pyrrol-2-ylcarbonyl)-8-methoxypyrazolo[1,5-a]quinazoline (11g)
3.14. Radioligand Binding Assay
3.15. General Methods for Electrophysiological Assays
3.15.1. Expression of Human Receptor Subunits
3.15.2. Electrophysiology
3.15.3. Statistics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| GABAAR | GABA receptors Type A |
| GABABR | GABA receptor Type B |
| GPCR | G-protein-coupled receptors |
| LGIC | ligand gated ion channel |
| PQ | pyrazoloquinazoline |
| PBTs | pyrazolobenzotriazines |
| PPs | pyrazolopyrimidines |
| PTs | pyrazolotriazines |
References
- Avoli, M.; D’Antuono, M.; Louvel, J.; Kohling, R.; Biagini, G.; Pumain, R.; D’Arcangelo, G.; Tancredi, V. Network and pharmacological mechanisms leading to epileptiform synchronization in the limbic system in vitro. Prog. Neurobiol. 2002, 68, 167–207. [Google Scholar] [CrossRef]
- Alexander, S.P.; Peters, J.A.; Kelly, E.; Marrion, N.V.; Faccenda, E.; Harding, S.D.; Pawson, A.J.; Sharman, J.L.; Southan, C.; Davies, J.A.; et al. THE CONCISE GUIDE TO PHARMACOLOGY 2017/18: Ligand-gated ion channels. Br. J. Pharmacol. 2017, 174 (Suppl. 1), S130–S159. [Google Scholar] [CrossRef]
- Chua, H.C.; Chebib, M. Chapter One—GABAA Receptors and the Diversity in their Structure and Pharmacology. Adv. Pharmacol. 2017, 79, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Sieghart, W.; Savić, M.M. International Union of Basic and Clinical Pharmacology. CVI: GABAA Receptor Subtype- and Function-selective Ligands: Key Issues in Translation to Humans. Pharmacol. Rev. 2018, 70, 836–878. [Google Scholar] [CrossRef] [PubMed]
- Krall, J.; Balle, T.; Krogsgaard-Larsen, N.; Sørensen, T.E.; Krogsgaard-Larsen, P.; Kristiansen, U.; Frølund, B. Chapter Eight—GABAA Receptor Partial Agonists and Antagonists: Structure, Binding Mode, and Pharmacology. Adv. Pharmacol. 2015, 72, 201–227. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.-J.; Forman, S.A. Comparison of αβδ and αβγ GABAA receptors: Allosteric modulation and identification of subunit arrangement by site-selective general anesthetics. Pharmacol. Res. 2018, 133, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Baur, R.; Tan, K.R.; Lüscher, B.P.; Gonthier, A.; Goeldner, M.; Sigel, E. Covalent modification of GABA A receptor isoforms by a diazepam analogue provides evidence for a novel benzodiazepine binding site that prevents modulation by these drugs. J. Neurochem. 2008, 106, 2353–2363. [Google Scholar] [CrossRef]
- Ramerstorfer, J.; Furtmüller, R.; Sarto-Jackson, I.; Varagic, Z.; Sieghart, W.; Ernst, M. The GABAA receptor alpha+beta- interface: A novel target for subtype selective drugs. J. Neurosci. 2011, 31, 870–877. [Google Scholar] [CrossRef]
- Sieghart, W.; Ramerstorfer, J.; Sarto-Jackson, I.; Varagic, Z.; Ernst, M. A novel GABAA receptor pharmacology: Drugs interacting with the α+β- interface. Br. J. Pharmacol. 2012, 166, 476–485. [Google Scholar] [CrossRef]
- Walters, R.J.; Hadley, S.H.; Morris, K.D.; Amin, J. Benzodiazepines act on GABAA receptors via two distinct and separable mechanisms. Nat. Neurosci. 2000, 3, 1274–1281. [Google Scholar] [CrossRef] [PubMed]
- Varagic, Z.; Wimmer, L.; Schnürch, M.; Mihovilovic, M.D.; Huang, S.; Rallapalli, S.; Cook, J.M.; Mirheydari, P.; Ecker, G.F.; Sieghart, W.; et al. Identification of novel positive allosteric modulators and null modulators at the GABAA receptor α+β- interface. Br. J. Pharmacol. 2013, 169, 371–383. [Google Scholar] [CrossRef] [PubMed]
- Simeone, X.; Siebert, D.C.B.; Bampali, K.; Varagic, Z.; Treven, M.; Rehman, S.; Pyszkowski, J.; Holzinger, R.; Steudle, F.; Scholze, P.; et al. Molecular tools for GABAAreceptors: High affinity ligands for β1-containing subtypes. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Mascia, M.P.; Ledda, G.; Orrù, A.; Marongiu, A.; Loriga, G.; Maciocco, E.; Biggio, G.; Ruiu, S. Differential modulation of GABAA receptor function by aryl pyrazoles. Eur. J. Pharmacol. 2014, 733, 1–6. [Google Scholar] [CrossRef]
- Varagic, Z.; Ramerstorfer, J.; Huang, S.; Rallapalli, S.; Sarto-Jackson, I.; Cook, J.; Sieghart, W.; Ernst, M. Subtype selectivity of α+β- site ligands of GABAA receptors: Identification of the first highly specific positive modulators at α6β2/3γ2 receptors. Br. J. Pharmacol. 2013, 169, 384–399. [Google Scholar] [CrossRef] [PubMed]
- Knutson, D.E.; Kodali, R.; Divović, B.; Treven, M.; Stephen, M.R.; Zahn, N.M.; Dobričić, V.; Huber, A.T.; Meirelles, M.A.; Verma, R.S.; et al. Design and Synthesis of Novel Deuterated Ligands Functionally Selective for the γ-Aminobutyric Acid Type A Receptor (GABA A R) α6 Subtype with Improved Metabolic Stability and Enhanced Bioavailability. J. Med. Chem. 2018, 61, 2422–2446. [Google Scholar] [CrossRef] [PubMed]
- Miller, P.S.; Aricescu, A.R. Crystal structure of a human GABAA receptor. Nature 2014, 512, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Noviello, C.M.; Teng, J.; Walsh, R.M.; Kim, J.J.; Hibbs, R.E. Structure of a human synaptic GABAA receptor. Nature 2018, 559, 67–88. [Google Scholar] [CrossRef]
- Guerrini, G.; Ciciani, G.; Crocetti, L.; Daniele, S.; Ghelardini, C.; Giovannoni, M.P.; Iacovone, A.; Di Cesare Mannelli, L.; Martini, C.; Vergelli, C. Identification of a New Pyrazolo[1,5-a ]quinazoline Ligand Highly Affine to γ-Aminobutyric Type A (GABAA ) Receptor Subtype with Anxiolytic-Like and Antihyperalgesic Activity. J. Med. Chem. 2017, 60, 9691–9702. [Google Scholar] [CrossRef]
- Lemon, M.; Strain, J.D.; Hegg, A.M.; Farver, D.K. Indiplon in the management of insomnia. Drug Des. Dev. Ther. 2009, 3, 131. [Google Scholar] [CrossRef]
- Skolnick, P. Anxioselective anxiolytics: On a quest for the Holy Grail. Trends Pharmacol. Sci. 2012, 33, 611–620. [Google Scholar] [CrossRef]
- Costanzo, A.; Guerrini, G.; Ciciani, G.; Bruni, F.; Selleri, S.; Costa, B.; Martini, C.; Lucacchini, A.; Malmberg Aiello, P.; Ipponi, A. Benzodiazepine receptor ligands. 4. Synthesis and pharmacological evaluation of 3-heteroaryl-8-chloropyrazolo[5,1-c][1,2,4]benzotriazine 5-oxides. J. Med. Chem. 1999, 42, 2218–2226. [Google Scholar] [CrossRef] [PubMed]
- Guerrini, G.; Ciciani, G.; Daniele, S.; Di Cesare Mannelli, L.; Ghelardini, C.; Martini, C.; Selleri, S. Synthesis and pharmacological evaluation of pyrazolo[1,5-a]pyrimidin-7(4H)-one derivatives as potential GABAA-R ligands. Bioorg. Med. Chem. 2017, 25, 1901–1906. [Google Scholar] [CrossRef] [PubMed]
- Guerrini, G.; Ciciani, G.; Daniele, S.; Martini, C.; Costagli, C.; Guarino, C.; Selleri, S. A new class of pyrazolo[5,1-c][1,2,4]triazines as γ-aminobutyric type A (GABAA) receptor subtype ligand: Synthesis and pharmacological evaluation. Bioorg. Med. Chem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Guerrini, G.; Ciciani, G.; Cambi, G.; Bruni, F.; Selleri, S.; Melani, F.; Montali, M.; Martini, C.; Ghelardini, C.; Norcini, M.; et al. Novel 3-aroylpyrazolo[5,1-c][1,2,4]benzotriazine 5-oxides 8-substituted, ligands at GABAA/benzodiazepine receptor complex: Synthesis, pharmacological and molecular modeling studies. Bioorg. Med. Chem. 2008, 16, 4471–4489. [Google Scholar] [CrossRef] [PubMed]
- Penning, T.D.; Thomas, S.A.; Hajduk, P.J.; Sauer, D.R.; Sarris, K.; Giranda, V.L. Potent PARP Inhibitors WO 2007149907 A2, 27 December 2007.
- Wongsamitkul, N.; Maldifassi, M.C.; Simeone, X.; Baur, R.; Ernst, M.; Sigel, E. α subunits in GABAA receptors are dispensable for GABA and diazepam action. Sci. Rep. 2017, 7, 15498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maldifassi, M.C.; Baur, R.; Sigel, E. Molecular mode of action of CGS 9895 at α1β2γ2GABAAreceptors. J. Neurochem. 2016, 3, 722–730. [Google Scholar] [CrossRef] [PubMed]
- Martini, C.; Lucacchini, A.; Ronca, G.; Hrelia, S.; Rossi, C.A. Isolation of Putative Benzodiazepine Receptors from Rat Brain Membranes by Affinity Chromatography. J. Neurochem. 1982, 38, 15–19. [Google Scholar] [CrossRef]
- Primofiore, G.; Da Settimo, F.; Taliani, S.; Marini, A.M.; Novellino, E.; Greco, G.; Lavecchia, A.; Besnard, F.; Trincavelli, L.; Costa, B.; et al. Novel N-(arylalkyl)indol-3-yl glyoxylylamides targeted as ligands of the benzodiazepine receptor: Synthesis, biological evaluation, and molecular modeling analysis of the structure activity relationships. J. Med. Chem. 2001, 44, 2286–2297. [Google Scholar] [CrossRef]
- Lowry, O.H.; Rosenbrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [CrossRef]
- Cheng, Y.; Prusoff, W.H. Relation between the inhibiton constant (Ki) and the concentration of inhibitor which causes 50 percent inhibiton (IC50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [CrossRef]
- Colman, A. Transcription and Translation: A Practical Approach; Hames, B.D., Higgins, S.J., Eds.; Oxford University Press: Washington, DC, USA, 1984; ISBN 9780904147520. [Google Scholar]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guerrini, G.; Vergelli, C.; Cantini, N.; Giovannoni, M.P.; Daniele, S.; Mascia, M.P.; Martini, C.; Crocetti, L. Synthesis of New GABAA Receptor Modulator with Pyrazolo[1,5-a]quinazoline (PQ) Scaffold. Int. J. Mol. Sci. 2019, 20, 1438. https://doi.org/10.3390/ijms20061438
Guerrini G, Vergelli C, Cantini N, Giovannoni MP, Daniele S, Mascia MP, Martini C, Crocetti L. Synthesis of New GABAA Receptor Modulator with Pyrazolo[1,5-a]quinazoline (PQ) Scaffold. International Journal of Molecular Sciences. 2019; 20(6):1438. https://doi.org/10.3390/ijms20061438
Chicago/Turabian StyleGuerrini, Gabriella, Claudia Vergelli, Niccolò Cantini, Maria Paola Giovannoni, Simona Daniele, Maria Paola Mascia, Claudia Martini, and Letizia Crocetti. 2019. "Synthesis of New GABAA Receptor Modulator with Pyrazolo[1,5-a]quinazoline (PQ) Scaffold" International Journal of Molecular Sciences 20, no. 6: 1438. https://doi.org/10.3390/ijms20061438