Secondary Bile Acids and Short Chain Fatty Acids in the Colon: A Focus on Colonic Microbiome, Cell Proliferation, Inflammation, and Cancer


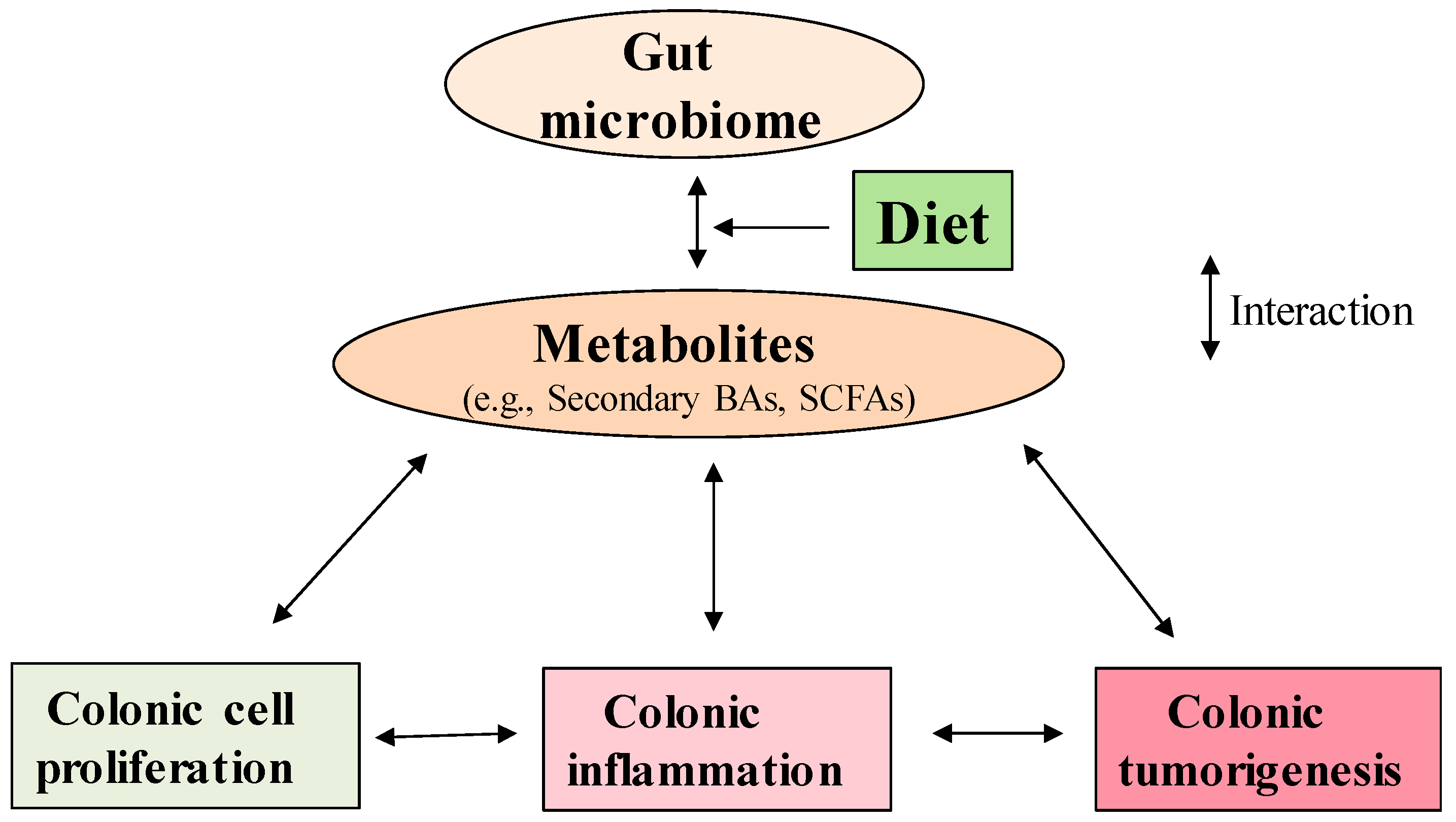{kind=link}
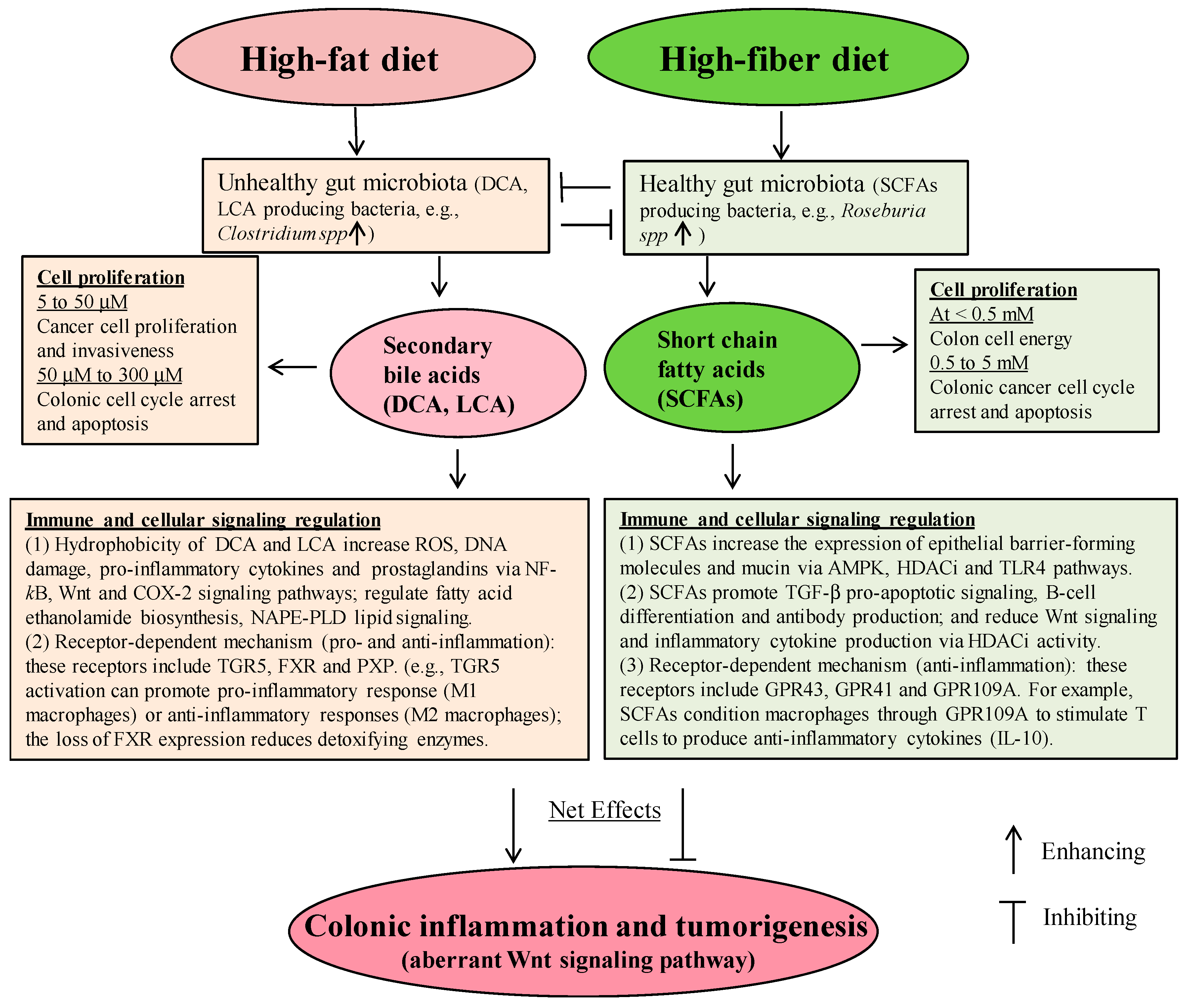{kind=link}
Abstract
:1. Introduction
2. Secondary BAs, SCFAs, and Microbiome
2.1. Secondary BAs
2.2. SCFAs
3. The Impact of Secondary BAs and SCFAs on Colonic Cell Proliferation
3.1. Secondary BAs and Colonic Cell Proliferation
3.2. SCFAs and Colonic Cell Proliferation
4. The Role of Secondary BAs and SCFAs in Colonic Inflammation
4.1. Secondary BAs and Colonic Inflammation
4.2. SCFAs and Colonic Inflammation
5. Secondary BAs, SCFAs, Oncomicrobes, and Colon Cancer
5.1. Secondary BAs and Colon Cancer
5.2. SCFAs and Colon Cancer
5.3. Oncomicrobes and Colon Cancer
6. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- American Cancer Society. Cancer Facts & Figures; American Cancer Society: Atlanta, GA, USA, 2016. [Google Scholar]
- Simon, K. Colorectal cancer development and advances in screening. Clin. Interv. Aging 2016, 11, 967–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Fedewa, S.A.; Ahnen, D.J.; Meester, R.G.S.; Barzi, A.; Jemal, A. Colorectal cancer statistics, 2017. CA Cancer J. Clin. 2017, 67, 177–193. [Google Scholar] [CrossRef] [PubMed]
- Raatz, S.K.; Conrad, Z.; Johnson, L.K.; Picklo, M.J.; Jahns, L. Relationship of the Reported Intakes of Fat and Fatty Acids to Body Weight in US Adults. Nutrients 2017, 9, 438. [Google Scholar] [CrossRef] [PubMed]
- Zacek, P.; Bukowski, M.; Mehus, A.; Johnson, L.; Zeng, H.; Raatz, S.; Idso, J.P.; Picklo, M. Dietary saturated fatty acid type impacts obesity-induced metabolic dysfunction and plasma lipidomic signatures in mice. J. Nutr. Biochem. 2019, 64, 32–44. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, A.M.; Burrington, C.M.; Gillaspie, E.A.; Lynch, D.T.; Horsman, M.J.; Greene, M.W. High-fat Western diet-induced obesity contributes to increased tumor growth in mouse models of human colon cancer. Nutr. Res. 2016, 36, 1325–1334. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Lazarova, D.L.; Bordonaro, M. Mechanisms linking dietary fiber, gut microbiota and colon cancer prevention. World J. Gastrointest. Oncol. 2014, 6, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, C.; Bernstein, H.; Garewal, H.; Dinning, P.; Jabi, R.; Sampliner, R.E.; McCuskey, M.K.; Panda, M.; Roe, D.J.; L’Heureux, L.; et al. A bile acid-induced apoptosis assay for colon cancer risk and associated quality control studies. Cancer Res. 1999, 59, 2353–2357. [Google Scholar] [PubMed]
- Weinberg, D.S.; Strom, B.L. Screening for colon cancer: A reviewof current and future strategies. Semin. Oncol. 1995, 22, 433–447. [Google Scholar] [PubMed]
- Zeng, H.; Claycombe, K.J.; Reindl, K.M. Butyrate and deoxycholic acid play common and distinct roles in HCT116 human colon cell proliferation. J. Nutr. Biochem. 2015, 26, 1022–1028. [Google Scholar] [CrossRef] [PubMed]
- Farhana, L.; Nangia-Makker, P.; Arbit, E.; Shango, K.; Sarkar, S.; Mahmud, H.; Hadden, T.; Yu, Y.; Majumdar, A.P. Bile acid: A potential inducer of colon cancer stem cells. Stem. Cell Res. Ther. 2016, 7, 181. [Google Scholar] [CrossRef] [PubMed]
- Stadler, J.; Stern, H.S.; Yeung, K.S.; McGuire, V.; Furrer, R.; Marcon, N.; Bruce, W.R. Effect of high fat consumption on cell proliferation activity of colorectal mucosa and on soluble faecal bile acids. Gut 1988, 29, 1326–1331. [Google Scholar] [CrossRef] [PubMed]
- Akin, H.; Tozun, N. Diet, microbiota, and colorectal cancer. J. Clin. Gastroenterol. 2014, 48 (Suppl. 1), S67–S69. [Google Scholar] [CrossRef] [PubMed]
- McNabney, S.M.; Henagan, T.M. Short Chain Fatty Acids in the Colon and Peripheral Tissues: A Focus on Butyrate, Colon Cancer, Obesity and Insulin Resistance. Nutrients 2017, 9, 1348. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Chassaing, B.; Singh, V.; Pellizzon, M.; Ricci, M.; Fythe, M.D.; Kumar, M.V.; Gewirtz, A.T. Fiber-Mediated Nourishment of Gut Microbiota Protects against Diet-Induced Obesity by Restoring IL-22-Mediated Colonic Health. Cell Host Microbe 2018, 23, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Makki, K.; Deehan, E.C.; Walter, J.; Backhed, F. The Impact of Dietary Fiber on Gut Microbiota in Host Health and Disease. Cell Host Microbe 2018, 23, 705–715. [Google Scholar] [CrossRef] [PubMed]
- Slavin, J.L. Position of the American Dietetic Association: Health implications of dietary fiber. J. Am. Diet. Assoc. 2008, 108, 1716–1731. [Google Scholar] [PubMed]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Floch, M.H. Intestinal microecology in health and wellness. J. Clin. Gastroenterol. 2011, 45, S108–S110. [Google Scholar] [CrossRef] [PubMed]
- Staley, C.; Weingarden, A.R.; Khoruts, A.; Sadowsky, M.J. Interaction of gut microbiota with bile acid metabolism and its influence on disease states. Appl. Microbiol. Biotechnol. 2017, 101, 47–64. [Google Scholar] [CrossRef] [PubMed]
- Dethlefsen, L.; Relman, D.A. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. 1), 4554–4561. [Google Scholar] [CrossRef]
- Schroeder, B.O.; Backhed, F. Signals from the gut microbiota to distant organs in physiology and disease. Nat. Med. 2016, 22, 1079–1089. [Google Scholar] [CrossRef] [PubMed]
- Walter, J.; Britton, R.A.; Roos, S. Host-microbial symbiosis in the vertebrate gastrointestinal tract and the Lactobacillus reuteri paradigm. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. 1), 4645–4652. [Google Scholar] [CrossRef]
- Hand, T.W.; Vujkovic-Cvijin, I.; Ridaura, V.K.; Belkaid, Y. Linking the Microbiota, Chronic Disease, and the Immune System. Trends Endocrinol. Metab. 2016, 27, 831–843. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.Y. Bile acids: Regulation of synthesis. J. Lipid Res. 2009, 50, 1955–1966. [Google Scholar] [CrossRef] [PubMed]
- Murakami, Y.; Tanabe, S.; Suzuki, T. High-fat Diet-induced Intestinal Hyperpermeability is Associated with Increased Bile Acids in the Large Intestine of Mice. J. Food Sci. 2016, 81, H216–H222. [Google Scholar] [CrossRef] [PubMed]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B. Bile salt biotransformations by human intestinal bacteria. J. Lipid Res. 2006, 47, 241–259. [Google Scholar] [CrossRef] [PubMed]
- Ridlon, J.M.; Harris, S.C.; Bhowmik, S.; Kang, D.J.; Hylemon, P.B. Consequences of bile salt biotransformations by intestinal bacteria. Gut Microbes 2016, 7, 22–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Islam, K.B.; Fukiya, S.; Hagio, M.; Fujii, N.; Ishizuka, S.; Ooka, T.; Ogura, Y.; Hayashi, T.; Yokota, A. Bile acid is a host factor that regulates the composition of the cecal microbiota in rats. Gastroenterology 2011, 141, 1773–1781. [Google Scholar] [CrossRef] [PubMed]
- Devkota, S.; Wang, Y.; Musch, M.W.; Leone, V.; Fehlner-Peach, H.; Nadimpalli, A.; Antonopoulos, D.A.; Jabri, B.; Chang, E.B. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10−/− mice. Nature 2012, 487, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Papathanasopoulos, A.; Camilleri, M. Dietary fiber supplements: Effects in obesity and metabolic syndrome and relationship to gastrointestinal functions. Gastroenterology 2010, 138, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Raninen, K.; Lappi, J.; Mykkanen, H.; Poutanen, K. Dietary fiber type reflects physiological functionality: Comparison of grain fiber, inulin, and polydextrose. Nutr. Rev. 2011, 69, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.H. Human Colonic Bacteria: Role in Nutrition, Physiology and Health; Gibson, G.R., Macfarlane, G.T., Eds.; CRC Press: Boca Raton, FL, USA, 1995; pp. 101–130. [Google Scholar]
- Louis, P.; Flint, H.J. Formation of propionate and butyrate by the human colonic microbiota. Environ. Microbiol. 2017, 19, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.H.; Pomare, E.W.; Branch, W.J.; Naylor, C.P.; Macfarlane, G.T. Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut 1987, 28, 1221–1227. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.L.; Wolin, M.J. Pathways of acetate, propionate, and butyrate formation by the human fecal microbial flora. Appl. Environ. Microbiol. 1996, 62, 1589–1592. [Google Scholar] [PubMed]
- Louis, P.; Hold, G.L.; Flint, H.J. The gut microbiota, bacterial metabolites and colorectal cancer. Nat. Rev. Microbiol. 2014, 12, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Koh, A.; De Vadder, F.; Kovatcheva-Datchary, P.; Backhed, F. From Dietary Fiber to Host Physiology: Short-Chain Fatty Acids as Key Bacterial Metabolites. Cell 2016, 165, 1332–1345. [Google Scholar] [CrossRef] [PubMed]
- Rey, F.E.; Faith, J.J.; Bain, J.; Muehlbauer, M.J.; Stevens, R.D.; Newgard, C.B.; Gordon, J.I. Dissecting the in vivo metabolic potential of two human gut acetogens. J. Biol. Chem. 2010, 285, 22082–22090. [Google Scholar] [CrossRef] [PubMed]
- Scott, K.P.; Martin, J.C.; Campbell, G.; Mayer, C.D.; Flint, H.J. Whole-genome transcription profiling reveals genes up-regulated by growth on fucose in the human gut bacterium “Roseburia inulinivorans”. J. Bacteriol. 2006, 188, 4340–4349. [Google Scholar] [CrossRef] [PubMed]
- Barcenilla, A.; Pryde, S.E.; Martin, J.C.; Duncan, S.H.; Stewart, C.S.; Henderson, C.; Flint, H.J. Phylogenetic relationships of butyrate-producing bacteria from the human gut. Appl. Environ. Microbiol. 2000, 66, 1654–1661. [Google Scholar] [CrossRef] [PubMed]
- Gunness, P.; Michiels, J.; Vanhaecke, L.; De Smet, S.; Kravchuk, O.; Van de Meene, A.; Gidley, M.J. Reduction in circulating bile acid and restricted diffusion across the intestinal epithelium are associated with a decrease in blood cholesterol in the presence of oat beta-glucan. FASEB J. 2016, 30, 4227–4238. [Google Scholar] [CrossRef] [PubMed]
- Gunness, P.; Williams, B.A.; Gerrits, W.J.; Bird, A.R.; Kravchuk, O.; Gidley, M.J. Circulating triglycerides and bile acids are reduced by a soluble wheat arabinoxylan via modulation of bile concentration and lipid digestion rates in a pig model. Mol. Nutr. Food Res. 2016, 60, 642–651. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, R. Role of dietary factors in cell replication and colon cancer. Am. J. Clin. Nutr. 1988, 48, 775–779. [Google Scholar] [CrossRef] [PubMed]
- Boman, B.M.; Fields, J.Z. An APC:WNT Counter-Current-Like Mechanism Regulates Cell Division Along the Human Colonic Crypt Axis: A Mechanism That Explains How APC Mutations Induce Proliferative Abnormalities That Drive Colon Cancer Development. Front. Oncol. 2013, 3, 244. [Google Scholar] [CrossRef] [PubMed]
- Lipkin, M.; Uehara, K.; Winawer, S.; Sanchez, A.; Bauer, C.; Phillips, R.; Lynch, H.T.; Blattner, W.A.; Fraumeni, J.F., Jr. Seventh-Day Adventist vegetarians have a quiescent proliferative activity in colonic mucosa. Cancer Lett. 1985, 26, 139–144. [Google Scholar] [CrossRef]
- Dermadi, D.; Valo, S.; Ollila, S.; Soliymani, R.; Sipari, N.; Pussila, M.; Sarantaus, L.; Linden, J.; Baumann, M.; Nystrom, M. Western Diet Deregulates Bile Acid Homeostasis, Cell Proliferation, and Tumorigenesis in Colon. Cancer Res. 2017, 77, 3352–3363. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Ishaq, S.L.; Zhao, F.Q.; Wright, A.G. Colonic inflammation accompanies an increase of beta-catenin signaling and Lachnospiraceae/Streptococcaceae bacteria in the hind gut of high-fat diet-fed mice. J. Nutr. Biochem. 2016, 35, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Ajouz, H.; Mukherji, D.; Shamseddine, A. Secondary bile acids: An underrecognized cause of colon cancer. World J. Surg. Oncol. 2014, 12, 164. [Google Scholar] [CrossRef] [PubMed]
- Nagengast, F.M.; Grubben, M.J.; van Munster, I.P. Role of bile acids in colorectal carcinogenesis. Eur. J. Cancer 1995, 31A, 1067–1070. [Google Scholar] [CrossRef]
- Hofmann, A.F.; Cravetto, C.; Molino, G.; Belforte, G.; Bona, B. Simulation of the metabolism and enterohepatic circulation of endogenous deoxycholic acid in humans using a physiologic pharmacokinetic model for bile acid metabolism. Gastroenterology 1987, 93, 693–709. [Google Scholar] [CrossRef]
- Degirolamo, C.; Modica, S.; Palasciano, G.; Moschetta, A. Bile acids and colon cancer: Solving the puzzle with nuclear receptors. Trends Mol. Med. 2011, 17, 564–572. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, C.; Holubec, H.; Bhattacharyya, A.K.; Nguyen, H.; Payne, C.M.; Zaitlin, B.; Bernstein, H. Carcinogenicity of deoxycholate, a secondary bile acid. Arch. Toxicol. 2011, 85, 863–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridlon, J.M.; Wolf, P.G.; Gaskins, H.R. Taurocholic acid metabolism by gut microbes and colon cancer. Gut Microbes 2016, 7, 201–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pai, R.; Tarnawski, A.S.; Tran, T. Deoxycholic acid activates beta-catenin signaling pathway and increases colon cell cancer growth and invasiveness. Mol. Biol. Cell 2004, 15, 2156–2163. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.; Raufman, J.P. Bile acid-induced proliferation of a human colon cancer cell line is mediated by transactivation of epidermal growth factor receptors. Biochem. Pharmacol. 2005, 70, 1035–1047. [Google Scholar] [CrossRef] [PubMed]
- Milovic, V.; Stein, J.; Odera, G.; Gilani, S.; Murphy, G.M. Low-dose deoxycholic acid stimulates putrescine uptake in colon cancer cells (Caco-2). Cancer Lett. 2000, 154, 195–200. [Google Scholar] [CrossRef]
- Brown, J.R.; DuBois, R.N. COX-2: A molecular target for colorectal cancer prevention. J. Clin. Oncol. 2005, 23, 2840–2855. [Google Scholar] [CrossRef] [PubMed]
- Amaral, J.D.; Viana, R.J.; Ramalho, R.M.; Steer, C.J.; Rodrigues, C.M. Bile acids: Regulation of apoptosis by ursodeoxycholic acid. J. Lipid Res. 2009, 50, 1721–1734. [Google Scholar] [CrossRef] [PubMed]
- Ha, Y.H.; Park, D.G. Effects of DCA on Cell Cycle Proteins in Colonocytes. J. Korean Soc. Coloproctol. 2010, 26, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Qiao, D.; Gaitonde, S.V.; Qi, W.; Martinez, J.D. Deoxycholic acid suppresses p53 by stimulating proteasome-mediated p53 protein degradation. Carcinogenesis 2001, 22, 957–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hylemon, P.B.; Zhou, H.; Pandak, W.M.; Ren, S.; Gil, G.; Dent, P. Bile acids as regulatory molecules. J. Lipid Res. 2009, 50, 1509–1520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernstein, H.; Bernstein, C.; Payne, C.M.; Dvorakova, K.; Garewal, H. Bile acids as carcinogens in human gastrointestinal cancers. Mutat. Res. 2005, 589, 47–65. [Google Scholar] [CrossRef] [PubMed]
- Perrin, P.; Pierre, F.; Patry, Y.; Champ, M.; Berreur, M.; Pradal, G.; Bornet, F.; Meflah, K.; Menanteau, J. Only fibres promoting a stable butyrate producing colonic ecosystem decrease the rate of aberrant crypt foci in rats. Gut 2001, 48, 53–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwiertz, A.; Taras, D.; Schafer, K.; Beijer, S.; Bos, N.A.; Donus, C.; Hardt, P.D. Microbiota and SCFA in lean and overweight healthy subjects. Obesity 2010, 18, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Van der Beek, C.M.; Canfora, E.E.; Kip, A.M.; Gorissen, S.H.M.; Olde Damink, S.W.M.; van Eijk, H.M.; Holst, J.J.; Blaak, E.E.; Dejong, C.H.C.; Lenaerts, K. The prebiotic inulin improves substrate metabolism and promotes short-chain fatty acid production in overweight to obese men. Metabolism 2018, 87, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, G.T.; Macfarlane, S. Fermentation in the human large intestine: Its physiologic consequences and the potential contribution of prebiotics. J. Clin. Gastroenterol. 2011, 45, S120–S127. [Google Scholar] [CrossRef] [PubMed]
- Riviere, A.; Selak, M.; Lantin, D.; Leroy, F.; De Vuyst, L. Bifidobacteria and Butyrate-Producing Colon Bacteria: Importance and Strategies for Their Stimulation in the Human Gut. Front. Microbiol. 2016, 7, 979. [Google Scholar] [CrossRef] [PubMed]
- Roediger, W.E. Role of anaerobic bacteria in the metabolic welfare of the colonic mucosa in man. Gut 1980, 21, 793–798. [Google Scholar] [CrossRef] [PubMed]
- Donohoe, D.R.; Garge, N.; Zhang, X.; Sun, W.; O’Connell, T.M.; Bunger, M.K.; Bultman, S.J. The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab. 2011, 13, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Donohoe, D.R.; Wali, A.; Brylawski, B.P.; Bultman, S.J. Microbial regulation of glucose metabolism and cell-cycle progression in mammalian colonocytes. PLoS ONE 2012, 7, e46589. [Google Scholar] [CrossRef] [PubMed]
- Hooper, L.V.; Midtvedt, T.; Gordon, J.I. How host-microbial interactions shape the nutrient environment of the mammalian intestine. Annu. Rev. Nutr. 2002, 22, 283–307. [Google Scholar] [CrossRef] [PubMed]
- Perego, S.; Sansoni, V.; Banfi, G.; Lombardi, G. Sodium butyrate has anti-proliferative, pro-differentiating, and immunomodulatory effects in osteosarcoma cells and counteracts the TNFalpha-induced low-grade inflammation. Int. J. Immunopathol. Pharmacol. 2018, 32. [Google Scholar] [CrossRef]
- Fung, K.Y.; Cosgrove, L.; Lockett, T.; Head, R.; Topping, D.L. A review of the potential mechanisms for the lowering of colorectal oncogenesis by butyrate. Br. J. Nutr. 2012, 108, 820–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, H.; Taussig, D.P.; Cheng, W.H.; Johnson, L.K.; Hakkak, R. Butyrate Inhibits Cancerous HCT116 Colon Cell Proliferation but to a Lesser Extent in Noncancerous NCM460 Colon Cells. Nutrients 2017, 9, 25. [Google Scholar] [CrossRef] [PubMed]
- Blottiere, H.M.; Buecher, B.; Galmiche, J.P.; Cherbut, C. Molecular analysis of the effect of short-chain fatty acids on intestinal cell proliferation. Proc. Nutr. Soc. 2003, 62, 101–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, H.; Briske-Anderson, M. Prolonged butyrate treatment inhibits the migration and invasion potential of HT1080 tumor cells. J. Nutr. 2005, 135, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Emenaker, N.J.; Calaf, G.M.; Cox, D.; Basson, M.D.; Qureshi, N. Short-chain fatty acids inhibit invasive human colon cancer by modulating uPA, TIMP-1, TIMP-2, mutant p53, Bcl-2, Bax, p21 and PCNA protein expression in an in vitro cell culture model. J. Nutr. 2001, 131, 3041S–3046S. [Google Scholar] [CrossRef] [PubMed]
- Akbar, A.N.; Salmon, M. Cellular environments and apoptosis: Tissue microenvironments control activated T-cell death. Immunol. Today 1997, 18, 72–76. [Google Scholar] [CrossRef]
- Jain, R.; Austin Pickens, C.; Fenton, J.I. The role of the lipidome in obesity-mediated colon cancer risk. J. Nutr. Biochem. 2018, 59, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, H.; Bording-Jorgensen, M.; Dijk, S.; Wine, E. The Complex Interplay between Chronic Inflammation, the Microbiome, and Cancer: Understanding Disease Progression and What We Can Do to Prevent It. Cancers 2018, 10, 83. [Google Scholar] [CrossRef] [PubMed]
- Hofmanova, J.; Strakova, N.; Vaculova, A.H.; Tylichova, Z.; Safarikova, B.; Skender, B.; Kozubik, A. Interaction of dietary fatty acids with tumour necrosis factor family cytokines during colon inflammation and cancer. Mediat. Inflamm. 2014, 2014, 848632. [Google Scholar] [CrossRef] [PubMed]
- Vaughn, B.P.; Kaiser, T.; Staley, C.; Hamilton, M.J.; Reich, J.; Graiziger, C.; Singroy, S.; Kabage, A.J.; Sadowsky, M.J.; Khoruts, A. A pilot study of fecal bile acid and microbiota profiles in inflammatory bowel disease and primary sclerosing cholangitis. Clin. Exp. Gastroenterol. 2019, 12, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Katsuma, S.; Hirasawa, A.; Tsujimoto, G. Bile acids promote glucagon-like peptide-1 secretion through TGR5 in a murine enteroendocrine cell line STC-1. Biochem. Biophys. Res. Commun. 2005, 329, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.Y.; Pathak, P.; Liu, H.; Donepudi, A.; Ferrell, J.; Boehme, S. Intestinal Farnesoid X Receptor and Takeda G Protein Couple Receptor 5 Signaling in Metabolic Regulation. Dig. Dis. 2017, 35, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Magotti, P.; Bauer, I.; Igarashi, M.; Babagoli, M.; Marotta, R.; Piomelli, D.; Garau, G. Structure of human N-acylphosphatidylethanolamine-hydrolyzing phospholipase D: Regulation of fatty acid ethanolamide biosynthesis by bile acids. Structure 2015, 23, 598–604. [Google Scholar] [CrossRef]
- Margheritis, E.; Castellani, B.; Magotti, P.; Peruzzi, S.; Romeo, E.; Natali, F.; Mostarda, S.; Gioiello, A.; Piomelli, D.; Garau, G. Bile Acid Recognition by NAPE-PLD. ACS Chem. Biol. 2016, 11, 2908–2914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gadaleta, R.M.; van Erpecum, K.J.; Oldenburg, B.; Willemsen, E.C.; Renooij, W.; Murzilli, S.; Klomp, L.W.; Siersema, P.D.; Schipper, M.E.; Danese, S.; et al. Farnesoid X receptor activation inhibits inflammation and preserves the intestinal barrier in inflammatory bowel disease. Gut 2011, 60, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Gadaleta, R.M.; Oldenburg, B.; Willemsen, E.C.; Spit, M.; Murzilli, S.; Salvatore, L.; Klomp, L.W.; Siersema, P.D.; van Erpecum, K.J.; van Mil, S.W. Activation of bile salt nuclear receptor FXR is repressed by pro-inflammatory cytokines activating NF-kappaB signaling in the intestine. Biochim. Biophys. Acta 2011, 1812, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Gadaleta, R.M.; van Mil, S.W.; Oldenburg, B.; Siersema, P.D.; Klomp, L.W.; van Erpecum, K.J. Bile acids and their nuclear receptor FXR: Relevance for hepatobiliary and gastrointestinal disease. Biochim. Biophys. Acta 2010, 1801, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Geurts, L.; Everard, A.; Van Hul, M.; Essaghir, A.; Duparc, T.; Matamoros, S.; Plovier, H.; Castel, J.; Denis, R.G.; Bergiers, M.; et al. Adipose tissue NAPE-PLD controls fat mass development by altering the browning process and gut microbiota. Nat. Commun. 2015, 6, 6495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, W.; Tian, W.; Hong, J.; Li, D.; Tavares, R.; Noble, L.; Moss, S.F.; Resnick, M.B. Expression of bile acid receptor TGR5 in gastric adenocarcinoma. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 304, G322–G327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, W.; Xie, G.; Jia, W. Bile acid-microbiota crosstalk in gastrointestinal inflammation and carcinogenesis. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 111–128. [Google Scholar] [CrossRef] [PubMed]
- West, A.C.; Jenkins, B.J. Inflammatory and non-inflammatory roles for Toll-like receptors in gastrointestinal cancer. Curr. Pharm. Des. 2015, 21, 2968–2977. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zhou, H.; Zhuang, L.; Auwerx, J.; Schoonjans, K.; Wang, X.; Feng, C.; Lu, L. Plasma membrane-bound G protein-coupled bile acid receptor attenuates liver ischemia/reperfusion injury via the inhibition of toll-like receptor 4 signaling in mice. Liver Transpl. 2017, 23, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.B.J.; Lajczak, N.K.; Kelly, O.B.; O’Dwyer, A.M.; Giddam, A.K.; Ni Gabhann, J.; Franco, P.; Tambuwala, M.M.; Jefferies, C.A.; Keely, S.; et al. Ursodeoxycholic acid and lithocholic acid exert anti-inflammatory actions in the colon. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, G550–G558. [Google Scholar] [CrossRef] [PubMed]
- Payne, C.M.; Bernstein, C.; Dvorak, K.; Bernstein, H. Hydrophobic bile acids, genomic instability, Darwinian selection, and colon carcinogenesis. Clin. Exp. Gastroenterol. 2008, 1, 19–47. [Google Scholar] [CrossRef] [PubMed]
- Rao, Y.P.; Stravitz, R.T.; Vlahcevic, Z.R.; Gurley, E.C.; Sando, J.J.; Hylemon, P.B. Activation of protein kinase C alpha and delta by bile acids: Correlation with bile acid structure and diacylglycerol formation. J. Lipid Res. 1997, 38, 2446–2454. [Google Scholar] [PubMed]
- Mencarelli, A.; Renga, B.; Migliorati, M.; Cipriani, S.; Distrutti, E.; Santucci, L.; Fiorucci, S. The bile acid sensor farnesoid X receptor is a modulator of liver immunity in a rodent model of acute hepatitis. J. Immunol. 2009, 183, 6657–6666. [Google Scholar] [CrossRef] [PubMed]
- Calmus, Y.; Poupon, R. Shaping macrophages function and innate immunity by bile acids: Mechanisms and implication in cholestatic liver diseases. Clin. Res. Hepatol. Gastroenterol. 2014, 38, 550–556. [Google Scholar] [CrossRef] [PubMed]
- Renga, B.; Mencarelli, A.; Cipriani, S.; D’Amore, C.; Carino, A.; Bruno, A.; Francisci, D.; Zampella, A.; Distrutti, E.; Fiorucci, S. The bile acid sensor FXR is required for immune-regulatory activities of TLR-9 in intestinal inflammation. PLoS ONE 2013, 8, e54472. [Google Scholar] [CrossRef] [PubMed]
- Shah, Y.M.; Ma, X.; Morimura, K.; Kim, I.; Gonzalez, F.J. Pregnane X receptor activation ameliorates DSS-induced inflammatory bowel disease via inhibition of NF-kappaB target gene expression. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G1114–G1122. [Google Scholar] [CrossRef] [PubMed]
- Bishop-Bailey, D.; Walsh, D.T.; Warner, T.D. Expression and activation of the farnesoid X receptor in the vasculature. Proc. Natl. Acad. Sci. USA 2004, 101, 3668–3673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.H. Immune regulation by microbiome metabolites. Immunology 2018, 154, 220–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kundu, J.K.; Surh, Y.J. Emerging avenues linking inflammation and cancer. Free Radic. Biol. Med. 2012, 52, 2013–2037. [Google Scholar] [CrossRef] [PubMed]
- Winer, D.A.; Luck, H.; Tsai, S.; Winer, S. The Intestinal Immune System in Obesity and Insulin Resistance. Cell. Metab. 2016, 23, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Li, Z.R.; Green, R.S.; Holzman, I.R.; Lin, J. Butyrate enhances the intestinal barrier by facilitating tight junction assembly via activation of AMP-activated protein kinase in Caco-2 cell monolayers. J. Nutr. 2009, 139, 1619–1625. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, M.; Singh, A.B.; Smith, J.J.; Sharma, A.; Chen, X.; Eschrich, S.; Yeatman, T.J.; Beauchamp, R.D.; Dhawan, P. HDAC inhibitors regulate claudin-1 expression in colon cancer cells through modulation of mRNA stability. Oncogene 2010, 29, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Kelly, C.J.; Battista, K.D.; Schaefer, R.; Lanis, J.M.; Alexeev, E.E.; Wang, R.X.; Onyiah, J.C.; Kominsky, D.J.; Colgan, S.P. Microbial-Derived Butyrate Promotes Epithelial Barrier Function through IL-10 Receptor-Dependent Repression of Claudin-2. J. Immunol. 2017, 199, 2976–2984. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.L.; Zou, J.Y.; Hu, E.D.; Chen, D.Z.; Chen, L.; Lu, F.B.; Xu, L.M.; Zheng, M.H.; Li, H.; Huang, Y.; et al. Sodium butyrate ameliorates S100/FCA-induced autoimmune hepatitis through regulation of intestinal tight junction and toll-like receptor 4 signaling pathway. Immunol. Lett. 2017, 190, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.J.; Goldsworthy, S.M.; Barnes, A.A.; Eilert, M.M.; Tcheang, L.; Daniels, D.; Muir, A.I.; Wigglesworth, M.J.; Kinghorn, I.; Fraser, N.J.; et al. The Orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J. Biol. Chem. 2003, 278, 11312–11319. [Google Scholar] [CrossRef] [PubMed]
- Thangaraju, M.; Cresci, G.A.; Liu, K.; Ananth, S.; Gnanaprakasam, J.P.; Browning, D.D.; Mellinger, J.D.; Smith, S.B.; Digby, G.J.; Lambert, N.A.; et al. GPR109A is a G-protein-coupled receptor for the bacterial fermentation product butyrate and functions as a tumor suppressor in colon. Cancer Res. 2009, 69, 2826–2832. [Google Scholar] [CrossRef] [PubMed]
- Pluznick, J.L.; Protzko, R.J.; Gevorgyan, H.; Peterlin, Z.; Sipos, A.; Han, J.; Brunet, I.; Wan, L.X.; Rey, F.; Wang, T.; et al. Olfactory receptor responding to gut microbiota-derived signals plays a role in renin secretion and blood pressure regulation. Proc. Natl. Acad. Sci. USA 2013, 110, 4410–4415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maslowski, K.M.; Vieira, A.T.; Ng, A.; Kranich, J.; Sierro, F.; Yu, D.; Schilter, H.C.; Rolph, M.S.; Mackay, F.; Artis, D.; et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 2009, 461, 1282–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luster, A.D.; Alon, R.; von Andrian, U.H. Immune cell migration in inflammation: Present and future therapeutic targets. Nat. Immunol. 2005, 6, 1182–1190. [Google Scholar] [CrossRef] [PubMed]
- Chandrakesan, P.; Ahmed, I.; Chinthalapally, A.; Singh, P.; Awasthi, S.; Anant, S.; Umar, S. Distinct compartmentalization of NF-kappaB activity in crypt and crypt-denuded lamina propria precedes and accompanies hyperplasia and/or colitis following bacterial infection. Infect Immun. 2012, 80, 753–767. [Google Scholar] [CrossRef] [PubMed]
- Melo, A.D.; Silveira, H.; Bortoluzzi, C.; Lara, L.J.; Garbossa, C.A.; Preis, G.; Costa, L.B.; Rostagno, M.H. Intestinal alkaline phosphatase and sodium butyrate may be beneficial in attenuating LPS-induced intestinal inflammation. Genet. Mol. Res. 2016, 15. [Google Scholar] [CrossRef] [PubMed]
- Simeoli, R.; Mattace Raso, G.; Pirozzi, C.; Lama, A.; Santoro, A.; Russo, R.; Montero-Melendez, T.; Berni Canani, R.; Calignano, A.; Perretti, M.; et al. An orally administered butyrate-releasing derivative reduces neutrophil recruitment and inflammation in dextran sulphate sodium-induced murine colitis. Br. J. Pharmacol. 2017, 174, 1484–1496. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Gurav, A.; Sivaprakasam, S.; Brady, E.; Padia, R.; Shi, H.; Thangaraju, M.; Prasad, P.D.; Manicassamy, S.; Munn, D.H.; et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity 2014, 40, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Du, M.; Yang, Q.; Zhu, M.J. Butyrate suppresses murine mast cell proliferation and cytokine production through inhibiting histone deacetylase. J. Nutr. Biochem. 2016, 27, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Zapolska-Downar, D.; Siennicka, A.; Kaczmarczyk, M.; Kolodziej, B.; Naruszewicz, M. Butyrate inhibits cytokine-induced VCAM-1 and ICAM-1 expression in cultured endothelial cells: The role of NF-κB and PPARalpha. J. Nutr. Biochem. 2004, 15, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Bultman, S.J. Molecular pathways: Gene-environment interactions regulating dietary fiber induction of proliferation and apoptosis via butyrate for cancer prevention. Clin. Cancer Res. 2014, 20, 799–803. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, M.A.; Singh, N.; Martin, P.M.; Thangaraju, M.; Ganapathy, V.; Waller, J.L.; Shi, H.; Robertson, K.D.; Munn, D.H.; Liu, K. Butyrate suppresses colonic inflammation through HDAC1-dependent Fas upregulation and Fas-mediated apoptosis of T cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G1405–G1415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.; Qie, Y.; Park, J.; Kim, C.H. Gut Microbial Metabolites Fuel Host Antibody Responses. Cell Host Microbe 2016, 20, 202–214. [Google Scholar] [CrossRef] [PubMed]
- Arpaia, N.; Campbell, C.; Fan, X.; Dikiy, S.; van der Veeken, J.; deRoos, P.; Liu, H.; Cross, J.R.; Pfeffer, K.; Coffer, P.J.; et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 2013, 504, 451–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin-Gallausiaux, C.; Beguet-Crespel, F.; Marinelli, L.; Jamet, A.; Ledue, F.; Blottiere, H.M.; Lapaque, N. Butyrate produced by gut commensal bacteria activates TGF-beta1 expression through the transcription factor SP1 in human intestinal epithelial cells. Sci. Rep. 2018, 8, 9742. [Google Scholar] [CrossRef] [PubMed]
- Kespohl, M.; Vachharajani, N.; Luu, M.; Harb, H.; Pautz, S.; Wolff, S.; Sillner, N.; Walker, A.; Schmitt-Kopplin, P.; Boettger, T.; et al. The Microbial Metabolite Butyrate Induces Expression of Th1-Associated Factors in CD4(+) T Cells. Front. Immunol. 2017, 8, 1036. [Google Scholar] [CrossRef] [PubMed]
- Bach Knudsen, K.E.; Laerke, H.N.; Hedemann, M.S.; Nielsen, T.S.; Ingerslev, A.K.; Gundelund Nielsen, D.S.; Theil, P.K.; Purup, S.; Hald, S.; Schioldan, A.G.; et al. Impact of Diet-Modulated Butyrate Production on Intestinal Barrier Function and Inflammation. Nutrients 2018, 10, 1499. [Google Scholar] [CrossRef] [PubMed]
- Morin, P.J.; Sparks, A.B.; Korinek, V.; Barker, N.; Clevers, H.; Vogelstein, B.; Kinzler, K.W. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science 1997, 275, 1787–1790. [Google Scholar] [CrossRef] [PubMed]
- Armaghany, T.; Wilson, J.D.; Chu, Q.; Mills, G. Genetic alterations in colorectal cancer. Gastrointest. Cancer Res. 2012, 5, 19–27. [Google Scholar] [PubMed]
- Willett, W.C.; Stampfer, M.J.; Colditz, G.A.; Rosner, B.A.; Speizer, F.E. Relation of meat, fat, and fiber intake to the risk of colon cancer in a prospective study among women. N. Engl. J. Med. 1990, 323, 1664–1672. [Google Scholar] [CrossRef] [PubMed]
- Bajor, A.; Gillberg, P.G.; Abrahamsson, H. Bile acids: Short and long term effects in the intestine. Scand. J. Gastroenterol. 2010, 45, 645–664. [Google Scholar] [CrossRef] [PubMed]
- Alok, A.; Lei, Z.; Jagannathan, N.S.; Kaur, S.; Harmston, N.; Rozen, S.G.; Tucker-Kellogg, L.; Virshup, D.M. Wnt proteins synergize to activate beta-catenin signaling. J. Cell Sci. 2017, 130, 1532–1544. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R.; Clevers, H. Wnt/beta-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef] [PubMed]
- Gadaleta, R.M.; Garcia-Irigoyen, O.; Moschetta, A. Bile acids and colon cancer: Is FXR the solution of the conundrum? Mol. Asp. Med. 2017, 56, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Sivaprakasam, S.; Bhutia, Y.D.; Ramachandran, S.; Ganapathy, V. Cell-Surface and Nuclear Receptors in the Colon as Targets for Bacterial Metabolites and Its Relevance to Colon Health. Nutrients 2017, 9, 856. [Google Scholar] [CrossRef] [PubMed]
- De Gottardi, A.; Touri, F.; Maurer, C.A.; Perez, A.; Maurhofer, O.; Ventre, G.; Bentzen, C.L.; Niesor, E.J.; Dufour, J.F. The bile acid nuclear receptor FXR and the bile acid binding protein IBABP are differently expressed in colon cancer. Dig. Dis. Sci. 2004, 49, 982–989. [Google Scholar] [CrossRef] [PubMed]
- Bailey, A.M.; Zhan, L.; Maru, D.; Shureiqi, I.; Pickering, C.R.; Kiriakova, G.; Izzo, J.; He, N.; Wei, C.; Baladandayuthapani, V.; et al. FXR silencing in human colon cancer by DNA methylation and KRAS signaling. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 306, G48–G58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Modica, S.; Murzilli, S.; Salvatore, L.; Schmidt, D.R.; Moschetta, A. Nuclear bile acid receptor FXR protects against intestinal tumorigenesis. Cancer Res. 2008, 68, 9589–9594. [Google Scholar] [CrossRef] [PubMed]
- Sohn, O.S.; Fiala, E.S. Effects of dietary restriction and fasting on selected rat liver enzymes of xenobiotic metabolism and on AOM-induced DNA guanine methylation in rat liver and colon. Nutr. Cancer 1995, 23, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Gupta, S. The multifaceted role of glutathione S-transferases in cancer. Cancer Lett. 2018, 433, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, D.R.; Schmidt, S.; Holmstrom, S.R.; Makishima, M.; Yu, R.T.; Cummins, C.L.; Mangelsdorf, D.J.; Kliewer, S.A. AKR1B7 is induced by the farnesoid X receptor and metabolizes bile acids. J. Biol. Chem. 2011, 286, 2425–2432. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Yuan, X.; Lu, D.; Guo, L.; Wu, B. Farnesoid X receptor regulates SULT1E1 expression through inhibition of PGC1alpha binding to HNF4alpha. Biochem. Pharmacol. 2017, 145, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.P.; Xie, G.; Raufman, J.P.; Hogan, S.; Griffin, T.L.; Packard, C.A.; Chatfield, D.A.; Hagey, L.R.; Steinbach, J.H.; Hofmann, A.F. Human cecal bile acids: Concentration and spectrum. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 293, G256–G263. [Google Scholar] [CrossRef] [PubMed]
- Van der Beek, C.M.; Dejong, C.H.C.; Troost, F.J.; Masclee, A.A.M.; Lenaerts, K. Role of short-chain fatty acids in colonic inflammation, carcinogenesis, and mucosal protection and healing. Nutr. Rev. 2017, 75, 286–305. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Hu, M.; Zhou, L.; Ling, S.; Li, Y.; Kong, B.; Huang, P. Dietary fiber intake and risks of proximal and distal colon cancers: A meta-analysis. Medicine 2018, 97, e11678. [Google Scholar] [CrossRef] [PubMed]
- Bordonaro, M.; Lazarova, D.L. CREB-binding protein, p300, butyrate, and Wnt signaling in colorectal cancer. World J. Gastroenterol. 2015, 21, 8238–8248. [Google Scholar] [CrossRef] [PubMed]
- Haque, S.; Morris, J.C. Transforming growth factor-beta: A therapeutic target for cancer. Hum. Vaccin. Immunother. 2017, 13, 1741–1750. [Google Scholar] [CrossRef] [PubMed]
- Lampropoulos, P.; Zizi-Sermpetzoglou, A.; Rizos, S.; Kostakis, A.; Nikiteas, N.; Papavassiliou, A.G. TGF-beta signalling in colon carcinogenesis. Cancer Lett. 2012, 314, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Gao, X.; Zhang, W.; Zhang, G.; Nguyen, A.K.; Liu, X.; Jimenez, F.; Cox, C.S., Jr.; Townsend, C.M., Jr.; Ko, T.C. Dietary fiber enhances TGF-beta signaling and growth inhibition in the gut. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G156–G164. [Google Scholar] [CrossRef] [PubMed]
- Lazarova, D.L.; Chiaro, C.; Wong, T.; Drago, E.; Rainey, A.; O’Malley, S.; Bordonaro, M. CBP Activity Mediates Effects of the Histone Deacetylase Inhibitor Butyrate on WNT Activity and Apoptosis in Colon Cancer Cells. J. Cancer 2013, 4, 481–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bordonaro, M.; Lazarova, D.L.; Sartorelli, A.C. Hyperinduction of Wnt activity: A new paradigm for the treatment of colorectal cancer? Oncol. Res. 2008, 17, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Donohoe, D.R.; Collins, L.B.; Wali, A.; Bigler, R.; Sun, W.; Bultman, S.J. The Warburg effect dictates the mechanism of butyrate-mediated histone acetylation and cell proliferation. Mol. Cell 2012, 48, 612–626. [Google Scholar] [CrossRef] [PubMed]
- Bordonaro, M.; Lazarova, D.L.; Sartorelli, A.C. Butyrate and Wnt signaling: A possible solution to the puzzle of dietary fiber and colon cancer risk? Cell Cycle 2008, 7, 1178–1183. [Google Scholar] [CrossRef] [PubMed]
- Bordonaro, M.; Tewari, S.; Cicco, C.E.; Atamna, W.; Lazarova, D.L. A switch from canonical to noncanonical Wnt signaling mediates drug resistance in colon cancer cells. PLoS ONE 2011, 6, e27308. [Google Scholar] [CrossRef]
- Zeng, H.; Ishaq, S.L.; Liu, Z.; Bukowski, M.R. Colonic aberrant crypt formation accompanies an increase of opportunistic pathogenic bacteria in C57BL/6 mice fed a high-fat diet. J. Nutr. Biochem. 2018, 54, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, I.; Roy, B.C.; Khan, S.A.; Septer, S.; Umar, S. Microbiome, Metabolome and Inflammatory Bowel Disease. Microorganisms 2016, 4. [Google Scholar] [CrossRef] [PubMed]
- Sears, C.L.; Garrett, W.S. Microbes, microbiota, and colon cancer. Cell Host Microbe 2014, 15, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Tjalsma, H.; Boleij, A.; Marchesi, J.R.; Dutilh, B.E. A bacterial driver-passenger model for colorectal cancer: Beyond the usual suspects. Nat. Rev. Microbiol. 2012, 10, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Raskov, H.; Burcharth, J.; Pommergaard, H.C. Linking Gut Microbiota to Colorectal Cancer. J. Cancer 2017, 8, 3378–3395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, J.R.; Johnston, B.; Kuskowski, M.A.; Nougayrede, J.P.; Oswald, E. Molecular epidemiology and phylogenetic distribution of the Escherichia coli pks genomic island. J. Clin. Microbiol. 2008, 46, 3906–3911. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Guo, B.; Gao, R.; Zhu, Q.; Qin, H. Microbiota disbiosis is associated with colorectal cancer. Front. Microbiol. 2015, 6, 20. [Google Scholar] [CrossRef] [PubMed]
- Sears, C.L.; Pardoll, D.M. Perspective: Alpha-bugs, their microbial partners, and the link to colon cancer. J. Infect. Dis. 2011, 203, 306–311. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, H.; Umar, S.; Rust, B.; Lazarova, D.; Bordonaro, M. Secondary Bile Acids and Short Chain Fatty Acids in the Colon: A Focus on Colonic Microbiome, Cell Proliferation, Inflammation, and Cancer. Int. J. Mol. Sci. 2019, 20, 1214. https://doi.org/10.3390/ijms20051214
Zeng H, Umar S, Rust B, Lazarova D, Bordonaro M. Secondary Bile Acids and Short Chain Fatty Acids in the Colon: A Focus on Colonic Microbiome, Cell Proliferation, Inflammation, and Cancer. International Journal of Molecular Sciences. 2019; 20(5):1214. https://doi.org/10.3390/ijms20051214
Chicago/Turabian StyleZeng, Huawei, Shahid Umar, Bret Rust, Darina Lazarova, and Michael Bordonaro. 2019. "Secondary Bile Acids and Short Chain Fatty Acids in the Colon: A Focus on Colonic Microbiome, Cell Proliferation, Inflammation, and Cancer" International Journal of Molecular Sciences 20, no. 5: 1214. https://doi.org/10.3390/ijms20051214