Molecular Characterization of Non-responders to Chemotherapy in Serous Ovarian Cancer



Abstract
:1. Introduction
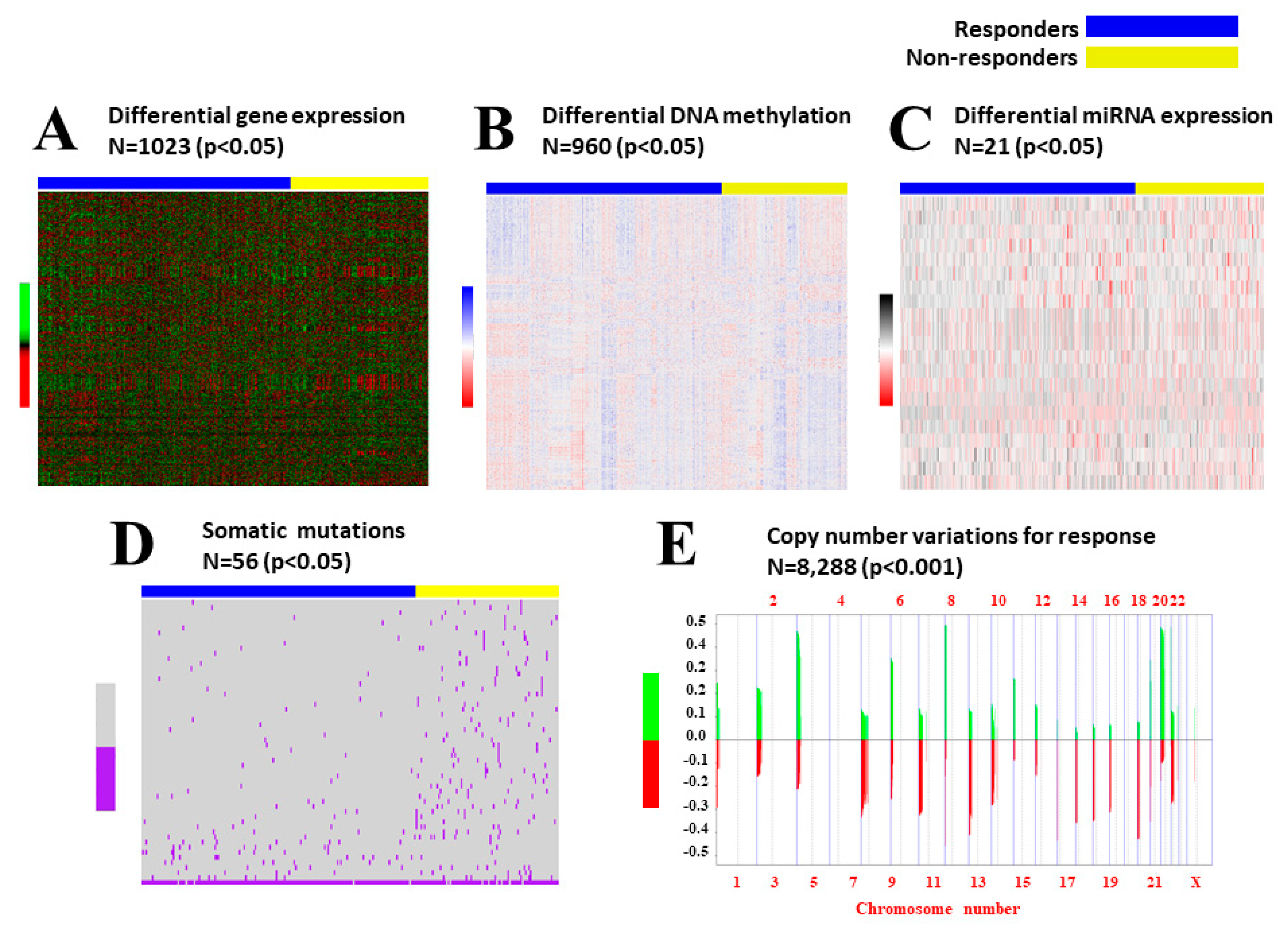
2. Results
2.1. Data Preprocessing
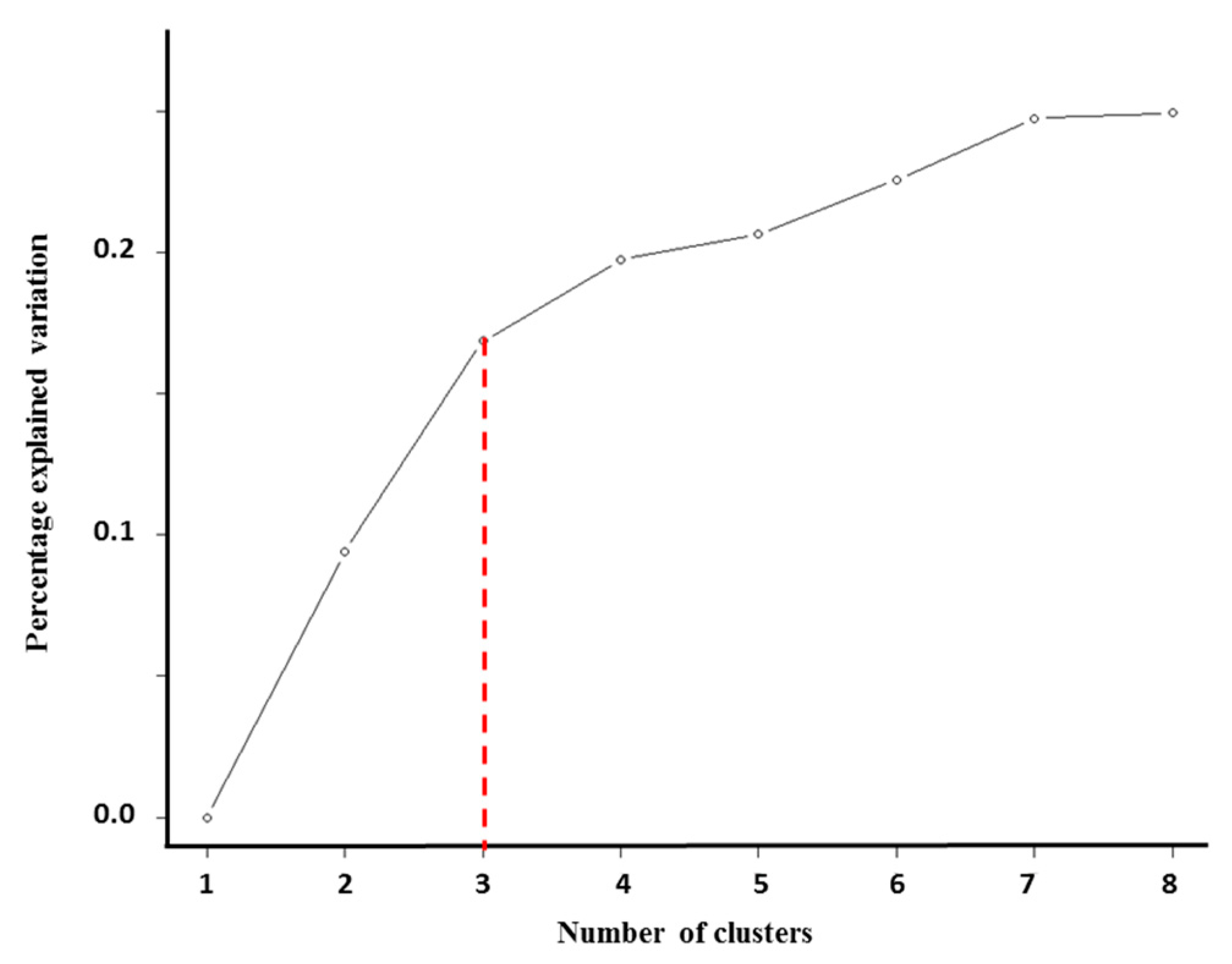
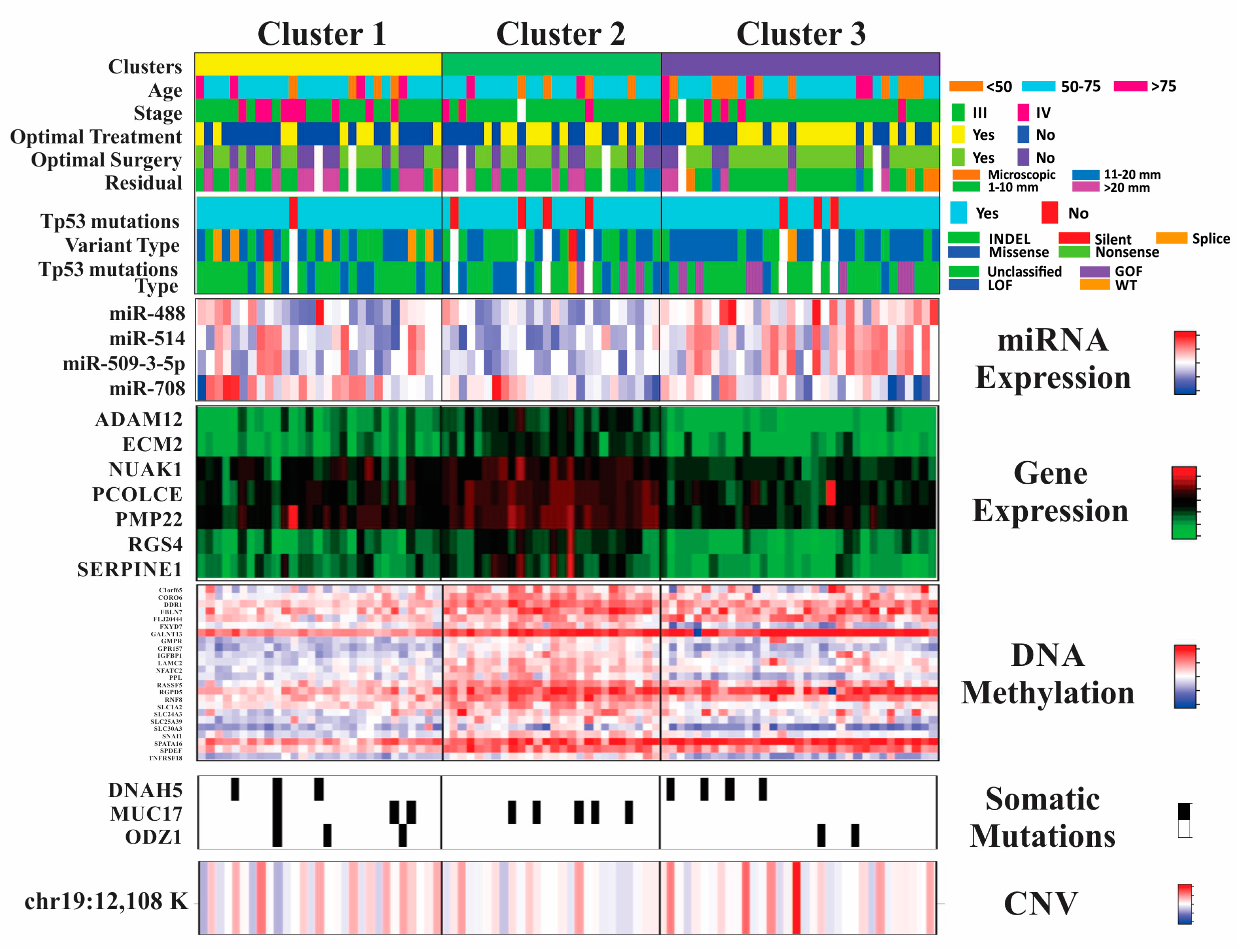
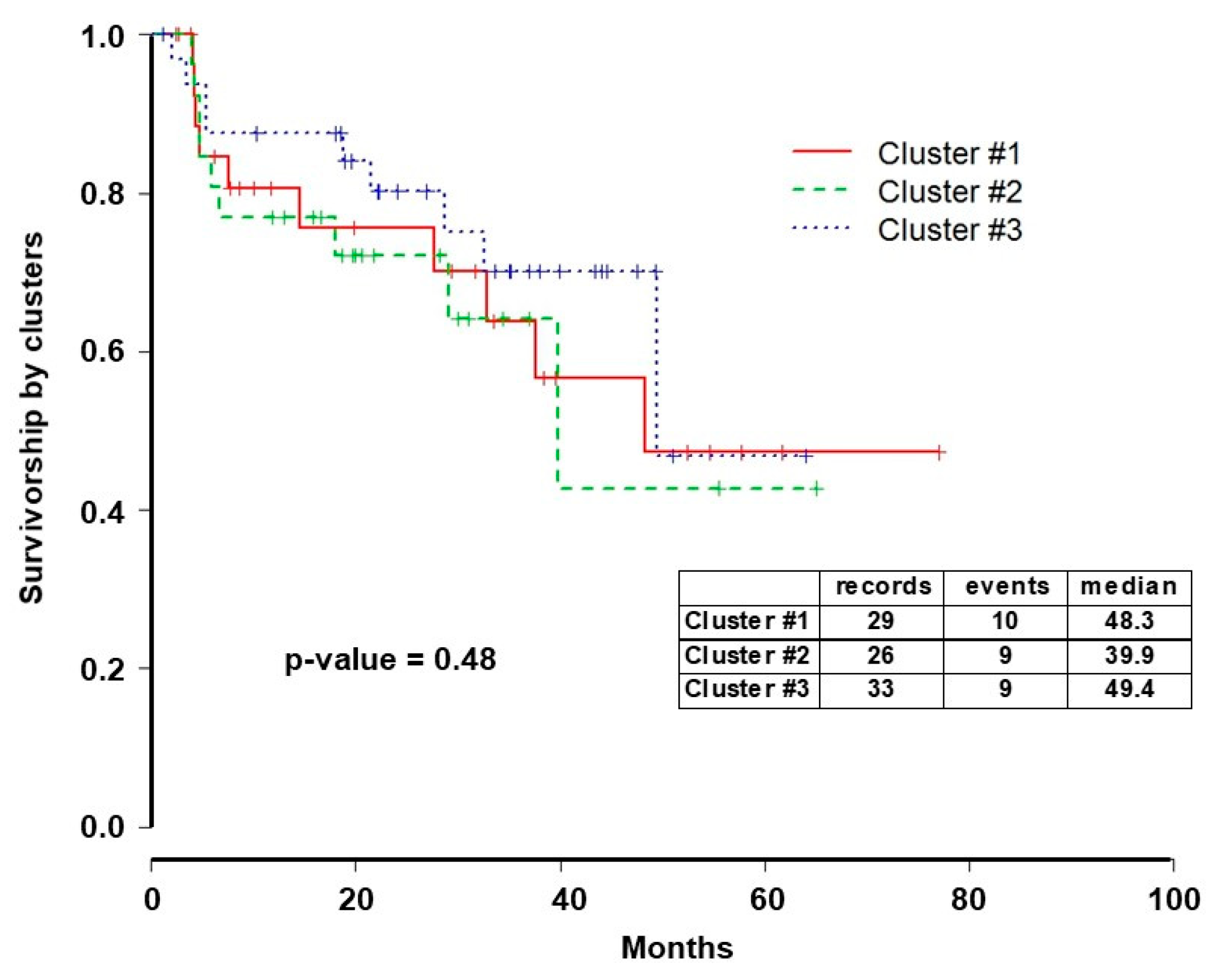
2.2. Integrative iClusterPlus Analysis
2.3. Pathway Enrichment Analysis
3. Discussion
4. Material and Methods
4.1. Outcomes Definition
4.2. Source of Data
4.2.1. Copy Number Alterations (CNA)
4.2.2. Mutation Analysis
4.2.3. Gene Expression
4.2.4. DNA Methylation
4.2.5. MicroRNA (miRNA) Expression
4.3. Statistical Analysis
4.3.1. Variable Selection
4.3.2. iClusterPlus Analysis
4.3.3. Pathway Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| HGSC | High-grade serous ovarian cancer |
| TCGA | The Cancer Genome Atlas |
| AUC | Area under the (receiver operating characteristic) curve |
| CNA | Gene copy number alteration |
| miRNA | Micro RNA |
| K | Clusters |
| PFS | Progression-free survival |
| OS | Overall survival |
| HDAC | Histone deacetylase |
| GOF | Gain-of-function |
| LOF | Loss-of-function |
| WT | Wildtype |
| GOG | Gynecologic Oncology Group |
| ICON | International Collaborative Ovarian Neoplasm |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical | Age |
| Stage | |
| Optimal treatment | |
| Optimal surgery | |
| Residual | |
| TP53 | Mutation status |
| Variant type | |
| TP53 Mutation type | |
| Gene expression | ADAM12 |
| ECM2 | |
| NUAK1 | |
| PCOLCE | |
| PMP22 | |
| RGS4 | |
| SERPINE1 | |
| Methylation | C1orf65 |
| CORO6 | |
| DDR1 | |
| FBLN7 | |
| FLJ20444 | |
| FXYD7 | |
| GALNT13 | |
| GMPR | |
| GPR157 | |
| IGFBP1 | |
| LAMC2 | |
| NFATC2 | |
| PPL | |
| RASSF5 | |
| RGPD5 | |
| RNF8 | |
| SLC1A2 | |
| SLC24A3 | |
| SLC25A39 | |
| SLC30A3 | |
| SNAI1 | |
| SPATA16 | |
| SPDEF | |
| TNFRSF18 | |
| Mutations | DNAH5 |
| MUC17 | |
| ODZ1 | |
| Copy number alteration | Chr19: 12,108,685-12,180,988 |
References
- American Cancer Society, Cancer Facts & Figures 2014; American Cancer Society: Atlanta, GA, USA, 2014.
- Levanon, K.; Crum, C.; Drapkin, R. New insights into the pathogenesis of serous ovarian cancer and its clinical impact. J. Clin. Oncol. 2008, 26, 5284–5293. [Google Scholar] [CrossRef] [PubMed]
- Alexandre, J.; Ray-Coquard, I.; Selle, F.; Floquet, A.; Cottu, P.; Weber, B.; Falandry, C.; Lebrun, D.; Pujade-Lauraine, E. Gineco, Mucinous advanced epithelial ovarian carcinoma: Clinical presentation and sensitivity to platinum-paclitaxel-based chemotherapy, the GINECO experience. Ann. Oncol. 2010, 21, 2377–2381. [Google Scholar] [CrossRef] [PubMed]
- Katagiri, A.; Nakayama, K.; Rahman, M.T.; Rahman, M.; Katagiri, H.; Nakayama, N.; Ishikawa, M.; Ishibashi, T.; Iida, K.; Kobayashi, H.; et al. Loss of ARID1A expression is related to shorter progression-free survival and chemoresistance in ovarian clear cell carcinoma. Mod. Pathol. 2012, 25, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Khan, S.; Sun, Y.; Hess, K.; Shmulevich, I.; Sood, A.K.; Zhang, W. Association of BRCA1 and BRCA2 mutations with survival, chemotherapy sensitivity, and gene mutator phenotype in patients with ovarian cancer. JAMA 2011, 306, 1557–1565. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Senovilla, L.; Eisenberg, T.; Carmona-Gutierrez, D.; Vacchelli, E.; Robert, T.; Ripoche, H.; Jagemann, N.; Paccard, C.; et al. Independent transcriptional reprogramming and apoptosis induction by cisplatin. Cell Cycle 2012, 11, 3472–3480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borst, P.; Rottenberg, S.; Jonkers, J. How do real tumors become resistant to cisplatin? Cell Cycle 2008, 7, 1353–1359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godwin, A.K.; Meister, A.; O’Dwyer, P.J.; Huang, C.S.; Hamilton, T.C.; Anderson, M.E. High resistance to cisplatin in human ovarian cancer cell lines is associated with marked increase of glutathione synthesis. Proc. Natl. Acad. Sci. USA 1992, 89, 3070–3074. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.W.; Laub, P.B.; Beesley, J.S.; Ozols, R.F.; Hamilton, T.C. Increased platinum-DNA damage tolerance is associated with cisplatin resistance and cross-resistance to various chemotherapeutic agents in unrelated human ovarian cancer cell lines. Cancer Res. 1997, 57, 850–856. [Google Scholar] [PubMed]
- Johnson, S.W.; Swiggard, P.A.; Handel, L.M.; Brennan, J.M.; Godwin, A.K.; Ozols, R.F.; Hamilton, T.C. Relationship between platinum-DNA adduct formation and removal and cisplatin cytotoxicity in cisplatin-sensitive and -resistant human ovarian cancer cells. Cancer Res. 1994, 54, 5911–5916. [Google Scholar] [PubMed]
- Gonzalez Bosquet, J.; Marchion, D.C.; Chon, H.; Lancaster, J.M.; Chanock, S. Analysis of chemotherapeutic response in ovarian cancers using publically available high-throughput data. Cancer Res. 2014, 74, 3902–3912. [Google Scholar] [CrossRef] [PubMed]
- Oikonomopoulou, K.; Li, L.; Zheng, Y.; Simon, I.; Wolfert, R.L.; Valik, D.; Nekulova, M.; Simickova, M.; Frgala, T.; Diamandis, E.P. Prediction of ovarian cancer prognosis and response to chemotherapy by a serum-based multiparametric biomarker panel. Br. J. Cancer 2008, 99, 1103–1113. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Katsaros, D.; Shan, S.J.; de la Longrais, I.R.; Porpiglia, M.; Scorilas, A.; Kim, N.W.; Wolfert, R.L.; Simon, I.; Li, L.; et al. A multiparametric panel for ovarian cancer diagnosis, prognosis, and response to chemotherapy. Clin. Cancer Res. 2007, 13, 6984–6992. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez Bosquet, J.; Newtson, A.M.; Chung, R.K.; Thiel, K.W.; Ginader, T.; Goodheart, M.J.; Leslie, K.K.; Smith, B.J. Prediction of chemo-response in serous ovarian cancer. Mol. Cancer 2016, 15, 66. [Google Scholar] [CrossRef] [PubMed]
- Shen, R.; Olshen, A.B.; Ladanyi, M. Integrative clustering of multiple genomic data types using a joint latent variable model with application to breast and lung cancer subtype analysis. Bioinformatics 2009, 25, 2906–2912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perren, T.J.; Swart, A.M.; Pfisterer, J.; Ledermann, J.A.; Pujade-Lauraine, E.; Kristensen, G.; Carey, M.S.; Beale, P.; Cervantes, A.; Kurzeder, C.; et al. A phase 3 trial of bevacizumab in ovarian cancer. N. Engl. J. Med. 2011, 365, 2484–2496. [Google Scholar] [CrossRef] [PubMed]
- Burger, R.A.; Brady, M.F.; Bookman, M.A.; Fleming, G.F.; Monk, B.J.; Huang, H.; Mannel, R.S.; Homesley, H.D.; Fowler, J.; Greer, B.E.; et al. Incorporation of bevacizumab in the primary treatment of ovarian cancer. N. Engl. J. Med. 2011, 365, 2473–2483. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Du Bois, A.; Kristensen, G.; Ray-Coquard, I.; Reuss, A.; Pignata, S.; Colombo, N.; Denison, U.; Vergote, I.; Del Campo, J.M.; Ottevanger, P.; et al. Standard first-line chemotherapy with or without nintedanib for advanced ovarian cancer (AGO-OVAR 12): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet Oncol. 2016, 17, 78–89. [Google Scholar] [CrossRef]
- Davidson, B.A.; Squatrito, R.; Duska, L.R.; Havrilesky, L.J.; Schwager, N.; McCollum, M.; Arapovic, S.; Secord, A.A. Phase II evaluation of nintedanib in the treatment of bevacizumab-resistant persistent/recurrent ovarian, fallopian tube, or primary peritoneal carcinoma. J. Clin. Oncol. 2016, 34 (Suppl. 15), TPS5601. [Google Scholar] [CrossRef]
- Ledermann, J.A.; Embleton, A.C.; Raja, F.; Perren, T.J.; Jayson, G.C.; Rustin, G.J.; Kaye, S.B.; Hirte, H.; Eisenhauer, E.; Vaughan, M.; et al. Cediranib in patients with relapsed platinum-sensitive ovarian cancer (ICON6): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet 2016, 387, 1066–1074. [Google Scholar] [CrossRef]
- Aziz, A.U.R.; Farid, S.; Qin, K.; Wang, H.; Liu, B. PIM Kinases and Their Relevance to the PI3K/AKT/mTOR Pathway in the Regulation of Ovarian Cancer. Biomolecules 2018, 8, 7. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Heo, J.H.; Park, J.Y.; Jeong, J.Y.; Cho, H.J.; Park, K.S.; Kim, S.H.; Moon, Y.W.; Kim, J.S.; An, H.J. A novel PI3K/mTOR dual inhibitor, CMG002, overcomes the chemoresistance in ovarian cancer. Gynecol. Oncol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Mabuchi, S.; Kuroda, H.; Takahashi, R.; Sasano, T. The PI3K/AKT/mTOR pathway as a therapeutic target in ovarian cancer. Gynecol. Oncol. 2015, 137, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.D.; Enders, G.H. Wnt-signaling and senescence: A tug of war in early neoplasia? Cancer Biol. Ther. 2008, 7, 1706–1711. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.C.; Rosen, L.S.; Chugh, R.; Goldman, J.W.; Xu, L.; Kapoun, A.; Brachmann, R.K.; Dupont, J.; Stagg, R.J.; Tolcher, A.W.; et al. First-in-human evaluation of the human monoclonal antibody vantictumab (OMP-18R5; anti-Frizzled) targeting the WNT pathway in a phase I study for patients with advanced solid tumors. J. Clin. Oncol. 2013, 31 (Suppl. 15), 2540. [Google Scholar]
- Combination Chemotherapy and Bevacizumab with Or without PRI-724 in Treating Patients with Newly Diagnosed Metastatic Colorectal Cancer. Available online: https://www.cancer.gov/about-cancer/treatment/clinical-trials/intervention/C95894 (accessed on 28 February 2019).
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Varga, A.; Piha-Paul, S.; Ott, P.A.; Mehnert, J.M.; Berton-Rigaud, D.; Morosky, A.; Yang, P.; Ruman, J.; Matei, D. Pembrolizumab in patients with programmed death ligand 1-positive advanced ovarian cancer: Analysis of KEYNOTE-028. Gynecol. Oncol. 2019, 152, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Hamanishi, J.; Mandai, M.; Ikeda, T.; Minami, M.; Kawaguchi, A.; Murayama, T.; Kanai, M.; Mori, Y.; Matsumoto, S.; Chikuma, S.; et al. Safety and Antitumor Activity of Anti-PD-1 Antibody, Nivolumab, in Patients With Platinum-Resistant Ovarian Cancer. J. Clin. Oncol. 2015, 33, 4015–4022. [Google Scholar] [CrossRef] [PubMed]
- Fridman, A.L.; Tainsky, M.A. Critical pathways in cellular senescence and immortalization revealed by gene expression profiling. Oncogene 2008, 27, 5975–5987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Marchenko, N.D.; Moll, U.M. SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis. Cell Death Differ. 2011, 18, 1904–1913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, W.; Liu, S.; Xu, E.; Zhang, J.; Zhang, Y.; Chen, X.; Chen, X. Histone deacetylase inhibitors suppress mutant p53 transcription via histone deacetylase 8. Oncogene 2013, 32, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Friedlander, M.L.; Stockler, M.R.; Butow, P.; King, M.T.; McAlpine, J.; Tinker, A.; Ledermann, J.A. Clinical trials of palliative chemotherapy in platinum-resistant or -refractory ovarian cancer: Time to think differently? J. Clin. Oncol. 2013, 31, 2362. [Google Scholar] [CrossRef] [PubMed]
- Therasse, P.; Arbuck, S.G.; Eisenhauer, E.A.; Wanders, J.; Kaplan, R.S.; Rubinstein, L.; Verweij, J.; Van Glabbeke, M.; van Oosterom, A.T.; Christian, M.C.; et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J. Natl. Cancer Inst. 2000, 92, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Friedlander, M.; Butow, P.; Stockler, M.; Gainford, C.; Martyn, J.; Oza, A.; Donovan, H.S.; Miller, B.; King, M. Symptom control in patients with recurrent ovarian cancer: Measuring the benefit of palliative chemotherapy in women with platinum refractory/resistant ovarian cancer. Int. J. Gynecol. Cancer 2009, 19 (Suppl. 2), S44–S48. [Google Scholar] [CrossRef] [PubMed]
- Olshen, A.B.; Venkatraman, E.S.; Lucito, R.; Wigler, M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics 2004, 5, 557–572. [Google Scholar] [CrossRef] [PubMed]
- Beroukhim, R.; Getz, G.; Nghiemphu, L.; Barretina, J.; Hsueh, T.; Linhart, D.; Vivanco, I.; Lee, J.C.; Huang, J.H.; Alexander, S.; et al. Assessing the significance of chromosomal aberrations in cancer: Methodology and application to glioma. Proc. Natl. Acad. Sci. USA 2007, 104, 20007–20012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mo, Q.; Wang, S.; Seshan, V.E.; Olshen, A.B.; Schultz, N.; Sander, C.; Powers, R.S.; Ladanyi, M.; Shen, R. Pattern discovery and cancer gene identification in integrated cancer genomic data. Proc. Natl. Acad. Sci. USA 2013, 110, 4245–4250. [Google Scholar] [CrossRef] [PubMed]
- Brachova, P.; Mueting, S.R.; Devor, E.J.; Leslie, K.K. Oncomorphic TP53 Mutations in Gynecologic Cancers Lose the Normal Protein:Protein Interactions with the microRNA Microprocessing Complex. J. Cancer 2014, 5, 506–516. [Google Scholar]
- Brachova, P.; Mueting, S.R.; Carlson, M.J.; Goodheart, M.J.; Button, A.M.; Mott, S.L.; Dai, D.; Thiel, K.W.; Devor, E.J.; Leslie, K.K. TP53 oncomorphic mutations predict resistance to platinum and taxanebased standard chemotherapy in patients diagnosed with advanced serous ovarian carcinoma. Int. J. Oncol. 2015, 46, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.; Hastie, T.; Tibshirani, R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J. Stat. Softw. 2010, 33, 1–22. [Google Scholar] [CrossRef] [PubMed]




| Cluster #1 | Cluster #2 | Cluster #3 | p-Value | |
|---|---|---|---|---|
| Number of Patients | 29 | 26 | 33 | |
| Average Age (years) | 61 | 59 | 57 | 0.149 |
| Grade | 0.744 | |||
| Grade 2 | 2 | 4 | 3 | |
| Grade 3 | 27 | 21 | 28 | |
| Stage | 0.081 | |||
| Stage II | 0 | 0 | 1 | |
| Stage III | 20 | 22 | 27 | |
| Stage IV | 9 | 3 | 5 | |
| Surgical Outcome | 0.079 | |||
| Optimal (<1 cm) | 15 | 13 | 24 | |
| Suboptimal (>1 cm) | 12 | 10 | 7 | |
| Residual Disease | 0.136 | |||
| Microscopic | 1 | 0 | 4 | |
| Macroscopic | 26 | 23 | 27 | |
| Optimal Treatment | 0.063 | |||
| Optimal (Surgery + 6 cycles) | 9 | 11 | 18 | |
| Suboptimal | 20 | 15 | 15 | |
| Chemotherapy | 0.151 | |||
| Platinum | 29 | 25 * | 31 ** | |
| Platinum +Taxane | 27 | 24 | 31 |
| KEGG ID | Description | p-Value | Gene ID |
|---|---|---|---|
| Cluster 1 | |||
| hsa04510 | Focal Adhesion | <0.001 | COL1A2/COL5A1/COMPATGA5/PDGFRA |
| hsa05214 | Glioma | 0.015 | PDGFR1/PDGFRB/IGF1 |
| hsa05218 | Melanoma | 0.019 | PDGFR1/PDGFRB/IGF1 |
| hsa05215 | Prostate Cancer | 0.034 | PDGFR1/PDGFRB/IGF1 |
| hsa04540 | Gap Junction | 0.034 | PDGFR1/PDGFRB/PRKX |
| hsa05414 | Dilated cardiomyopathy | 0.034 | ITGA5/PRKX/IGF1 |
| hsa04512 | ECM-Receptor Interaction | 0.005 | COL1A2/COL5A1/COMP/ITGA5 |
| hsa04270 | Vascular Smooth Muscle Contraction | 0.013 | ACTG2/CALD1/EDNRA/PRKX |
| Cluster 2 | |||
| hsa04218 | Cellular senescence | 0.001 | NFATC2/RASSF5/SERPINE1/FBXW11 |
| hsa04066 | HIF-1 signaling pathway | 0.040 | SERPINE1/EGLN1 |
| hsa00450 | Seleno-compound metabolism | 0.053 * | TXNRD2 |
| hsa0 1040 | Biosynthesis of unsaturated fatty acids | 0.083 * | SCD5 |
| hsa04390 | Hippo signaling pathway | 0.086 * | SERPINE1/FBXW11 |
| hsa04310 | Wnt signaling pathway | 0.089 * | NFATC2/FBXW11 |
| Cluster 3 | |||
| hsa04218 | Cellular senescence | 0.003 | NFATC2/RASSF5/SERPINE1 |
| hsa00512 | Mucin type O-glycan biosynthesis | 0.056 * | GALNT13 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McDonald, M.E.; Salinas, E.A.; Devor, E.J.; Newtson, A.M.; Thiel, K.W.; Goodheart, M.J.; Bender, D.P.; Smith, B.J.; Leslie, K.K.; Gonzalez-Bosquet, J. Molecular Characterization of Non-responders to Chemotherapy in Serous Ovarian Cancer. Int. J. Mol. Sci. 2019, 20, 1175. https://doi.org/10.3390/ijms20051175
McDonald ME, Salinas EA, Devor EJ, Newtson AM, Thiel KW, Goodheart MJ, Bender DP, Smith BJ, Leslie KK, Gonzalez-Bosquet J. Molecular Characterization of Non-responders to Chemotherapy in Serous Ovarian Cancer. International Journal of Molecular Sciences. 2019; 20(5):1175. https://doi.org/10.3390/ijms20051175
Chicago/Turabian StyleMcDonald, Megan E., Erin A. Salinas, Eric J. Devor, Andreea M. Newtson, Kristina W. Thiel, Michael J. Goodheart, David P. Bender, Brian J. Smith, Kimberly K. Leslie, and Jesus Gonzalez-Bosquet. 2019. "Molecular Characterization of Non-responders to Chemotherapy in Serous Ovarian Cancer" International Journal of Molecular Sciences 20, no. 5: 1175. https://doi.org/10.3390/ijms20051175