Transcriptome and Proteome-Based Network Analysis Reveals a Model of Gene Activation in Wheat Resistance to Stripe Rust

 ,
,
Abstract
:
1. Introduction
2. Results
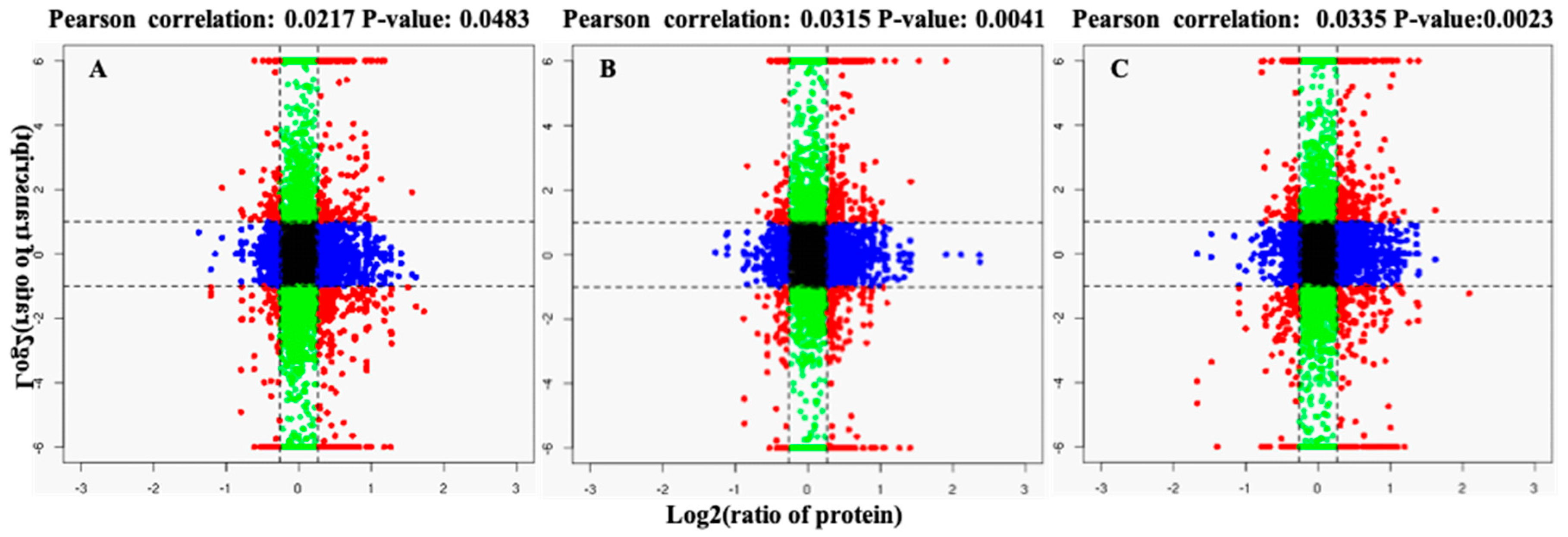
2.1. Global Analysis of iTRAQ Data
2.2. PPI Network Construction for DAPs Induced by Pst Stress
2.3. Co-Expression Network Analysis of Wheat Resistance to Stripe Rust
2.4. The Pst-Induced Module Transiently Expresses Genes Enriched in Splicing and Transcription Activity
2.5. Screening for Disease-Resistance Priming Genes in N9134 Using Transcriptome-Proteome-Associated Analysis
3. Discussion
3.1. Splicing Activity May Play an Important Regulatory Role in Plant Responses to Fungi
3.2. Resistance Gene Regulators May Be More Important in Understanding the Pathogen Defense Mechanism in Wheat
4. Material and Methods
4.1. Plant Materials and Pathogen Stress Treatment
4.2. Protein Extraction, iTRAQ Labeling, and LC-ESI-MS/MS Analysis
4.3. Peptide and Protein Identification
4.4. Protein–Protein Interaction (PPI) Network Construction
4.5. Weighted Gene Correlation Network Analysis (WGCNA) and Transcriptome-Proteome-Associated Analysis
4.6. k-Means Clustering Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- McIntosh, R.; Yamazaki, Y.; Dubcovsky, J.; Rogers, J.; Morris, C.; Appels, R.; Xia, X. Catalogue of gene symbols for wheat 2013–2014 supplement. In Proceedings of the 12th International Wheat Genetic Symposium, Yokohama, Japan, 8–13 September 2013. [Google Scholar]
- Zhao, J.; Wang, M.; Chen, X.; Kang, Z. Role of Alternate Hosts in Epidemiology and Pathogen Variation of Cereal Rusts. Annu. Rev. Phytopathol. 2016, 54, 207–228. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, L.; Wang, C.; Wang, Y.; Zhou, X.; Lv, S.; Liu, X.; Kang, Z.; Ji, W. Molecular mapping and marker development for the Triticum dicoccoides-derived stripe rust resistance gene YrSM139-1B in bread wheat cv. Shaanmai 139. Theor. Appl. Genet. 2016, 129, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.N.; Costa Alves, G.S.; Van Sluys, M.A. Plant immunity: Unravelling the complexity of plant responses to biotic stresses. Ann. Bot. 2017, 119, 681–687. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yang, Y.; Wang, C.; Liu, M.; Li, H.; Fu, Y.; Wang, Y.; Nie, Y.; Liu, X.; Ji, W. Large-scale transcriptome comparison reveals distinct gene activations in wheat responding to stripe rust and powdery mildew. BMC Genom. 2014, 15, 898. [Google Scholar] [CrossRef] [PubMed]
- Dobon, A.; Bunting, D.C.; Cabrera-Quio, L.E.; Uauy, C.; Saunders, D.G. The host-pathogen interaction between wheat and yellow rust induces temporally coordinated waves of gene expression. BMC Genom. 2016, 17, 380. [Google Scholar] [CrossRef] [PubMed]
- Coram, T.E.; Huang, X.; Zhan, G.; Settles, M.L.; Chen, X. Meta-analysis of transcripts associated with race-specific resistance to stripe rust in wheat demonstrates common induction of blue copper-binding protein, heat-stress transcription factor, pathogen-induced WIR1A protein, and ent-kaurene synthase transcripts. Funct. Integr. Genom. 2010, 10, 383–392. [Google Scholar] [CrossRef]
- Feng, H.; Wang, B.; Zhang, Q.; Fu, Y.; Huang, L.; Wang, X.; Kang, Z. Exploration of microRNAs and their targets engaging in the resistance interaction between wheat and stripe rust. Front. Plant Sci. 2015, 6, 469. [Google Scholar] [CrossRef] [PubMed]
- Jupe, F.; Witek, K.; Verweij, W.; Sliwka, J.; Pritchard, L.; Etherington, G.J.; Maclean, D.; Cock, P.J.; Leggett, R.M.; Bryan, G.J.; et al. Resistance gene enrichment sequencing (RenSeq) enables reannotation of the NB-LRR gene family from sequenced plant genomes and rapid mapping of resistance loci in segregating populations. Plant J. 2013, 76, 530–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janke, C.; Bulinski, J.C. Post-translational regulation of the microtubule cytoskeleton: Mechanisms and functions. Nat. Rev. Mol. Cell Biol. 2011, 12, 773–786. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.S.; Marquez, Y.; Kalyna, M.; Barta, A. Complexity of the alternative splicing landscape in plants. Plant Cell 2013, 25, 3657–3683. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Zhang, X.; Yang, X.; Li, G.; Tang, S.; Wang, S.; Ding, Y.; Liu, Z. Proteomic analysis of proteins related to rice grain chalkiness using iTRAQ and a novel comparison system based on a notched-belly mutant with white-belly. BMC Plant Biol. 2014, 14, 163. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.Y.; Fang, Z.T.; Lin, J.K.; Sun, Y.; Du, Z.Z.; Guo, Y.L.; Liu, J.H.; Liang, Y.R.; Ye, J.H. Complementary iTRAQ Proteomic and Transcriptomic Analyses of Leaves in Tea Plant (Camellia sinensis L.) with Different Maturity and Regulatory Network of Flavonoid Biosynthesis. J. Proteome Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Zhang, H.; Mandal, S.N.; Wang, C.; Chen, C.; Ji, W. Quantitative proteomics reveals the central changes of wheat in response to powdery mildew. J. Proteom. 2016, 130, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Hollender, C.A.; Kang, C.; Darwish, O.; Geretz, A.; Matthews, B.F.; Slovin, J.; Alkharouf, N.; Liu, Z. Floral transcriptomes in woodland strawberry uncover developing receptacle and anther gene networks. Plant Physiol. 2014, 165, 1062–1075. [Google Scholar] [CrossRef] [PubMed]
- Xue, F.; Ji, W.; Wang, C.; Zhang, H.; Yang, B. High-density mapping and marker development for the powdery mildew resistance gene PmAS846 derived from wild emmer wheat (Triticum turgidum var. dicoccoides). Theor. Appl. Genet. 2012, 124, 1549–1560. [Google Scholar] [CrossRef] [PubMed]
- Dangl, J.L.; Horvath, D.M.; Staskawicz, B.J. Pivoting the plant immune system from dissection to deployment. Science 2013, 341, 746–751. [Google Scholar] [CrossRef] [PubMed]
- Baggs, E.; Dagdas, G.; Krasileva, K.V. NLR diversity, helpers and integrated domains: Making sense of the NLR IDentity. Curr. Opin. Plant Biol. 2017, 38, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Ajuh, P.; Kuster, B.; Panov, K.; Zomerdijk, J.C.; Mann, M.; Lamond, A.I. Functional analysis of the human CDC5L complex and identification of its components by mass spectrometry. EMBO J. 2000, 19, 6569–6581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, P.; Xu, R. Structure and assembly of the SF3a splicing factor complex of U2 snRNP. EMBO J. 2012, 31, 1579–1590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scotti, M.M.; Swanson, M.S. RNA mis-splicing in disease. Nat. Rev. Genet. 2016, 17, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Hardin, J.W.; Warnasooriya, C.; Kondo, Y.; Nagai, K.; Rueda, D. Assembly and dynamics of the U4/U6 di-snRNP by single-molecule FRET. Nucleic Acids Res. 2015, 43, 10963–10974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makarov, E.M.; Makarova, O.V.; Achsel, T.; Lührmann, R. The human homologue of the yeast splicing factor prp6p contains multiple TPR elements and is stably associated with the U5 snRNP via protein-protein interactions. J. Mol. Biol. 2000, 298, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Tanackovic, G.; Ransijn, A.; Ayuso, C.; Harper, S.; Berson, E.L.; Rivolta, C. A missense mutation in PRPF6 causes impairment of pre-mRNA splicing and autosomal-dominant retinitis pigmentosa. Am. J. Hum. Genet. 2011, 88, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Virbasius, C.M.A.; Wagner, S.; Green, M.R. A human nuclear-localized chaperone that regulates dimerization, DNA binding, and transcriptional activity of bZIP proteins. Mol. Cell 1999, 4, 219–228. [Google Scholar] [CrossRef]
- Rondon, A.G.; Jimeno, S.; Garcia-Rubio, M.; Aguilera, A. Molecular evidence that the eukaryotic THO/TREX complex is required for efficient transcription elongation. J. Biol. Chem. 2003, 278, 39037–39043. [Google Scholar] [CrossRef] [PubMed]
- Katahira, J. mRNA export and the TREX complex. Biochim. Biophys. Acta 2012, 1819, 507–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopytova, D.V.; Orlova, A.V.; Krasnov, A.N.; Gurskiy, D.Y.; Nikolenko, J.V.; Nabirochkina, E.N.; Shidlovskii, Y.V.; Georgieva, S.G. Multifunctional factor ENY2 is associated with the THO complex and promotes its recruitment onto nascent mRNA. Genes Dev. 2010, 24, 86–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoning, J.C.; Streitner, C.; Meyer, I.M.; Gao, Y.; Staiger, D. Reciprocal regulation of glycine-rich RNA-binding proteins via an interlocked feedback loop coupling alternative splicing to nonsense-mediated decay in Arabidopsis. Nucleic Acids Res. 2008, 36, 6977–6987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, Y.; Wang, T.; Wang, K.; Wang, X.; Fu, Y.; Huang, L.; Kang, Z. Transcriptome Analysis Provides Insights into the Mechanisms Underlying Wheat Plant Resistance to Stripe Rust at the Adult Plant Stage. PLoS ONE 2016, 11, e0150717. [Google Scholar] [CrossRef] [PubMed]
- Graumann, K.; Runions, J.; Evans, D.E. Characterization of SUN-domain proteins at the higher plant nuclear envelope. Plant J. 2010, 61, 134–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, S.P.; Simmons, C.R.; Bass, H.W. Structure and expression of the maize (Zea mays L.) SUN-domain protein gene family: Evidence for the existence of two divergent classes of SUN proteins in plants. BMC Plant Biol. 2010, 10, 269. [Google Scholar] [CrossRef] [PubMed]
- Coates, M.E.; Beynon, J.L. Hyaloperonospora Arabidopsidis as a pathogen model. Annu. Rev. Phytopathol. 2010, 48, 329–345. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Graumann, K.; Meier, I. The plant nuclear envelope as a multifunctional platform LINCed by SUN and KASH. J. Exp. Bot. 2015, 66, 1649–1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felts, S.J.; Toft, D.O. p23, a simple protein with complex activities. Cell Stress Chaperones 2003, 8, 108–113. [Google Scholar] [CrossRef]
- Takahashi, A.; Casais, C.; Ichimura, K.; Shirasu, K. HSP90 interacts with RAR1 and SGT1 and is essential for RPS2-mediated disease resistance in Arabidopsis. Proc. Natl. Acad. Sci. USA 2003, 100, 11777–11782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.F.; Wei, X.; Fan, R.; Zhou, H.; Wang, X.; Yu, C.; Dong, L.; Dong, Z.; Wang, X.; Kang, Z.; et al. Molecular analysis of common wheat genes encoding three types of cytosolic heat shock protein 90 (Hsp90): Functional involvement of cytosolic Hsp90s in the control of wheat seedling growth and disease resistance. New Phytol. 2011, 191, 418–431. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Eckert, C.; Anschutz, U.; Scholz, M.; Held, K.; Waadt, R.; Reyer, A.; Hippler, M.; Becker, D.; Kudla, J. Phosphorylation of calcineurin B-like (CBL) calcium sensor proteins by their CBL-interacting protein kinases (CIPKs) is required for full activity of CBL-CIPK complexes toward their target proteins. J. Biol. Chem. 2012, 287, 7956–7968. [Google Scholar] [CrossRef] [PubMed]
- Kurusu, T.; Hamada, J.; Nokajima, H.; Kitagawa, Y.; Kiyoduka, M.; Takahashi, A.; Hanamata, S.; Ohno, R.; Hayashi, T.; Okada, K.; et al. Regulation of Microbe-Associated Molecular Pattern-Induced Hypersensitive Cell Death, Phytoalexin Production, and Defense Gene Expression by Calcineurin B-Like Protein-Interacting Protein Kinases, OsCIPK14/15, in Rice Cultured Cells. Plant Physiol. 2010, 153, 678–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luan, S. The CBL-CIPK network in plant calcium signaling. Trends Plant Sci. 2009, 14, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Garcia, A.; Cayla, X.; Guergnon, J.; Dessauge, F.; Hospital, V.; Rebollo, M.P.; Fleischer, A.; Rebollo, A. Serine/threonine protein phosphatases PP1 and PP2A are key players in apoptosis. Biochimie 2003, 85, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Lan, W.Z.; Lee, S.C.; Che, Y.F.; Jiang, Y.Q.; Luan, S. Mechanistic analysis of AKT1 regulation by the CBL-CIPK-PP2CA interactions. Mol. Plant 2011, 4, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Ballare, C.L. Light regulation of plant defense. Annu. Rev. Plant Biol. 2014, 65, 335–363. [Google Scholar] [CrossRef] [PubMed]
- Hornitschek, P.; Kohnen, M.V.; Lorrain, S.; Rougemont, J.; Ljung, K.; Lopez-Vidriero, I.; Franco-Zorrilla, J.M.; Solano, R.; Trevisan, M.; Pradervand, S.; et al. Phytochrome interacting factors 4 and 5 control seedling growth in changing light conditions by directly controlling auxin signaling. Plant J. 2012, 71, 699–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karpinski, S.; Gabrys, H.; Mateo, A.; Karpinska, B.; Mullineaux, P.M. Light perception in plant disease defence signalling. Curr. Opin. Plant Biol. 2003, 6, 390–396. [Google Scholar] [CrossRef]
- Uauy, C.; Wulff, B.B.H.; Dubcovsky, J. Combining Traditional Mutagenesis with New High-Throughput Sequencing and Genome Editing to Reveal Hidden Variation in Polyploid Wheat. Annu. Rev. Genet. 2017, 51, 435–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagudah, E.S.; Krattinger, S.G.; Herrera-Foessel, S.; Singh, R.P.; Huerta-Espino, J.; Spielmeyer, W.; Brown-Guedira, G.; Selter, L.L.; Keller, B. Gene-specific markers for the wheat gene Lr34/Yr18/Pm38 which confers resistance to multiple fungal pathogens. Theor. Appl. Genet. 2009, 119, 889–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Hendrickson, D.G.; Sauvageau, M.; Goff, L.; Rinn, J.L.; Pachter, L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 2013, 31, 46–53. [Google Scholar] [CrossRef] [PubMed]
- International Wheat Genome Sequencing Consortium. A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science 2014, 345, 1251788. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Ernst, J.; Bar-Joseph, Z. STEM: A tool for the analysis of short time series gene expression data. BMC Bioinform. 2006, 7, 191. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway ID | Pathway Description | DAP Number | P/Q Value | ||||
|---|---|---|---|---|---|---|---|
| 24 hpi | 48 hpi | 72 hpi | 24 hpi | 48 hpi | 72 hpi | ||
| ko03010 | Ribosome | 297 (31.5%) | 218 (21.9%) | 309 (31.7%) | 0*/0 | 0*/0 | 0*/0 |
| ko00190 | Oxidative phosphorylation | 124 (13.2%) | 192 (19.3%) | 112 (11.5%) | 0*/0 | 0*/0 | 0*/0 |
| ko00195 | Photosynthesis | 133 (14.1%) | 95 (9. 6%) | 50 (5.1%) | 0*/0 | 0*/0 | 0.076/0.411 |
| ko04626 | Plant-pathogen interaction | 33 (3.5%) | 27 (2.7%) | 30 (3.1%) | 0*/0 | 0.002*/0.015 | 0*/0.002 |
| ko04145 | Phagosome | 26 (2.8%) | 26 (2.6%) | 20 (2.1%) | 0.007*/0.067 | 0.013*/0.061 | 0.189/0.758 |
| ko00260 | Glycine, serine and threonine metabolism | 32 (3.4%) | 27 (2.7%) | 49 (5.0%) | 0.001*/0.119 | 0.041*/0.160 | 0*/0 |
| ko00740 | Riboflavin metabolism | – | 26 (2.6%) | 11 (1.1%) | – | 0*/0 | 0.012*/0.107 |
| ko01200 | Carbon metabolism | 87 (9.2%) | 116 (11.7%) | 110 (11.3%) | 0.085/0.661 | 0*/0 | 0*/0.001 |
| ko00941 | Flavonoid biosynthesis | 13 (1.4%) | 13 (1.3%) | – | 0*/0 | 0*/0 | – |
| ko00450 | Selenocompound metabolism | 5 (0.5%) | 23 (2.3%) | 4 (0.4%) | 0.721/1 | 0*/0 | 0.875/1 |
| ko00261 | Monobactam biosynthesis | – | 22 (2.2%) | – | – | 0*/0 | – |
| ko03022 | Basal transcription factors | – | 5 (0.5%) | 5 (0.5%) | – | 0*/0 | 0*/0 |
| ko00020 | Citrate cycle (TCA cycle) | 16 (1.7%) | 39 (3.9%) | 35 (3.6%) | 0.684/1 | 0*/0 | 0*/0.001 |
| ko00630 | Glyoxylate and dicarboxylate metabolism | 35 (3.7%) | 50 (5.0%) | 59 (6.0%) | 0.193/1 | 0.001*/0.006 | 0*/0 |
| ko00250 | Alanine, aspartate, and glutamate metabolism | 13 (1.4%) | 22 (2.2%) | 19 (1.9%) | 0.248/1 | 0.001*/0.009 | 0.012/0.107 |
| ko00920 | Sulfur metabolism | 6 (0.6%) | 24 (2.4%) | 6 (0.6%) | 0.989/1 | 0.003*/0.019 | 0.992/1 |
| ko01230 | Biosynthesis of amino acids | 48 (5.1%) | 82 (8.3%) | 76 (7.8%) | 0.933/1 | 0.003*/0.019 | 0.018/0.143 |
| ko04712 | Circadian rhythm-plant | 15 (1.6%) | 13 (1.3%) | – | 0*/0.006 | 0.006*/0.032 | – |
| ko00480 | Glutathione metabolism | 25 (2.6%) | 35 (3.5%) | 26 (2.7%) | 0.24192/1 | 0.006*/0.032 | 0.230*/0.826 |
| ko00670 | One carbon pool by folate | 1 (0.1%) | 11 (1.1%) | 10 (1.0%) | 0.997/1 | 0.031*/0.131 | 0.061/0.376 |
| ko00330 | Arginine and proline metabolism | 4 (0.4%) | 14 (1.4%) | 10 (1.0%) | 0.956/1 | 0.032*/0.131 | 0.278/0.919 |
| ko00380 | Tryptophan metabolism | 6 (0.6%) | – | 8 (0.8%) | 0.180/1 | – | 0.041*/0.297 |
| KEGG Pathway | Gene ID | Connectivity | Description |
|---|---|---|---|
| Nuclear envelope | Traes_1BS_7101D8D63 | 1385.04 | SUN domain-containing protein 3-like |
| Ribosome | Traes_1BS_71C6FDF1A | 975.60 | Ribosomal protein L28 |
| Ribosome | Traes_1BS_DD122AEA6 | 41.87 | Ribosomal protein S18 |
| Ribosome | Traes_1BS_262C70465 | – | Ribosomal protein S19 (chloroplast) |
| Ribosome | Traes_1BS_F2A8D87C5 | – | Ribosomal protein L2 (chloroplast) |
| Spliceosome | Traes_1BS_68CB9BCB3 | 23.14 | U1 small nuclear ribonucleoprotein A |
| Spliceosome | Traes_1BS_D86496196 | 890.18 | mRNA transport factor |
| Protein processing in ER | Traes_1BS_C2166CFB6 | 726.51 | Protein disulfide isomerase-like 2-1 |
| Basal transcription factors | Traes_1BS_30B4D96B4 | 284.64 | Transcription initiation factor TFIID subunit 13 |
| Transcription factors | Traes_1BS_351490964 | - | Transcription factor BTF3-like |
| Plant hormone signal transduction | Traes_1BS_D1FCBFBE8 | 342.10 | Transcription factor PIF3 |
| Plant hormone signal transduction | Traes_1BS_8A19C460B | 292.68 | Auxin-responsive protein IAA15 |
| Plant hormone signal transduction | Traes_1BS_F1838F858 | 123.94 | Abscisic acid receptor PYL8-like |
| Arachidonic acid metabolism | Traes_1BS_A6A616FA0 | 399.75 | p23 (Hsp90 co-chaperone) |
| Insulin resistance | Traes_1BS_BF71914E7 | 122.99 | Ser/thr-protein phosphatase PP1 isozyme 9 |
| Fatty acid elongation | Traes_1BS_F54A0F09C | 122.75 | 3-Ketoacyl-CoA synthase 12 |
| Carotenoid biosynthesis | Traes_1BS_5963FC5FC | 109.15 | Prolycopene isomerase, chloroplastic-like |
| Regulation of autophagy | Traes_1BS_C385DE03B | 90.79 | CBL-interacting protein kinase 17 |
| Flavonoid biosynthesis. | Traes_1BS_B00164D10 | 47.44 | Chalcone-flavonone isomerase 1B-1 |
| Defense | Traes_1BS_2BCFB8A20 | 182.20 | Subtilisin-chymotrypsin inhibitor-2A |
| Selenocompound metabolism | Traes_1BS_B67548763 | 35.60 | Methionine S-methyltransferase |
| Val, leu and isoleucine degradation | Traes_1BS_190C56A4E | 30.76 | Dihydrolipoyl dehydrogenase 1, mitochondrial |
| Peroxisome | Traes_1BS_A344E8CC2 | 24.82 | Peroxisomal membrane protein 2 |
| Peroxisome | Traes_1BS_40116F0E7 | 10.18 | Peroxisomal membrane protein PEX14-like |
| Glutathione metabolism | Traes_1BS_F226DF9B4 | 368.85 | Dehydroascorbate reductase |
| Oxidative phosphorylation | Traes_1BS_6F8E6B9B6 | – | ATP synthase protein atp8-1(3 copes in 1BS) |
| Cell cycle | Traes_1BS_4E2FA3101 | – | TPA: histone H4.3 |
| Fructose and mannose metabolism | Traes_1BS_0039045F7 | 215.75 | Fructose-2, 6-biphosphatase 1 |
| Pyruvate metabolism | Traes_1BS_478039FB4 | 201.84 | Probable lactoylglutathione lyase, chloroplast |
| Porphyrin metabolism | Traes_1BS_ABD495014 | 154.21 | Glutamyl tRNA reductase |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.; Fu, Y.; Guo, H.; Zhang, L.; Wang, C.; Song, W.; Yan, Z.; Wang, Y.; Ji, W. Transcriptome and Proteome-Based Network Analysis Reveals a Model of Gene Activation in Wheat Resistance to Stripe Rust. Int. J. Mol. Sci. 2019, 20, 1106. https://doi.org/10.3390/ijms20051106
Zhang H, Fu Y, Guo H, Zhang L, Wang C, Song W, Yan Z, Wang Y, Ji W. Transcriptome and Proteome-Based Network Analysis Reveals a Model of Gene Activation in Wheat Resistance to Stripe Rust. International Journal of Molecular Sciences. 2019; 20(5):1106. https://doi.org/10.3390/ijms20051106
Chicago/Turabian StyleZhang, Hong, Ying Fu, Huan Guo, Lu Zhang, Changyou Wang, Weining Song, Zhaogui Yan, Yajuan Wang, and Wanquan Ji. 2019. "Transcriptome and Proteome-Based Network Analysis Reveals a Model of Gene Activation in Wheat Resistance to Stripe Rust" International Journal of Molecular Sciences 20, no. 5: 1106. https://doi.org/10.3390/ijms20051106