Inhibition of Triple-Negative Breast Cancer Cell Aggressiveness by Cathepsin D Blockage: Role of Annexin A1


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
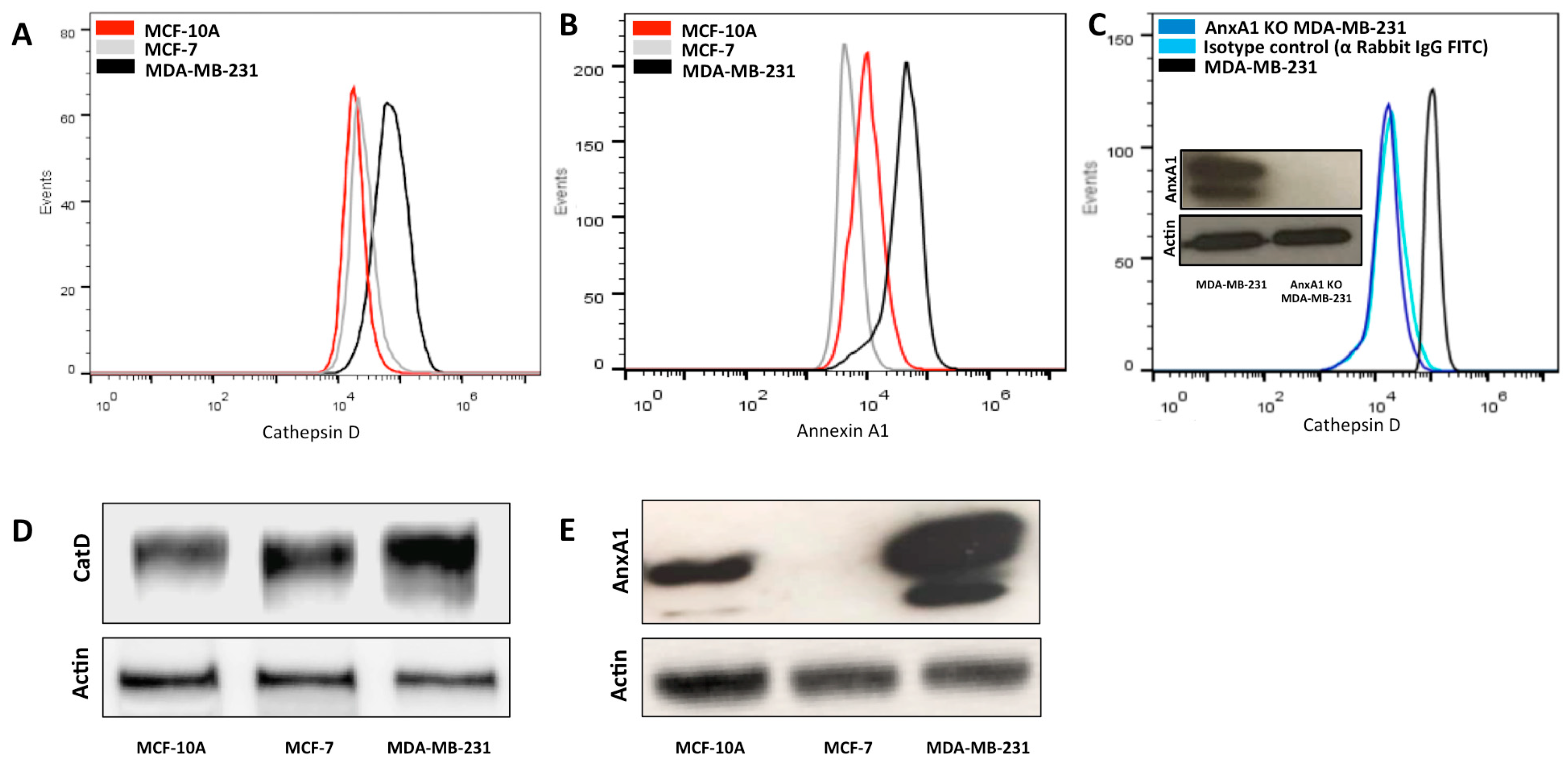
2.1. High Expression of AnxA1 Is Associated with CatD Expression in TNBC Cells
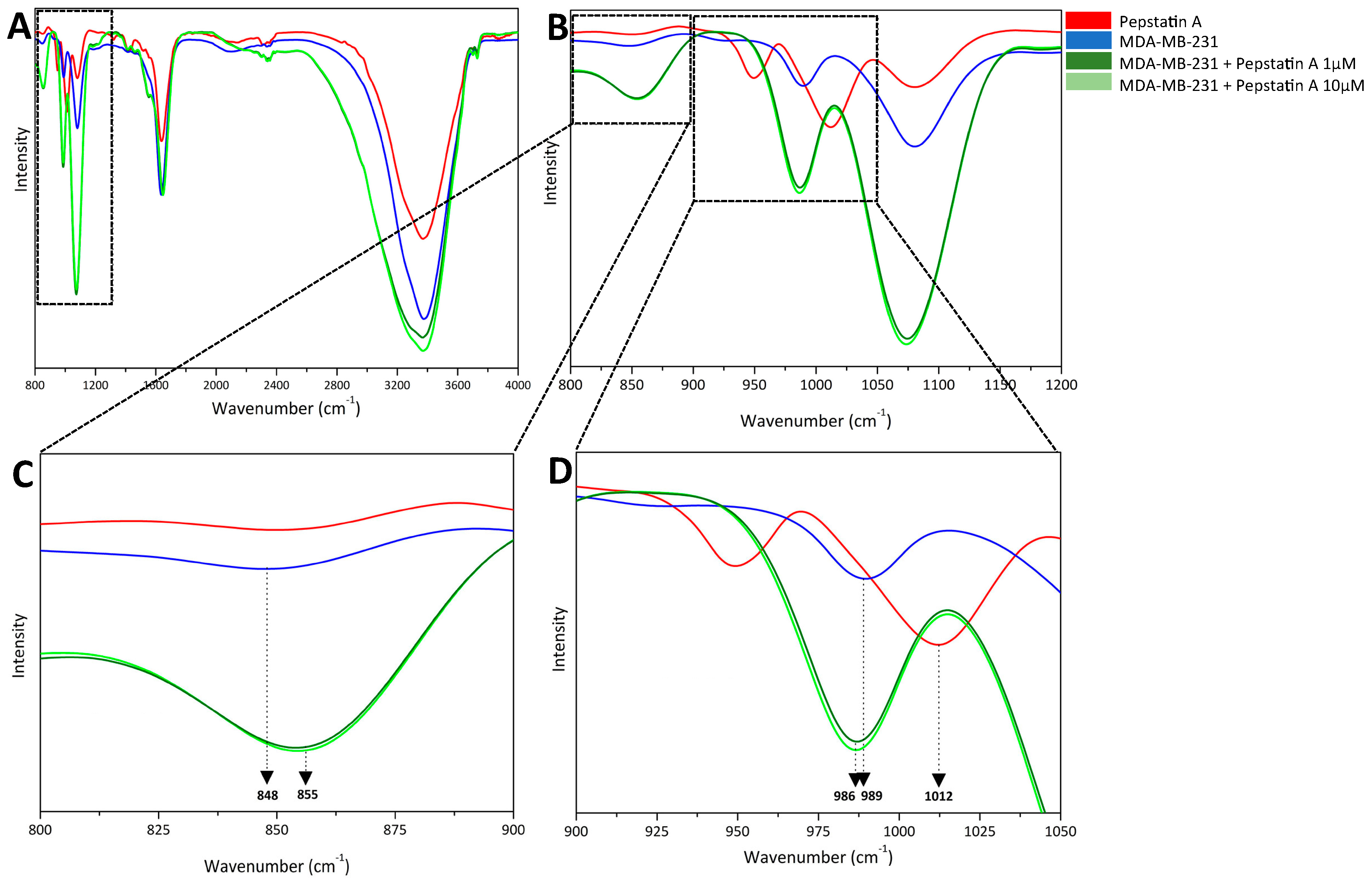
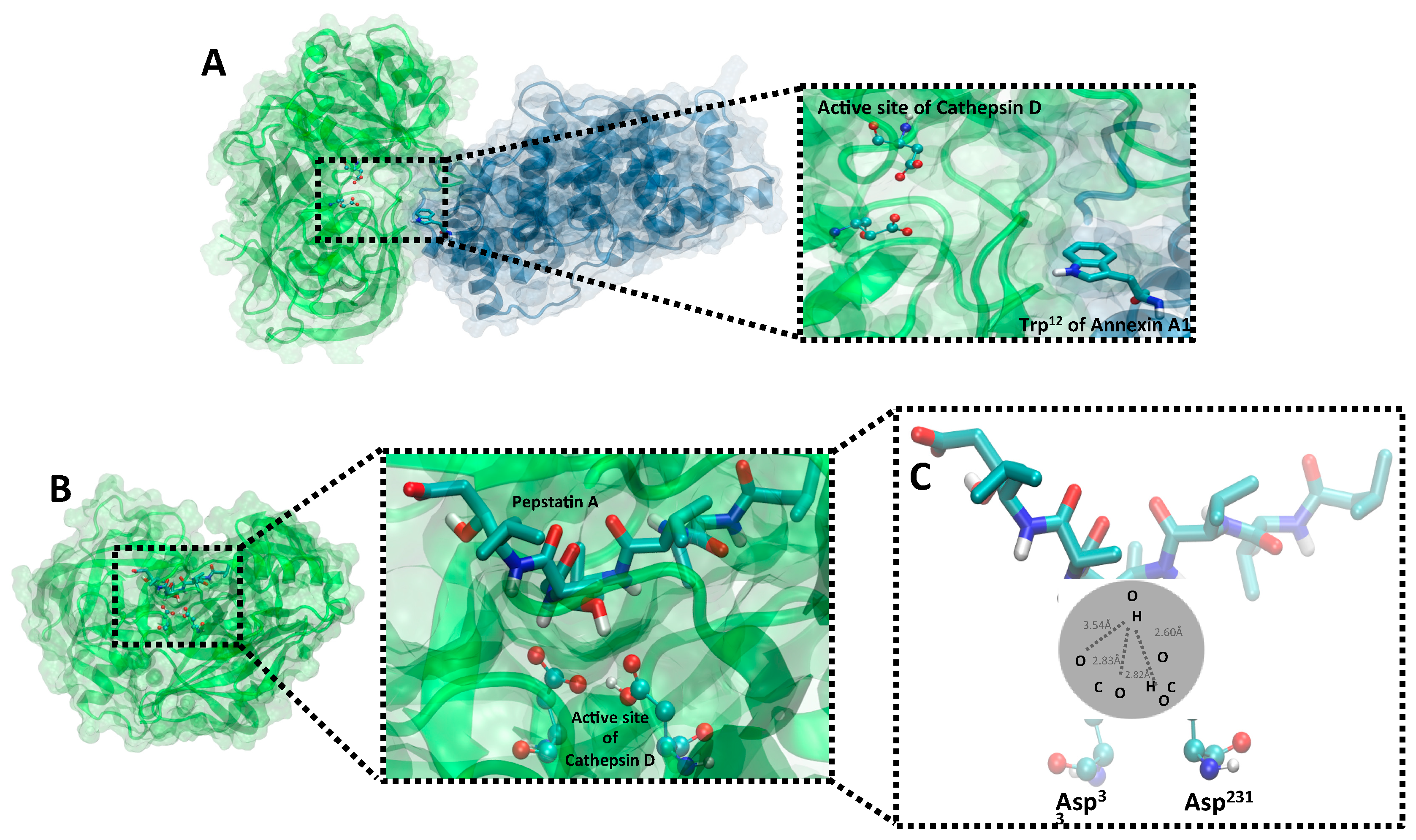
2.2. PepA Inhibts CatD through Carboxylic Acids from Catalytic Aspartate Dyad
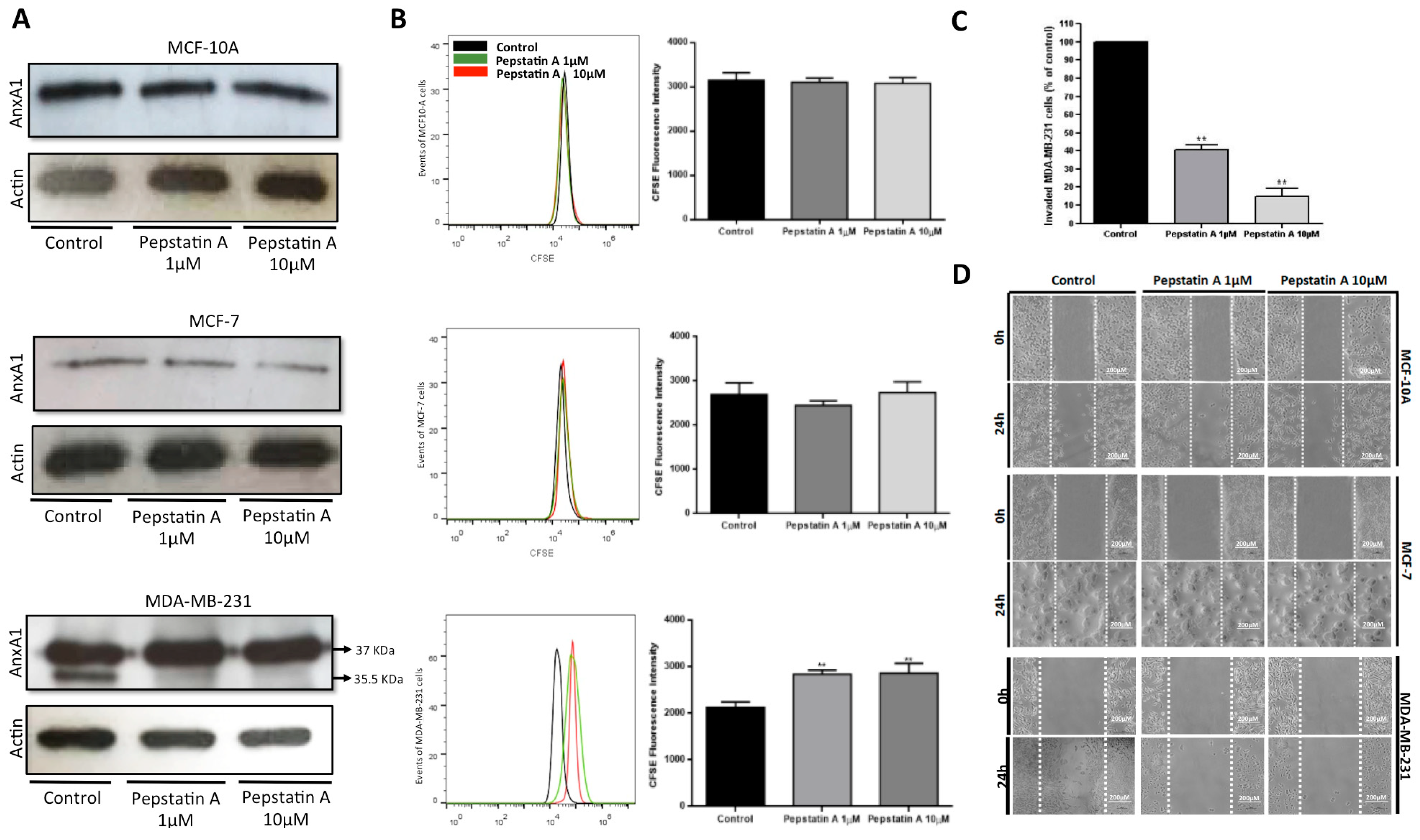
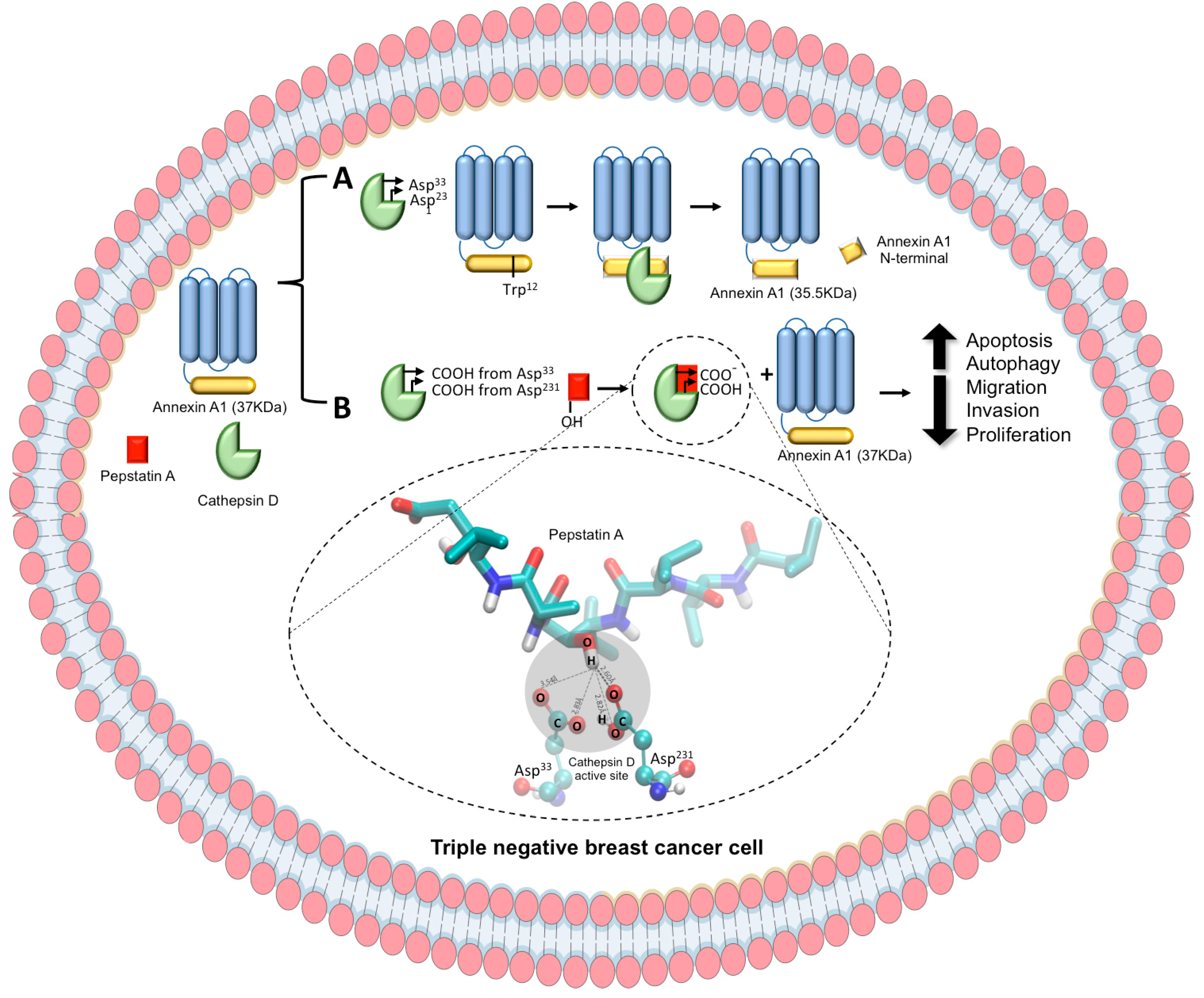
2.3. CatD Cleaves AnxA1: A Crucial Event in TNBC Cells Proliferation and Invasion
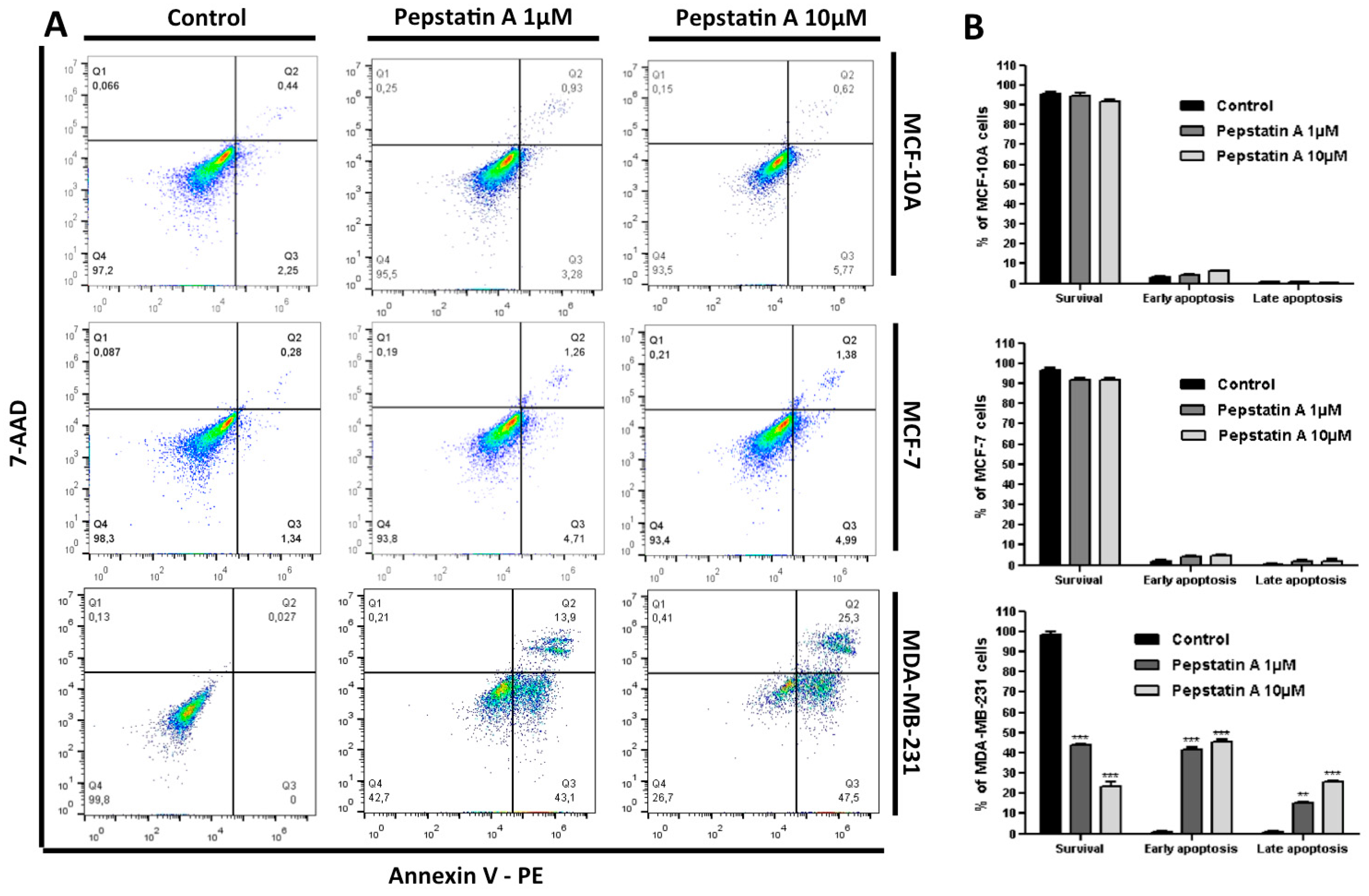
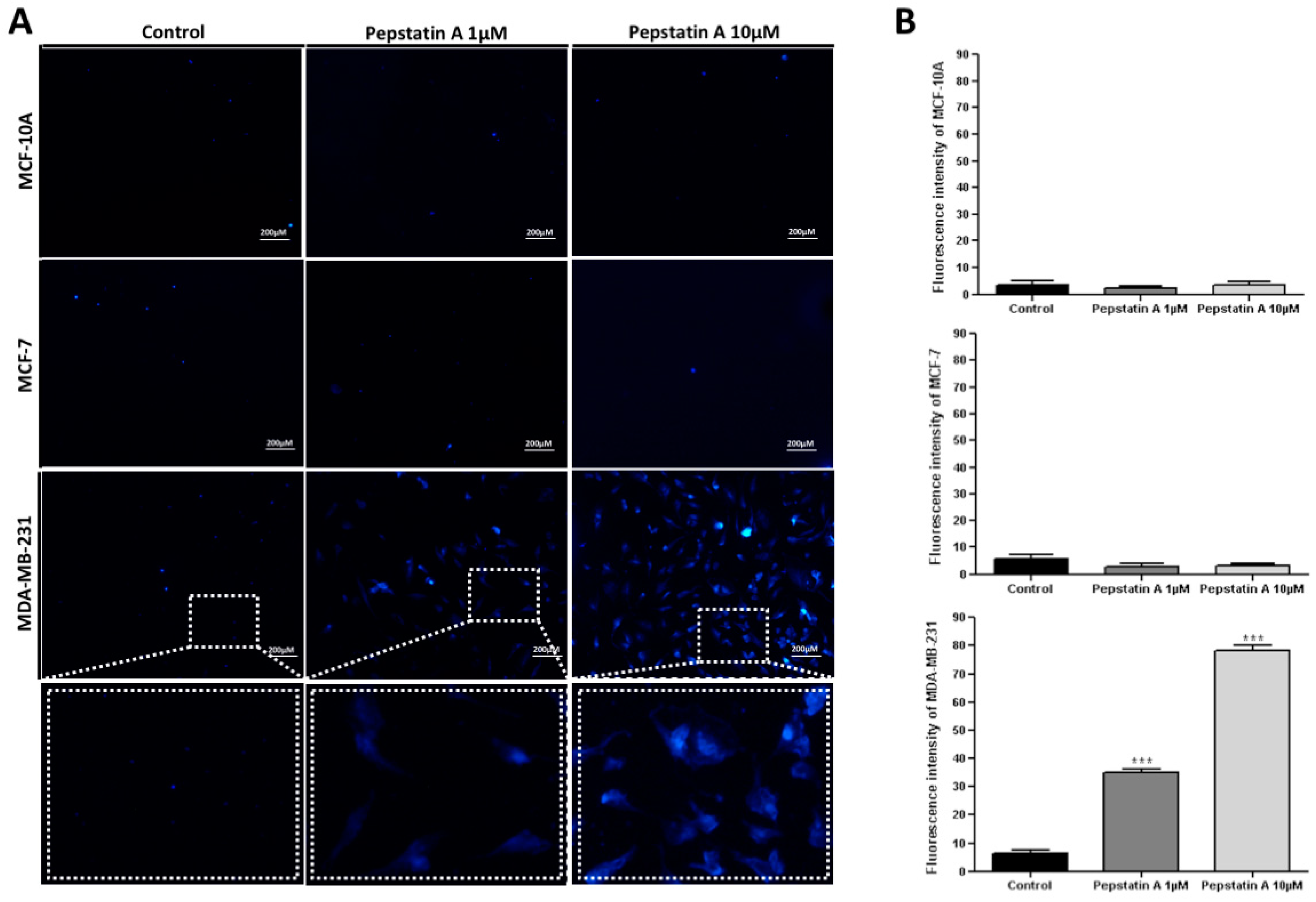
2.4. CatD Inhibition also Induces Apoptosis and Autophagy Processes in TNBC Cells
2.5. Validation of Structural Models of CatD/AnxA1 and CatD/PepA Interactions
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Generation of AnxA1 Knockdown MDA-MB-231 Cell Clones
4.3. Antibodies and Reagents
4.4. Protein Extraction and Western Blot
4.5. Apoptosis Assay
4.6. Autophagy Assay
4.7. Proliferation Assay
4.8. Matrigel Invasion Assay
4.9. Wound-Healing Migration Assay
4.10. FTIR Analysis
4.11. Molecular Modeling Docking Analysis
4.12. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AnxA1 | Annexin A1 |
| BC | Breast cancer |
| CatD | Cathepsin D |
| CFSE | Carboxylfluorescein succinimidyl ester |
| ER | Estrogen receptor |
| FTIR | Fourier transform infrared |
| HER2 | Human epidermal growth factor receptor 2 |
| MDC | Monodansylcadaverine |
| PepA | Pepstatin A |
| PR | Progesterone receptor |
| TNBC | Triple-negative breast cancer |
References
- The Lancet Oncology. New interventions offer prostate cancer hope. Lancet Oncol. 2018, 19, 427. [Google Scholar] [CrossRef]
- Wyatt, A.W.; Mo, F.; Wang, Y.; Collins, C.C. The diverse heterogeneity of molecular alterations in prostate cancer identified through next-generation sequencing. Asian J. Androl. 2013, 15, 301–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.; Zapatero-Rodriguez, J.; O’Kennedy, R. Prostate cancer diagnostics: Clinical challenges and the ongoing need for disruptive and effective diagnostic tools. Biotechnol. Adv. 2017, 35, 135–149. [Google Scholar] [CrossRef]
- Kadoch, C.; Crabtree, G.R. Mammalian SWI/SNF chromatin remodeling complexes and cancer: Mechanistic insights gained from human genomics. Sci. Adv. 2015, 1, e1500447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, C.; Allis, C.D. SWI/SNF complex in cancer. Nat. Genet. 2017, 49, 178–179. [Google Scholar] [CrossRef] [PubMed]
- Hodges, C.; Kirkland, J.G.; Crabtree, G.R. The Many Roles of BAF (mSWI/SNF) and PBAF Complexes in Cancer. Cold Spring Harb. Perspect. Med. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.G.; Roberts, C.W. SWI/SNF nucleosome remodellers and cancer. Nat. Rev. Cancer 2011, 11, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.N.; Roberts, C.W. ARID1A mutations in cancer: Another epigenetic tumor suppressor? Cancer Discov. 2013, 3, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Oike, T.; Ogiwara, H.; Nakano, T.; Yokota, J.; Kohno, T. Inactivating mutations in SWI/SNF chromatin remodeling genes in human cancer. Jpn. J. Clin. Oncol. 2013, 43, 849–855. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M. Polybromo-1: The chromatin targeting subunit of the PBAF complex. Biochimie 2009, 91, 309–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horikawa, I.; Barrett, J.C. cDNA cloning of the human polybromo-1 gene on chromosome 3p21. DNA Seq. 2002, 13, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Qu, Y.; Dai, B.; Zhu, Y.; Shi, G.; Zhu, Y.; Shen, Y.; Zhang, H.; Ye, D. PBRM1 regulates proliferation and the cell cycle in renal cell carcinoma through a chemokine/chemokine receptor interaction pathway. PLoS ONE 2017, 12, e0180862. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.; Nagase, S.; Montia, A.G.; Kalachikov, S.M.; Keniry, M.; Su, T.; Memeo, L.; Hibshoosh, H.; Parsons, R. BAF180 is a critical regulator of p21 induction and a tumor suppressor mutated in breast cancer. Cancer Res. 2008, 68, 1667–1674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brownlee, P.M.; Chambers, A.L.; Cloney, R.; Bianchi, A.; Downs, J.A. BAF180 promotes cohesion and prevents genome instability and aneuploidy. Cell Rep. 2014, 6, 973–981. [Google Scholar] [CrossRef]
- Lee, H.; Dai, F.; Zhuang, L.; Xiao, Z.D.; Kim, J.; Zhang, Y.; Ma, L.; You, M.J.; Wang, Z.; Gan, B. BAF180 regulates cellular senescence and hematopoietic stem cell homeostasis through p21. Oncotarget 2016, 7, 19134–19146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burrows, A.E.; Smogorzewska, A.; Elledge, S.J. Polybromo-associated BRG1-associated factor components BRD7 and BAF180 are critical regulators of p53 required for induction of replicative senescence. Proc. Natl. Acad. Sci. USA 2010, 107, 14280–14285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niimi, A.; Hopkins, S.R.; Downs, J.A.; Masutani, C. The BAH domain of BAF180 is required for PCNA ubiquitination. Mutat. Res. 2015, 779, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Hopson, S.; Thompson, M.J. BAF180: Its Roles in DNA Repair and Consequences in Cancer. ACS Chem. Boil. 2017, 12, 2482–2490. [Google Scholar] [CrossRef] [PubMed]
- Varela, I.; Tarpey, P.; Raine, K.; Huang, D.; Ong, C.K.; Stephens, P.; Davies, H.; Jones, D.; Lin, M.L.; Teague, J.; et al. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature 2011, 469, 539–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.; Peng, Y.; Zhong, G.; Xie, W.; Dong, W.; Wang, B.; Chen, X.; Gu, P.; He, W.; Wu, S.; et al. PBRM1 suppresses bladder cancer by cyclin B1 induced cell cycle arrest. Oncotarget 2015, 6, 16366–16378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shu, X.S.; Zhao, Y.; Sun, Y.; Zhong, L.; Cheng, Y.; Zhang, Y.; Ning, K.; Tao, Q.; Wang, Y.; Ying, Y. The epigenetic modifier PBRM1 restricts the basal activity of the innate immune system by repressing retinoic acid-inducible gene-I-like receptor signalling and is a potential prognostic biomarker for colon cancer. J. Pathol. 2018, 244, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Sekine, I.; Sato, M.; Sunaga, N.; Toyooka, S.; Peyton, M.; Parsons, R.; Wang, W.; Gazdar, A.F.; Minna, J.D. The 3p21 candidate tumor suppressor gene BAF180 is normally expressed in human lung cancer. Oncogene 2005, 24, 2735–2738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Link, K.A.; Balasubramaniam, S.; Sharma, A.; Comstock, C.E.; Godoy-Tundidor, S.; Powers, N.; Cao, K.H.; Haelens, A.; Claessens, F.; Revelo, M.P.; et al. Targeting the BAF57 SWI/SNF subunit in prostate cancer: A novel platform to control androgen receptor activity. Cancer Res. 2008, 68, 4551–4558. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Powers, N.; Saini, N.; Comstock, C.E.; Sharma, A.; Weaver, K.; Revelo, M.P.; Gerald, W.; Williams, E.; Jessen, W.J.; et al. The SWI/SNF ATPase Brm is a gatekeeper of proliferative control in prostate cancer. Cancer Res. 2008, 68, 10154–10162. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.B.; Sun, A.J.; Wang, C.; Chen, L.R. Expression of BRG1 and BRM proteins in prostatic cancer. Zhonghua Bing Li Xue Za Zhi = Chin. J. Pathol. 2010, 39, 591–594. [Google Scholar]
- Hansen, R.L.; Heeboll, S.; Ottosen, P.D.; Dyrskjot, L.; Borre, M. Smarcc1 expression: A significant predictor of disease-specific survival in patients with clinically localized prostate cancer treated with no intention to cure. Scand. J. Urol. Nephrol. 2011, 45, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Prensner, J.R.; Iyer, M.K.; Sahu, A.; Asangani, I.A.; Cao, Q.; Patel, L.; Vergara, I.A.; Davicioni, E.; Erho, N.; Ghadessi, M.; et al. The long noncoding RNA SChLAP1 promotes aggressive prostate cancer and antagonizes the SWI/SNF complex. Nat. Genet. 2013, 45, 1392–1398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, A.K.; Barton, M.C. Bromodomain Histone Readers and Cancer. J. Mol. Boil. 2017, 429, 2003–3010. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Picaud, S.; Mangos, M.; Keates, T.; Lambert, J.P.; Barsyte-Lovejoy, D.; Felletar, I.; Volkmer, R.; Muller, S.; Pawson, T.; et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 2012, 149, 214–231. [Google Scholar] [CrossRef]
- Zeng, L.; Zhou, M.M. Bromodomain: An acetyl-lysine binding domain. FEBS Lett. 2002, 513, 124–128. [Google Scholar] [CrossRef]
- Barbieri, I.; Cannizzaro, E.; Dawson, M.A. Bromodomains as therapeutic targets in cancer. Brief. Funct. Genom. 2013, 12, 219–230. [Google Scholar] [CrossRef] [Green Version]
- Ghoshal, A.; Yugandhar, D.; Srivastava, A.K. BET inhibitors in cancer therapeutics: A patent review. Expert Opin. Ther. Pat. 2016, 26, 505–522. [Google Scholar] [CrossRef] [PubMed]
- Fung, J.J.; Kosaka, A.; Shan, X.; Danet-Desnoyers, G.; Gormally, M.; Owen, K.; Reproducibility Project: Cancer, B.; Reproducibility Project Cancer, B. Registered report: Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukemia. eLife 2015, 4, e08997. [Google Scholar] [CrossRef]
- Stuhlmiller, T.J.; Miller, S.M.; Zawistowski, J.S.; Nakamura, K.; Beltran, A.S.; Duncan, J.S.; Angus, S.P.; Collins, K.A.; Granger, D.A.; Reuther, R.A.; et al. Inhibition of Lapatinib-Induced Kinome Reprogramming in ERBB2-Positive Breast Cancer by Targeting BET Family Bromodomains. Cell Rep. 2015, 11, 390–404. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Nahar, S.; Nakagawa, A.; Fernandez-Barrena, M.G.; Mertz, J.A.; Bryant, B.M.; Adams, C.E.; Mino-Kenudson, M.; Von Alt, K.N.; Chang, K.; et al. Regulation of GLI Underlies a Role for BET Bromodomains in Pancreatic Cancer Growth and the Tumor Microenvironment. Clin. Cancer Res. 2016, 22, 4259–4270. [Google Scholar] [CrossRef] [PubMed]
- Porter, E.G.; Dykhuizen, E.C. Individual Bromodomains of Polybromo-1 Contribute to Chromatin Association and Tumor Suppression in Clear Cell Renal Carcinoma. J. Boil. Chem. 2017, 292, 2601–2610. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, P.M.; Chambers, A.L.; Oliver, A.W.; Downs, J.A. Cancer and the bromodomains of BAF180. Biochem. Soc. Trans. 2012, 40, 364–369. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Zhou, B.; Yang, C.Y.; Ji, J.; McEachern, D.; Przybranowski, S.; Jiang, H.; Hu, J.; Xu, F.; Zhao, Y.; et al. Targeted Degradation of BET Proteins in Triple-Negative Breast Cancer. Cancer Res. 2017, 77, 2476–2487. [Google Scholar] [CrossRef]
- Kaur, G.; Reinhart, R.A.; Monks, A.; Evans, D.; Morris, J.; Polley, E.; Teicher, B.A. Bromodomain and hedgehog pathway targets in small cell lung cancer. Cancer Lett. 2016, 371, 225–239. [Google Scholar] [CrossRef]
- Pawlowski, R.; Muhl, S.M.; Sulser, T.; Krek, W.; Moch, H.; Schraml, P. Loss of PBRM1 expression is associated with renal cell carcinoma progression. Int. J. Cancer 2013, 132, E11–E17. [Google Scholar] [CrossRef]
- Mo, D.; Li, C.; Liang, J.; Shi, Q.; Su, N.; Luo, S.; Zeng, T.; Li, X. Low PBRM1 identifies tumor progression and poor prognosis in breast cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 9307–9313. [Google Scholar] [PubMed]
- Kim, J.Y.; Lee, S.H.; Moon, K.C.; Kwak, C.; Kim, H.H.; Keam, B.; Kim, T.M.; Heo, D.S. The Impact of PBRM1 Expression as a Prognostic and Predictive Marker in Metastatic Renal Cell Carcinoma. J. Urol. 2015, 194, 1112–1119. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Xu, R.M. Structure and function of the BAH domain in chromatin biology. Crit. Rev. Biochem. Mol. Boil. 2013, 48, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, G.H.; Nicolas, R.H. The BAH domain, polybromo and the RSC chromatin remodelling complex. Gene 2001, 268, 1–7. [Google Scholar] [CrossRef]
- Zhu, H.; Wan, X.; Li, J.; Han, L.; Bo, X.; Chen, W.; Lu, C.; Shen, Z.; Xu, C.; Chen, L.; et al. Computational Prediction and Validation of BAHD1 as a Novel Molecule for Ulcerative Colitis. Sci. Rep. 2015, 5, 12227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monte, E.; Rosa-Garrido, M.; Karbassi, E.; Chen, H.; Lopez, R.; Rau, C.D.; Wang, J.; Nelson, S.F.; Wu, Y.; Stefani, E.; et al. Reciprocal Regulation of the Cardiac Epigenome by Chromatin Structural Proteins Hmgb and Ctcf: IMPLICATIONS FOR TRANSCRIPTIONAL REGULATION. J. Boil. Chem. 2016, 291, 15428–15446. [Google Scholar] [CrossRef]
- Ueda, T.; Yoshida, M. HMGB proteins and transcriptional regulation. Biochim. Biophys. Acta 2010, 1799, 114–118. [Google Scholar] [CrossRef]
- Peluso, S.; Chiappetta, G. High-Mobility Group A (HMGA) Proteins and Breast Cancer. Breast Care 2010, 5, 81–85. [Google Scholar] [CrossRef] [Green Version]
- Li, X.L.; Eishi, Y.; Bai, Y.Q.; Sakai, H.; Akiyama, Y.; Tani, M.; Takizawa, T.; Koike, M.; Yuasa, Y. Expression of the SRY-related HMG box protein SOX2 in human gastric carcinoma. Int. J. Oncol. 2004, 24, 257–263. [Google Scholar] [CrossRef]
- Wang, H.; Li, Z.; Sun, Y.; Xu, Z.; Han, J.; Song, B.; Song, W.; Qin, C.; Yin, L. Relationship between high-mobility group box 1 overexpression in ovarian cancer tissue and serum: A meta-analysis. Oncotargets Ther. 2015, 8, 3523–3531. [Google Scholar] [CrossRef]
- Valenta, T.; Lukas, J.; Korinek, V. HMG box transcription factor TCF-4’s interaction with CtBP1 controls the expression of the Wnt target Axin2/Conductin in human embryonic kidney cells. Nucleic Acids Res. 2003, 31, 2369–2380. [Google Scholar] [CrossRef] [PubMed]
- Barreiro-Alonso, A.; Lamas-Maceiras, M.; Rodriguez-Belmonte, E.; Vizoso-Vazquez, A.; Quindos, M.; Cerdan, M.E. High Mobility Group B Proteins, Their Partners, and Other Redox Sensors in Ovarian and Prostate Cancer. Oxidative Med. Cell. Longev. 2016, 2016, 5845061. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Shin, N.; Song, M.; Kang, U.B.; Yeom, J.; Lee, C.; Ahn, Y.H.; Yoo, J.S.; Paik, Y.K.; Kim, H. Analysis of nuclear high mobility group box 1 (HMGB1)-binding proteins in colon cancer cells: Clustering with proteins involved in secretion and extranuclear function. J. Proteome Res. 2010, 9, 4661–4670. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ma, T.; Yang, S.; Xia, M.; Xu, J.; An, H.; Yang, Y.; Li, S. High-mobility group A1 proteins enhance the expression of the oncogenic miR-222 in lung cancer cells. Mol. Cell. Biochem. 2011, 357, 363–371. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zóia, M.A.P.; Azevedo, F.V.P.; Vecchi, L.; Mota, S.T.S.; Rodovalho, V.d.R.; Cordeiro, A.O.; Correia, L.I.V.; Silva, A.C.A.; Ávila, V.d.M.R.; Araújo, T.G.d.; et al. Inhibition of Triple-Negative Breast Cancer Cell Aggressiveness by Cathepsin D Blockage: Role of Annexin A1. Int. J. Mol. Sci. 2019, 20, 1337. https://doi.org/10.3390/ijms20061337
Zóia MAP, Azevedo FVP, Vecchi L, Mota STS, Rodovalho VdR, Cordeiro AO, Correia LIV, Silva ACA, Ávila VdMR, Araújo TGd, et al. Inhibition of Triple-Negative Breast Cancer Cell Aggressiveness by Cathepsin D Blockage: Role of Annexin A1. International Journal of Molecular Sciences. 2019; 20(6):1337. https://doi.org/10.3390/ijms20061337
Chicago/Turabian StyleZóia, Mariana Alves Pereira, Fernanda Van Petten Azevedo, Lara Vecchi, Sara Teixeira Soares Mota, Vinícius de Rezende Rodovalho, Antonielle Oliveira Cordeiro, Lucas Ian Veloso Correia, Anielle Christine Almeida Silva, Veridiana de Melo Rodrigues Ávila, Thaise Gonçalves de Araújo, and et al. 2019. "Inhibition of Triple-Negative Breast Cancer Cell Aggressiveness by Cathepsin D Blockage: Role of Annexin A1" International Journal of Molecular Sciences 20, no. 6: 1337. https://doi.org/10.3390/ijms20061337