Interleukin-1 Receptor Antagonist Modulates Liver Inflammation and Fibrosis in Mice in a Model-Dependent Manner

 , , ,
, , ,
Abstract
1. Introduction
2. Results
2.1. Interleukin-1 Receptor Antagonist and Liver Fibrosis
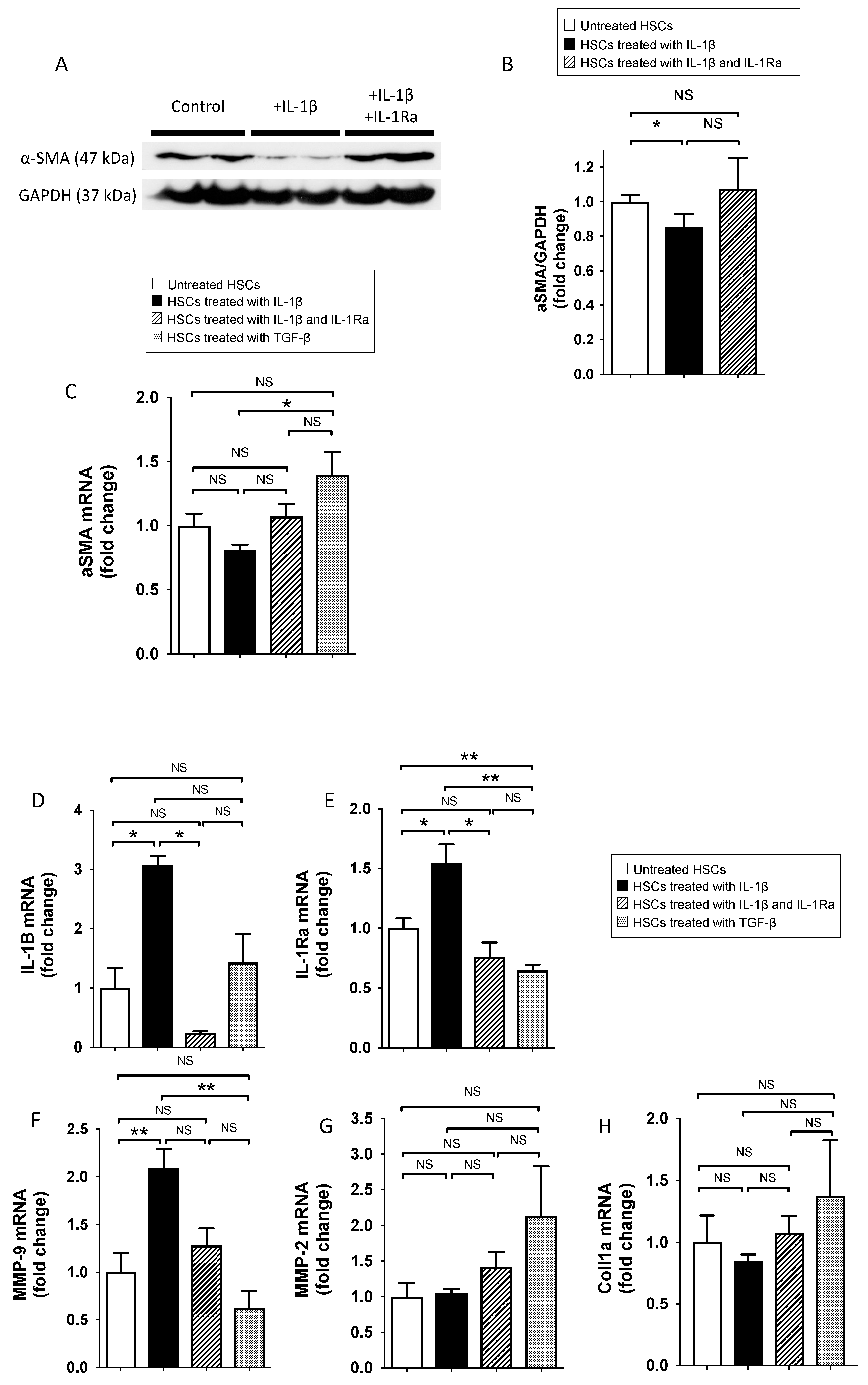
2.1.1. IL-1β Reduce Stellate Cell Activation In Vitro
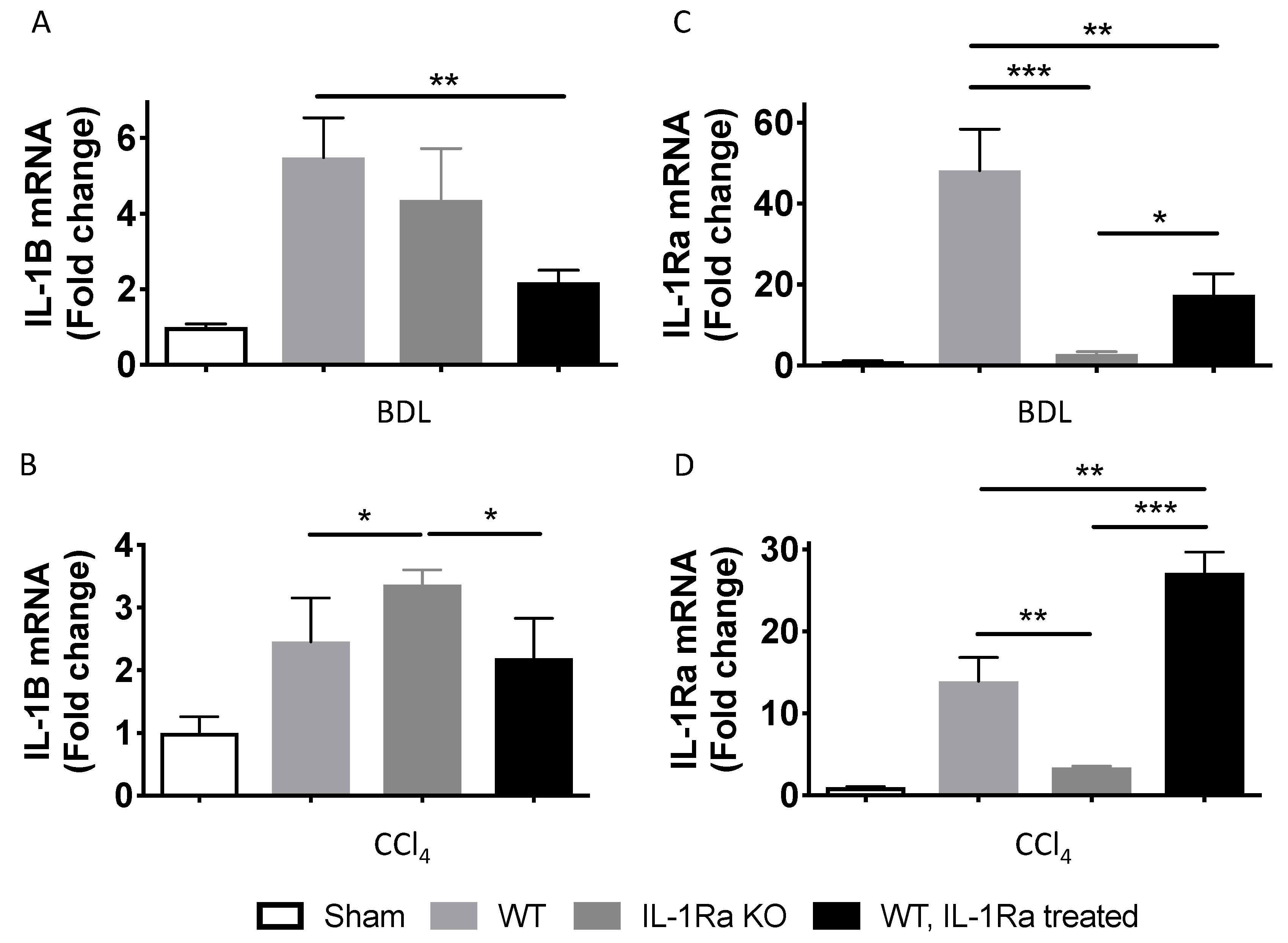
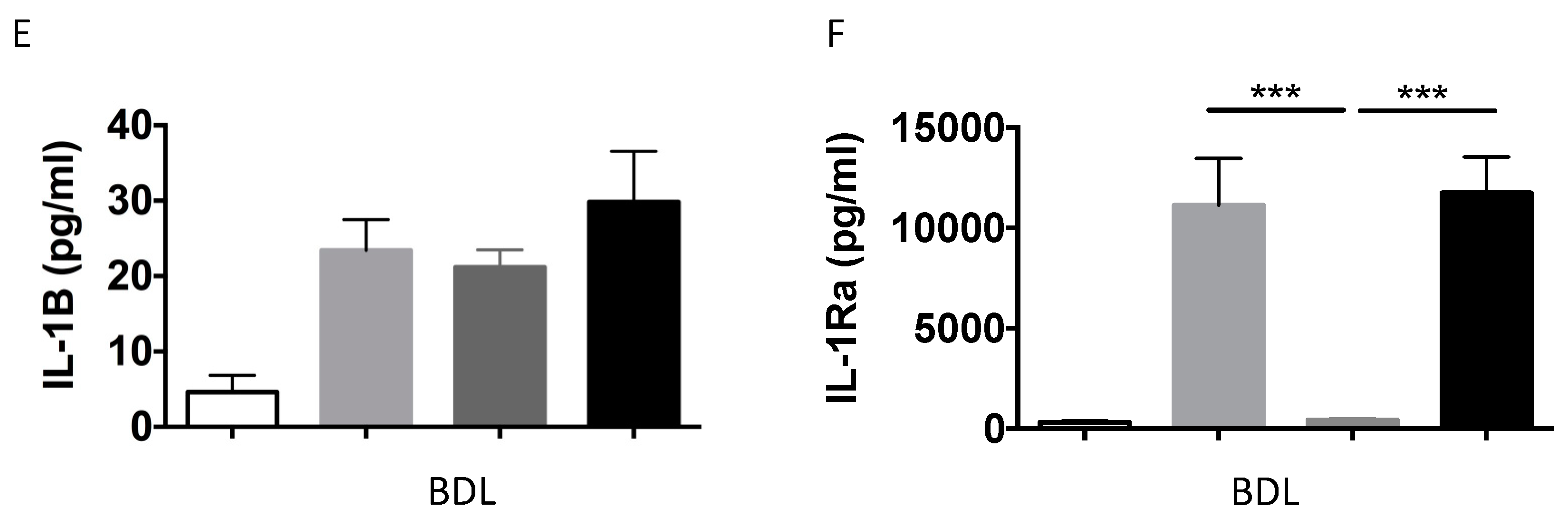
2.1.2. IL-1β and IL-1Ra Expression Levels are Upregulated in Mice after BDL or CCl-4-Induced Liver Fibrosis
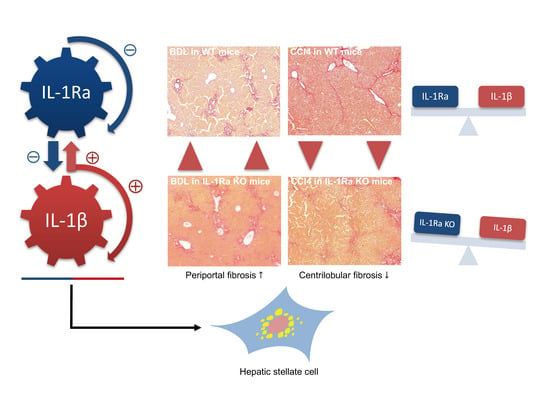
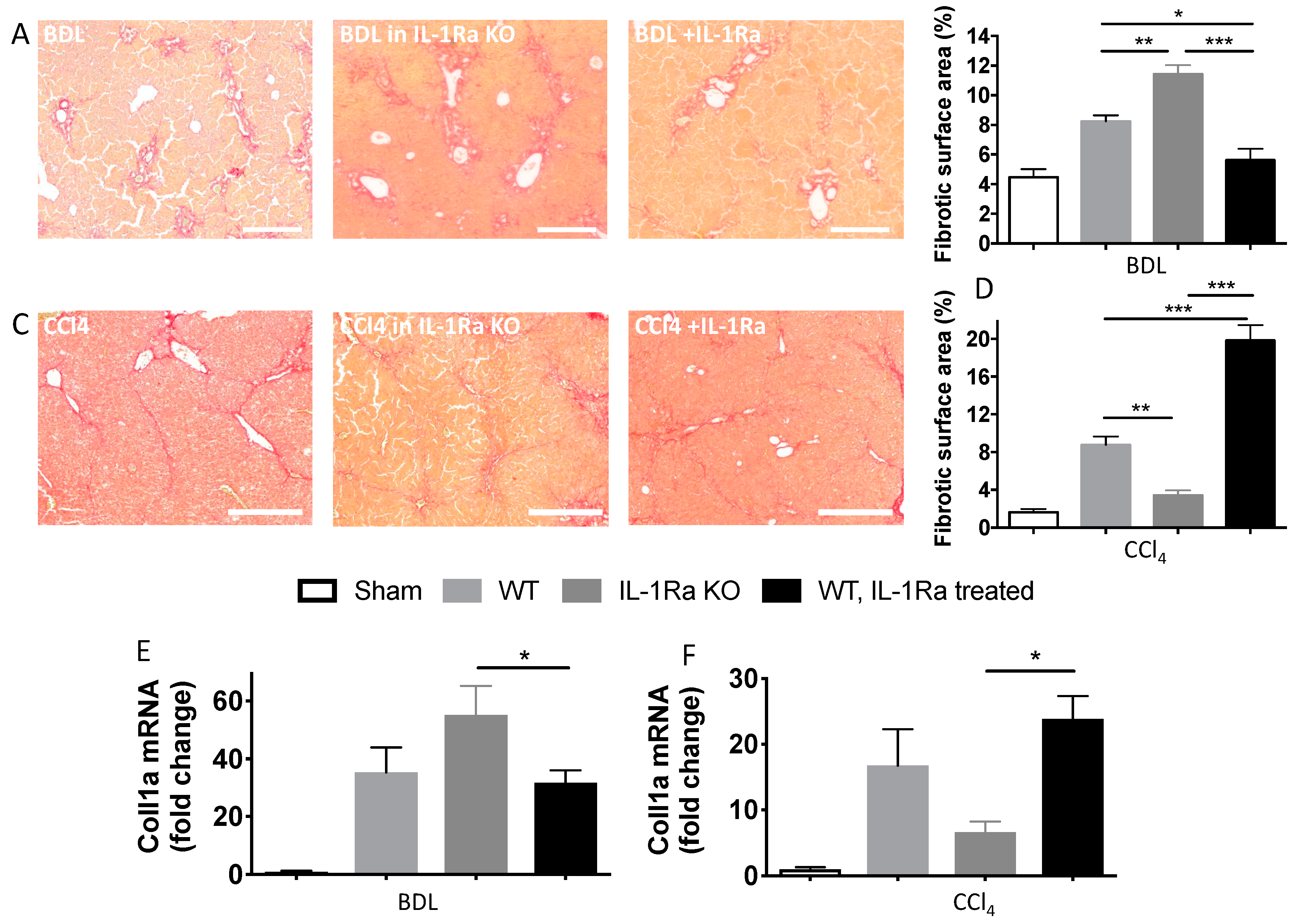
2.1.3. IL-1Ra Has Bivalent Effects on Liver Fibrosis in BDL and CCl-4-Induced Liver Fibrosis
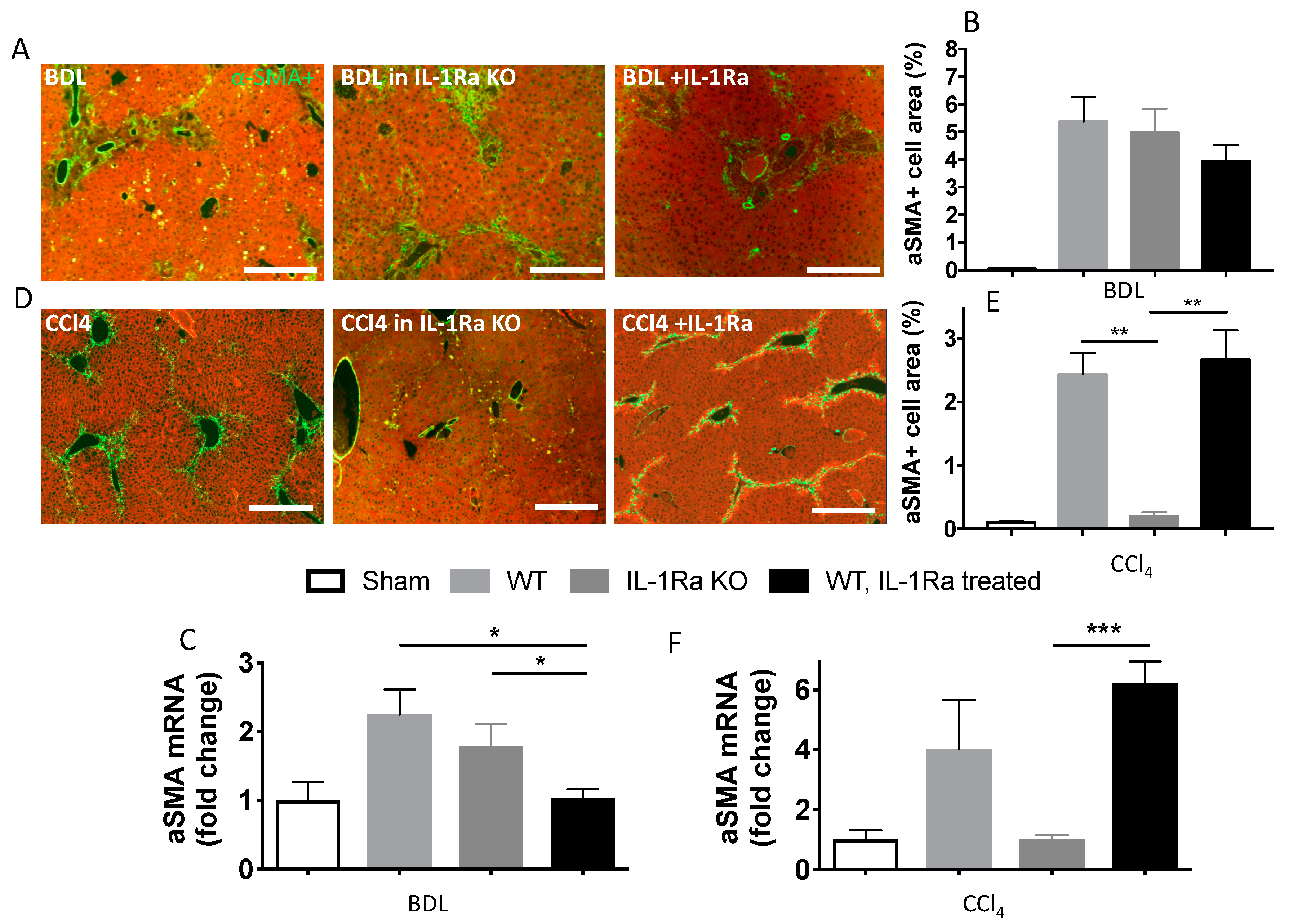
2.1.4. Hepatic Stellate Cell Activation
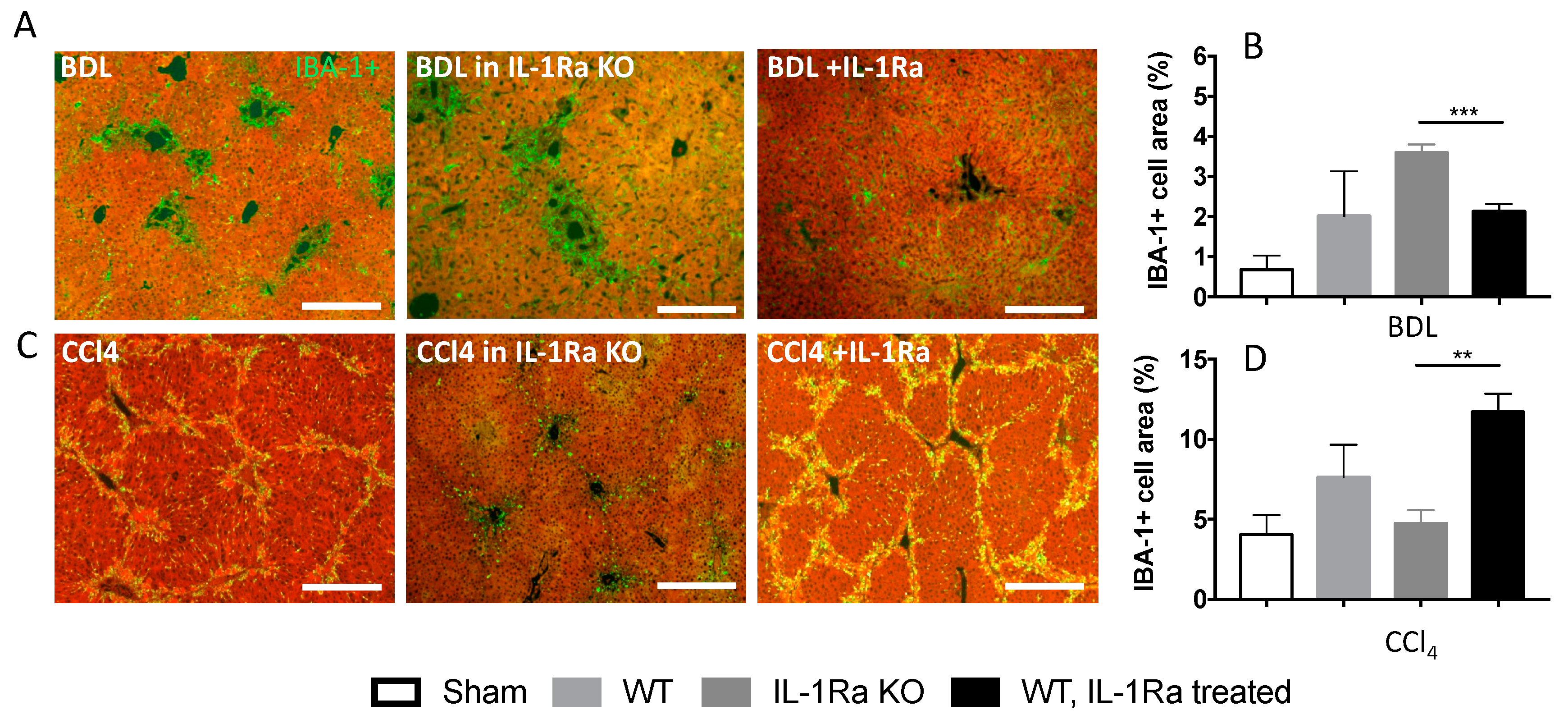
2.1.5. Kupffer Cell Activation
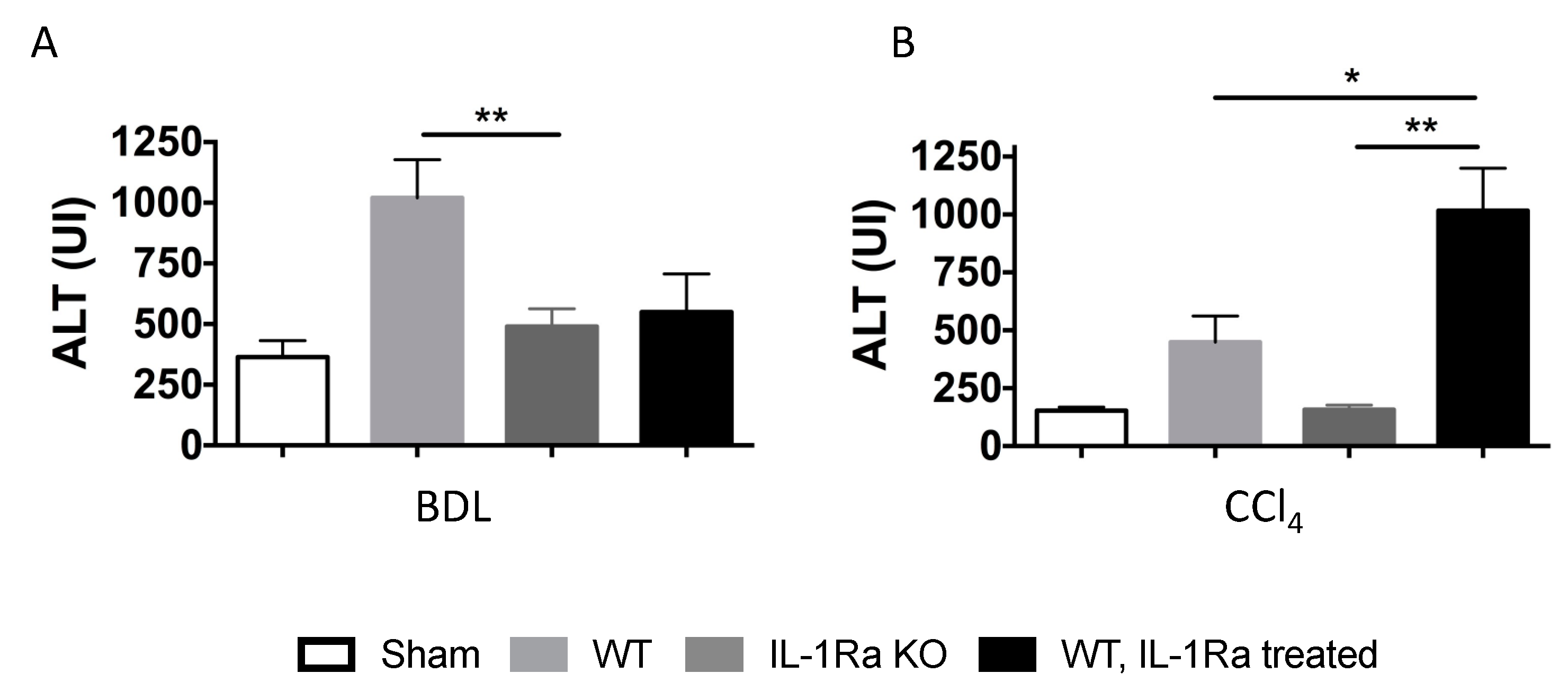
2.1.6. Liver Enzymes
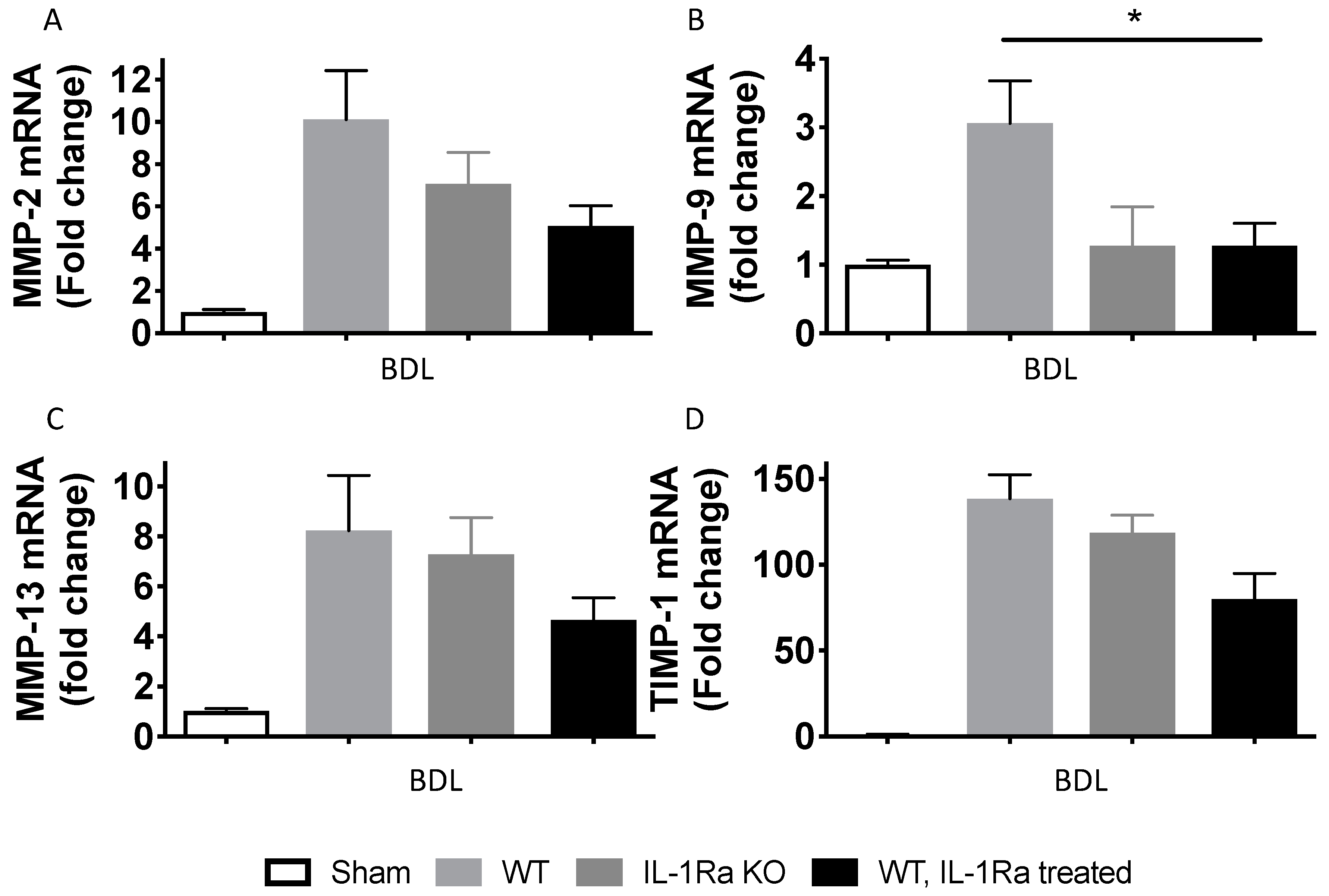
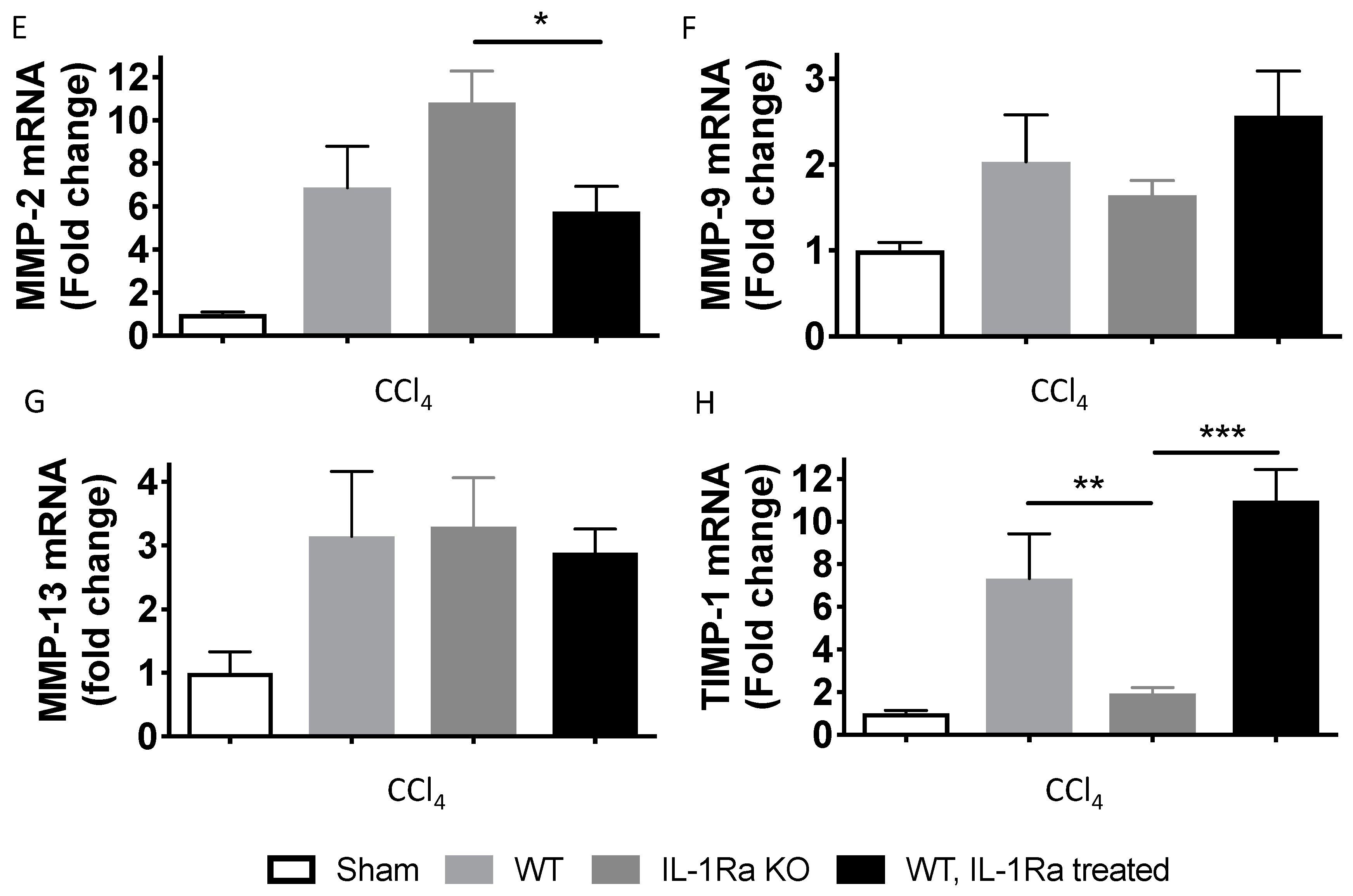
2.1.7. Matrix Metalloproteinases
3. Discussion
4. Materials and Methods
4.1. HSC Isolation and Culture
4.2. Electrophoretic and Immunoblot Analysis
4.3. Animals
4.4. Fibrosis Induction in Mice
4.5. Assessment of Hepatic Fibrosis
4.6. Immunostaining for Alpha-Smooth Muscle Actin (α-SMA) and Ionized Calcium Binding Adaptor Molecule 1 (IBA-1)
4.7. Real-Time Polymerase Chain Reaction (RT-PCR)
4.8. Serum Assays
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ALT | Alanine transaminase |
| BDL | Bile duct ligation |
| CCl-4 | Carbon tetrachloride |
| HSC | Hepatic stellate cells |
| IL-1Ra | Interleukin-1 receptor antagonist |
| IL-1β | Interleukin-1β |
| KO | Knockout |
| MMP | Matrix metalloproteinase |
| MSCs | Mesenchymal stem cells |
| TIMP-1 | Tissue inhibitor of matrix metalloproteinases 1 |
| TGF-β1 | Transforming growth factor-β1 |
| TNF-α | Tumor necrosis factor-α |
| WT | Wild type |
References
- Dinarello, C.A. Interleukin-1beta and the autoinflammatory diseases. New Engl. J. Med. 2009, 360, 2467–2470. [Google Scholar] [CrossRef]
- Gabay, C.; Lamacchia, C.; Palmer, G. IL-1 pathways in inflammation and human diseases. Nat. Rev. Rheumatol. 2010, 6, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Seckinger, P.; Williamson, K.; Balavoine, J.F.; Mach, B.; Mazzei, G.; Shaw, A.; Dayer, J.M. A urine inhibitor of interleukin 1 activity affects both interleukin 1 alpha and 1 beta but not tumor necrosis factor alpha. J. Immunol. 1987, 139, 1541–1545. [Google Scholar]
- Seckinger, P.; Lowenthal, J.W.; Williamson, K.; Dayer, J.M.; MacDonald, H.R. A urine inhibitor of interleukin 1 activity that blocks ligand binding. J. Immunol. 1987, 139, 1546–1549. [Google Scholar]
- Dinarello, C.A. The interleukin-1 family: 10 years of discovery. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1994, 8, 1314–1325. [Google Scholar] [CrossRef]
- Ortiz, L.A.; Dutreil, M.; Fattman, C.; Pandey, A.C.; Torres, G.; Go, K.; Phinney, D.G. Interleukin 1 receptor antagonist mediates the antiinflammatory and antifibrotic effect of mesenchymal stem cells during lung injury. Proc. Natl. Acad. Sci. USA 2007, 104, 11002–11007. [Google Scholar] [CrossRef] [PubMed]
- Meier, R.P.; Muller, Y.D.; Morel, P.; Gonelle-Gispert, C.; Buhler, L.H. Transplantation of mesenchymal stem cells for the treatment of liver diseases, is there enough evidence? Stem Cell Res. 2013, 11, 1348–1364. [Google Scholar] [CrossRef]
- Gieling, R.G.; Wallace, K.; Han, Y.P. Interleukin-1 participates in the progression from liver injury to fibrosis. American journal of physiology. Gastrointest. Liver Physiol. 2009, 296, G1324–G1331. [Google Scholar] [CrossRef] [PubMed]
- McClain, C.J.; Cohen, D.A.; Dinarello, C.A.; Cannon, J.G.; Shedlofsky, S.I.; Kaplan, A.M. Serum interleukin-1 (IL-1) activity in alcoholic hepatitis. Life Sci. 1986, 39, 1479–1485. [Google Scholar] [CrossRef]
- Ludwiczek, O.; Vannier, E.; Moschen, A.; Salazar-Montes, A.; Borggraefe, I.; Gabay, C.; Enrich, B.; Kaser, A.; Siegmund, B.; Dinarello, C.; et al. Impaired counter-regulation of interleukin-1 by the soluble IL-1 receptor type II in patients with chronic liver disease. Scand. J. Gastroenterol. 2008, 43, 1360–1365. [Google Scholar] [CrossRef]
- Tilg, H.; Vogel, W.; Wiedermann, C.J.; Shapiro, L.; Herold, M.; Judmaier, G.; Dinarello, C.A. Circulating interleukin-1 and tumor necrosis factor antagonists in liver disease. Hepatology 1993, 18, 1132–1138. [Google Scholar] [CrossRef]
- Tilg, H.; Wilmer, A.; Vogel, W.; Herold, M.; Nolchen, B.; Judmaier, G.; Huber, C. Serum levels of cytokines in chronic liver diseases. Gastroenterology 1992, 103, 264–274. [Google Scholar] [CrossRef]
- Kamari, Y.; Shaish, A.; Vax, E.; Shemesh, S.; Kandel-Kfir, M.; Arbel, Y.; Olteanu, S.; Barshack, I.; Dotan, S.; Voronov, E.; et al. Lack of interleukin-1alpha or interleukin-1beta inhibits transformation of steatosis to steatohepatitis and liver fibrosis in hypercholesterolemic mice. J. Hepatol. 2011, 55, 1086–1094. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R.; Szabo, G. Interleukin-1 and inflammasomes in alcoholic liver disease/acute alcoholic hepatitis and nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology 2016, 64, 955–965. [Google Scholar] [CrossRef]
- Alcaraz-Quiles, J.; Titos, E.; Casulleras, M.; Pavesi, M.; Lopez-Vicario, C.; Rius, B.; Lopategi, A.; de Gottardi, A.; Graziadei, I.; Gronbaek, H.; et al. Polymorphisms in the IL-1 gene cluster influence systemic inflammation in patients at risk for acute-on-chronic liver failure. Hepatology 2017, 65, 202–216. [Google Scholar] [CrossRef]
- Sgroi, A.; Gonelle-Gispert, C.; Morel, P.; Baertschiger, R.M.; Niclauss, N.; Mentha, G.; Majno, P.; Serre-Beinier, V.; Buhler, L. Interleukin-1 receptor antagonist modulates the early phase of liver regeneration after partial hepatectomy in mice. PloS ONE 2011, 6, e25442. [Google Scholar] [CrossRef]
- Tan, Q.; Hu, J.; Yu, X.; Guan, W.; Lu, H.; Yu, Y.; Yu, Y.; Zang, G.; Tang, Z. The Role of IL-1 Family Members and Kupffer Cells in Liver Regeneration. Biomed. Res. Int. 2016, 2016, 6495793. [Google Scholar] [CrossRef]
- Gehrke, N.; Hovelmeyer, N.; Waisman, A.; Straub, B.K.; Weinmann-Menke, J.; Worns, M.A.; Galle, P.R.; Schattenberg, J.M. Hepatocyte-specific deletion of IL1-RI attenuates liver injury by blocking IL-1 driven autoinflammation. J. Hepatol. 2018, 68, 986–995. [Google Scholar] [CrossRef]
- Meier, R.P.H.; Mahou, R.; Morel, P.; Meyer, J.; Montanari, E.; Muller, Y.D.; Christofilopoulos, P.; Wandrey, C.; Gonelle-Gispert, C.; Bühler, L.H. Microencapsulated human mesenchymal stem cells decrease liver fibrosis in mice. J. Hepatol. 2015, 62, 634–641. [Google Scholar] [CrossRef]
- Lee, K.C.; Lin, H.C.; Huang, Y.H.; Hung, S.C. Allo-transplantation of mesenchymal stem cells attenuates hepatic injury through IL1Ra dependent macrophage switch in a mouse model of liver disease. J. Hepatol. 2015, 63, 1405–1412. [Google Scholar] [CrossRef]
- Sang, J.F.; Shi, X.L.; Han, B.; Huang, X.; Huang, T.; Ren, H.Z.; Ding, Y.T. Combined mesenchymal stem cell transplantation and interleukin-1 receptor antagonism after partial hepatectomy. World J. Gastroenterol. WJG 2016, 22, 4120–4135. [Google Scholar] [CrossRef] [PubMed]
- Mancini, R.; Benedetti, A.; Jezequel, A.M. An interleukin-1 receptor antagonist decreases fibrosis induced by dimethylnitrosamine in rat liver. Virchows Arch. Int. J. Pathol. 1994, 424, 25–31. [Google Scholar] [CrossRef]
- Friedman, S.L. Pathogenesis of Hepatic Fibrosis; Runyon, B.A., Robson, K.M., Eds.; UpToDate Inc.: Waltham, MA, USA; Available online: https://www.uptodate.com (accessed on 1 January 2019).
- Pradere, J.-P.; Kluwe, J.; De Minicis, S.; Jiao, J.-J.; Gwak, G.-Y.; Dapito, D.H.; Jang, M.-K.; Guenther, N.D.; Mederacke, I.; Friedman, R.; et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology 2013, 58, 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.P.; Yan, C.; Zhou, L.; Qin, L.; Tsukamoto, H. A matrix metalloproteinase-9 activation cascade by hepatic stellate cells in trans-differentiation in the three-dimensional extracellular matrix. J. Biol. Chem. 2007, 282, 12928–12939. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.P.; Zhou, L.; Wang, J.; Xiong, S.; Garner, W.L.; French, S.W.; Tsukamoto, H. Essential role of matrix metalloproteinases in interleukin-1-induced myofibroblastic activation of hepatic stellate cell in collagen. J. Biol. Chem. 2004, 279, 4820–4828. [Google Scholar] [CrossRef]
- Imai, Y.; Ibata, I.; Ito, D.; Ohsawa, K.; Kohsaka, S. A novel gene iba1 in the major histocompatibility complex class III region encoding an EF hand protein expressed in a monocytic lineage. Biochem. Biophys. Res. Commun. 1996, 224, 855–862. [Google Scholar] [CrossRef]
- Ito, D.; Imai, Y.; Ohsawa, K.; Nakajima, K.; Fukuuchi, Y.; Kohsaka, S. Microglia-specific localisation of a novel calcium binding protein, Iba1. Brain research. Mol. Brain Res. 1998, 57, 1–9. [Google Scholar] [CrossRef]
- Pihlajamaki, J.; Kuulasmaa, T.; Kaminska, D.; Simonen, M.; Karja, V.; Gronlund, S.; Kakela, P.; Paakkonen, M.; Kainulainen, S.; Punnonen, K.; et al. Serum interleukin 1 receptor antagonist as an independent marker of non-alcoholic steatohepatitis in humans. J. Hepatol. 2012, 56, 663–670. [Google Scholar] [CrossRef]
- Muto, Y.; Nouri-Aria, K.T.; Meager, A.; Alexander, G.J.; Eddleston, A.L.; Williams, R. Enhanced tumour necrosis factor and interleukin-1 in fulminant hepatic failure. Lancet 1988, 2, 72–74. [Google Scholar] [CrossRef]
- Sekiyama, K.D.; Yoshiba, M.; Thomson, A.W. Circulating proinflammatory cytokines (IL-1 beta, TNF-alpha, and IL-6) and IL-1 receptor antagonist (IL-1Ra) in fulminant hepatic failure and acute hepatitis. Clin. Exp. Immunol. 1994, 98, 71–77. [Google Scholar] [CrossRef]
- Gramantieri, L.; Casali, A.; Trere, D.; Gaiani, S.; Piscaglia, F.; Chieco, P.; Cola, B.; Bolondi, L. Imbalance of IL-1 beta and IL-1 receptor antagonist mRNA in liver tissue from hepatitis C virus (HCV)-related chronic hepatitis. Clin. Exp. Immunol. 1999, 115, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Imaeda, A.B.; Watanabe, A.; Sohail, M.A.; Mahmood, S.; Mohamadnejad, M.; Sutterwala, F.S.; Flavell, R.A.; Mehal, W.Z. Acetaminophen-induced hepatotoxicity in mice is dependent on Tlr9 and the Nalp3 inflammasome. J. Clin. Investig. 2009, 119, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Ksontini, R.; Colagiovanni, D.B.; Josephs, M.D.; Edwards, C.K., 3rd; Tannahill, C.L.; Solorzano, C.C.; Norman, J.; Denham, W.; Clare-Salzler, M.; MacKay, S.L.; et al. Disparate roles for TNF-alpha and Fas ligand in concanavalin A-induced hepatitis. J. Immunol. 1998, 160, 4082–4089. [Google Scholar] [PubMed]
- Miura, K.; Kodama, Y.; Inokuchi, S.; Schnabl, B.; Aoyama, T.; Ohnishi, H.; Olefsky, J.M.; Brenner, D.A.; Seki, E. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology 2010, 139, 323–334.e7. [Google Scholar] [CrossRef] [PubMed]
- Mizuhara, H.; O’Neill, E.; Seki, N.; Ogawa, T.; Kusunoki, C.; Otsuka, K.; Satoh, S.; Niwa, M.; Senoh, H.; Fujiwara, H. T cell activation-associated hepatic injury: Mediation by tumor necrosis factors and protection by interleukin 6. J. Exp. Med. 1994, 179, 1529–1537. [Google Scholar] [CrossRef] [PubMed]
- Petrasek, J.; Bala, S.; Csak, T.; Lippai, D.; Kodys, K.; Menashy, V.; Barrieau, M.; Min, S.Y.; Kurt-Jones, E.A.; Szabo, G. IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. J. Clin. Investig. 2012, 122, 3476–3489. [Google Scholar] [CrossRef]
- Petrasek, J.; Dolganiuc, A.; Csak, T.; Kurt-Jones, E.A.; Szabo, G. Type I interferons protect from Toll-like receptor 9-associated liver injury and regulate IL-1 receptor antagonist in mice. Gastroenterology 2011, 140, 697–708.e4. [Google Scholar] [CrossRef]
- Zhu, R.Z.; Xiang, D.; Xie, C.; Li, J.J.; Hu, J.J.; He, H.L.; Yuan, Y.S.; Gao, J.; Han, W.; Yu, Y. Protective effect of recombinant human IL-1Ra on CCl-4-induced acute liver injury in mice. World J. Gastroenterol. WJG 2010, 16, 2771–2779. [Google Scholar] [CrossRef]
- Gabay, C.; Smith, M.F.; Eidlen, D.; Arend, W.P. Interleukin 1 receptor antagonist (IL-1Ra) is an acute-phase protein. J. Clin. Investig. 1997, 99, 2930–2940. [Google Scholar] [CrossRef]
- Lamacchia, C.; Rodriguez, E.; Palmer, G.; Vesin, C.; Seemayer, C.A.; Rubbia-Brandt, L.; Gabay, C. Mice deficient in hepatocyte-specific IL-1Ra show delayed resolution of concanavalin A-induced hepatitis. Eur. J. Immunol. 2012, 42, 1294–1303. [Google Scholar] [CrossRef]
- Yanguas, S.C.; Cogliati, B.; Willebrords, J.; Maes, M.; Colle, I.; van den Bossche, B.; de Oliveira, C.; Andraus, W.; Alves, V.A.F.; Leclercq, I.; et al. Experimental models of liver fibrosis. Arch. Toxicol. 2016, 90, 1025–1048. [Google Scholar] [CrossRef]
- Chobert, M.N.; Couchie, D.; Fourcot, A.; Zafrani, E.S.; Laperche, Y.; Mavier, P.; Brouillet, A. Liver precursor cells increase hepatic fibrosis induced by chronic carbon tetrachloride intoxication in rats. Lab. Investig. 2012, 92, 135–150. [Google Scholar] [CrossRef]
- Friedman, S.L. Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev. 2008, 88, 125–172. [Google Scholar] [CrossRef]
- Matsuoka, M.; Pham, N.T.; Tsukamoto, H. Differential effects of interleukin-1 alpha, tumor necrosis factor alpha, and transforming growth factor beta 1 on cell proliferation and collagen formation by cultured fat-storing cells. Liver 1989, 9, 71–78. [Google Scholar] [CrossRef]
- Tiggelman, A.M.; Boers, W.; Linthorst, C.; Sala, M.; Chamuleau, R.A. Collagen synthesis by human liver (myo)fibroblasts in culture: Evidence for a regulatory role of IL-1 beta, IL-4, TGF beta and IFN gamma. J. Hepatol. 1995, 23, 307–317. [Google Scholar] [CrossRef]
- Higashiyama, R.; Inagaki, Y.; Hong, Y.Y.; Kushida, M.; Nakao, S.; Niioka, M.; Watanabe, T.; Okano, H.; Matsuzaki, Y.; Shiota, G.; et al. Bone marrow-derived cells express matrix metalloproteinases and contribute to regression of liver fibrosis in mice. Hepatology 2007, 45, 213–222. [Google Scholar] [CrossRef]
- Koyama, Y.; Brenner, D.A. Liver inflammation and fibrosis. J. Clin. Investig. 2017, 127, 55–64. [Google Scholar] [CrossRef]
- Gabay, C.; Gigley, J.; Sipe, J.; Arend, W.P.; Fantuzzi, G. Production of IL-1 receptor antagonist by hepatocytes is regulated as an acute-phase protein in vivo. Eur. J. Immunol. 2001, 31, 490–499. [Google Scholar] [CrossRef]
- Donaldson, P.; Agarwal, K.; Craggs, A.; Craig, W.; James, O.; Jones, D. HLA and interleukin 1 gene polymorphisms in primary biliary cirrhosis: Associations with disease progression and disease susceptibility. Gut 2001, 48, 397–402. [Google Scholar] [CrossRef]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef]
- Watashi, K.; Liang, G.; Iwamoto, M.; Marusawa, H.; Uchida, N.; Daito, T.; Kitamura, K.; Muramatsu, M.; Ohashi, H.; Kiyohara, T.; et al. Interleukin-1 and tumor necrosis factor-alpha trigger restriction of hepatitis B virus infection via a cytidine deaminase activation-induced cytidine deaminase (AID). J. Biol. Chem. 2013, 288, 31715–31727. [Google Scholar] [CrossRef]
- Zhu, H.; Liu, C. Interleukin-1 inhibits hepatitis C virus subgenomic RNA replication by activation of extracellular regulated kinase pathway. J. Virol. 2003, 77, 5493–5498. [Google Scholar] [CrossRef]
- Aksentijevich, I.; Masters, S.L.; Ferguson, P.J.; Dancey, P.; Frenkel, J.; van Royen-Kerkhoff, A.; Laxer, R.; Tedgard, U.; Cowen, E.W.; Pham, T.H.; et al. An autoinflammatory disease with deficiency of the interleukin-1-receptor antagonist. New Engl. J. Med. 2009, 360, 2426–2437. [Google Scholar] [CrossRef]
- Garcia-Compean, D.; Jaquez-Quintana, J.O.; Maldonado-Garza, H. Hepatogenous diabetes. Current views of an ancient problem. Ann. Hepatol. 2009, 8, 13–20. [Google Scholar]
- Larsen, C.M.; Faulenbach, M.; Vaag, A.; Volund, A.; Ehses, J.A.; Seifert, B.; Mandrup-Poulsen, T.; Donath, M.Y. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. New Engl. J. Med. 2007, 356, 1517–1526. [Google Scholar] [CrossRef]
- Bataller, R.; Schwabe, R.F.; Choi, Y.H.; Yang, L.; Paik, Y.H.; Lindquist, J.; Qian, T.; Schoonhoven, R.; Hagedorn, C.H.; Lemasters, J.J.; et al. NADPH oxidase signal transduces angiotensin II in hepatic stellate cells and is critical in hepatic fibrosis. J. Clin. Investig. 2003, 112, 1383–1394. [Google Scholar] [CrossRef]
- Clement, S.; Pascarella, S.; Conzelmann, S.; Gonelle-Gispert, C.; Guilloux, K.; Negro, F. The hepatitis C virus core protein indirectly induces alpha-smooth muscle actin expression in hepatic stellate cells via interleukin-8. J. Hepatol. 2010, 52, 635–643. [Google Scholar] [CrossRef]
- Skalli, O.; Ropraz, P.; Trzeciak, A.; Benzonana, G.; Gillessen, D.; Gabbiani, G. A monoclonal antibody against alpha-smooth muscle actin: A new probe for smooth muscle differentiation. J. Cell Biol. 1986, 103, 2787–2796. [Google Scholar] [CrossRef]
- Hirsch, E.; Irikura, V.M.; Paul, S.M.; Hirsh, D. Functions of interleukin 1 receptor antagonist in gene knockout and overproducing mice. Proc. Natl. Acad. Sci. USA 1996, 93, 11008–11013. [Google Scholar] [CrossRef]
- Palmer, G.; Talabot-Ayer, D.; Kaya, G.; Gabay, C. Type I IL-1 receptor mediates IL-1 and intracellular IL-1 receptor antagonist effects in skin inflammation. J. Investig. Dermatol. 2007, 127, 1938–1946. [Google Scholar] [CrossRef]
- Puchtler, H.; Waldrop, F.S.; Valentine, L.S. Polarization microscopic studies of connective tissue stained with picro-sirius red FBA. Beitr. Pathol. 1973, 150, 174–187. [Google Scholar] [CrossRef]
- Vandesompele, J.; De Paepe, A.; Speleman, F. Elimination of primer-dimer artifacts and genomic coamplification using a two-step SYBR green I real-time RT-PCR. Anal. Biochem. 2002, 303, 95–98. [Google Scholar] [CrossRef]
- Carta, S.; Tassi, S.; Pettinati, I.; Delfino, L.; Dinarello, C.A.; Rubartelli, A. The rate of interleukin-1beta secretion in different myeloid cells varies with the extent of redox response to Toll-like receptor triggering. J. Biol. Chem. 2011, 286, 27069–27080. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Primer-Probe Sequence 5′-3′ | |
|---|---|---|
| Forward | Reverse | |
| Mice | ||
| IL-1β | ACT CCT TAG TCC TCG GCC A | TGG TTT CTT GTG ACC CTG AGC |
| IL-1Ra | CTG CAC TTC CAC AGT CCA GA | ATA TGT GAT GCC CTG GTG GT |
| Collagen type I alpha 1 | GCA TGG CCA AGA AGA CAT CC | CCT CGG GTT TCC ACG TCT C |
| MMP-2 | TGG GGG AGA TTC TCA CTT TG | CAT CAC TGC GAC CAG TGT CT |
| MMP-9 | AGT TGC CCC TAC TGG AAG GT | GTG GAT AGC TCG GTG GTG TT |
| MMP-13 | AGT TGA CAG GCT CCG AGA AA | AGT TCG TTT GGG ACC ATT TG |
| TIMP-1 | GCA TCT CTG GCA TCT GGC ATC | GAA GGC TGT CTG TGG GTG GG1 [8] |
| Human | ||
| IL-1β | TCC AGG GAC AGG ATA TGG AG | TCT TTC AAC ACG CAG GAC AG [64] |
| IL-1Ra | AAG ACC AGT CCA TGA GGG AG | CTC CCC GAA AGA ACA TAA TCT C |
| α-SMA | CAT CTA TGA GGG CTA TGC CTT G | GTG AAG GAA TAG CCA CGC TC |
| MMP-9 | TTG ACA GCG ACA AGA AGT GG | GCC ATT CAC GTC GTC CTT AT |
| MMP-2 | Ref: QT00088396 | Ref: QT00088396 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meier, R.P.H.; Meyer, J.; Montanari, E.; Lacotte, S.; Balaphas, A.; Muller, Y.D.; Clément, S.; Negro, F.; Toso, C.; Morel, P.; et al. Interleukin-1 Receptor Antagonist Modulates Liver Inflammation and Fibrosis in Mice in a Model-Dependent Manner. Int. J. Mol. Sci. 2019, 20, 1295. https://doi.org/10.3390/ijms20061295
Meier RPH, Meyer J, Montanari E, Lacotte S, Balaphas A, Muller YD, Clément S, Negro F, Toso C, Morel P, et al. Interleukin-1 Receptor Antagonist Modulates Liver Inflammation and Fibrosis in Mice in a Model-Dependent Manner. International Journal of Molecular Sciences. 2019; 20(6):1295. https://doi.org/10.3390/ijms20061295
Chicago/Turabian StyleMeier, Raphael P. H., Jeremy Meyer, Elisa Montanari, Stephanie Lacotte, Alexandre Balaphas, Yannick D. Muller, Sophie Clément, Francesco Negro, Christian Toso, Philippe Morel, and et al. 2019. "Interleukin-1 Receptor Antagonist Modulates Liver Inflammation and Fibrosis in Mice in a Model-Dependent Manner" International Journal of Molecular Sciences 20, no. 6: 1295. https://doi.org/10.3390/ijms20061295
APA StyleMeier, R. P. H., Meyer, J., Montanari, E., Lacotte, S., Balaphas, A., Muller, Y. D., Clément, S., Negro, F., Toso, C., Morel, P., & Buhler, L. H. (2019). Interleukin-1 Receptor Antagonist Modulates Liver Inflammation and Fibrosis in Mice in a Model-Dependent Manner. International Journal of Molecular Sciences, 20(6), 1295. https://doi.org/10.3390/ijms20061295