Refining the Phenotype of Recurrent Rearrangements of Chromosome 16

 , , ,
, , , 
Abstract
:1. Introduction
2. Results
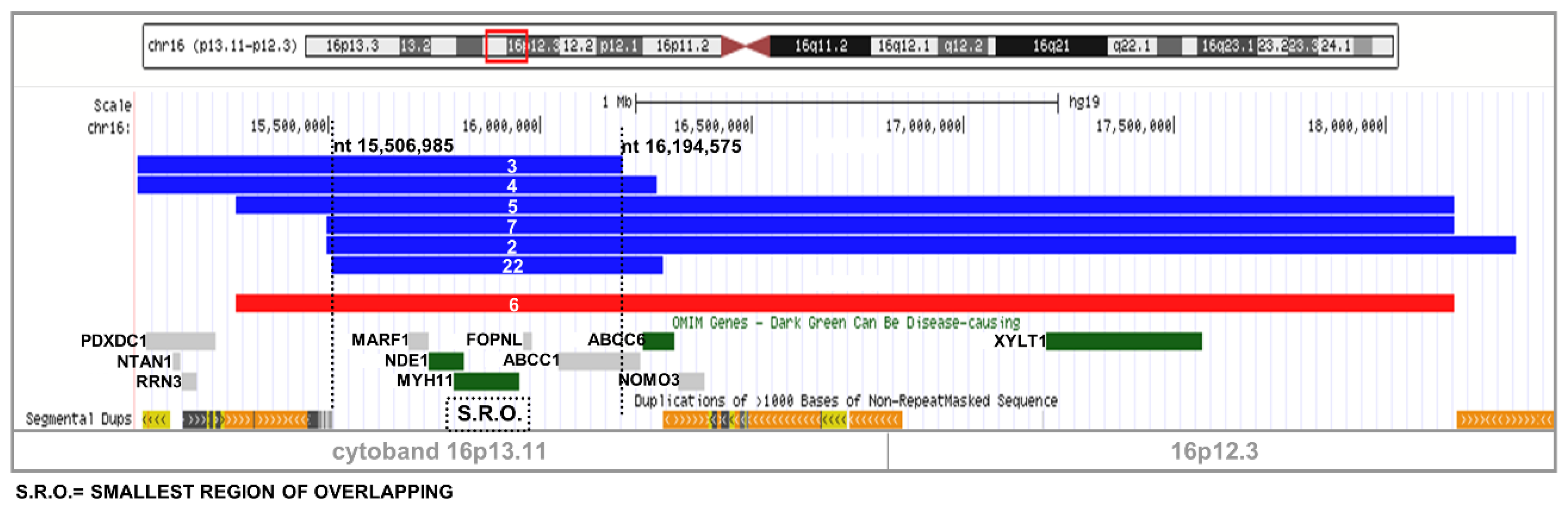
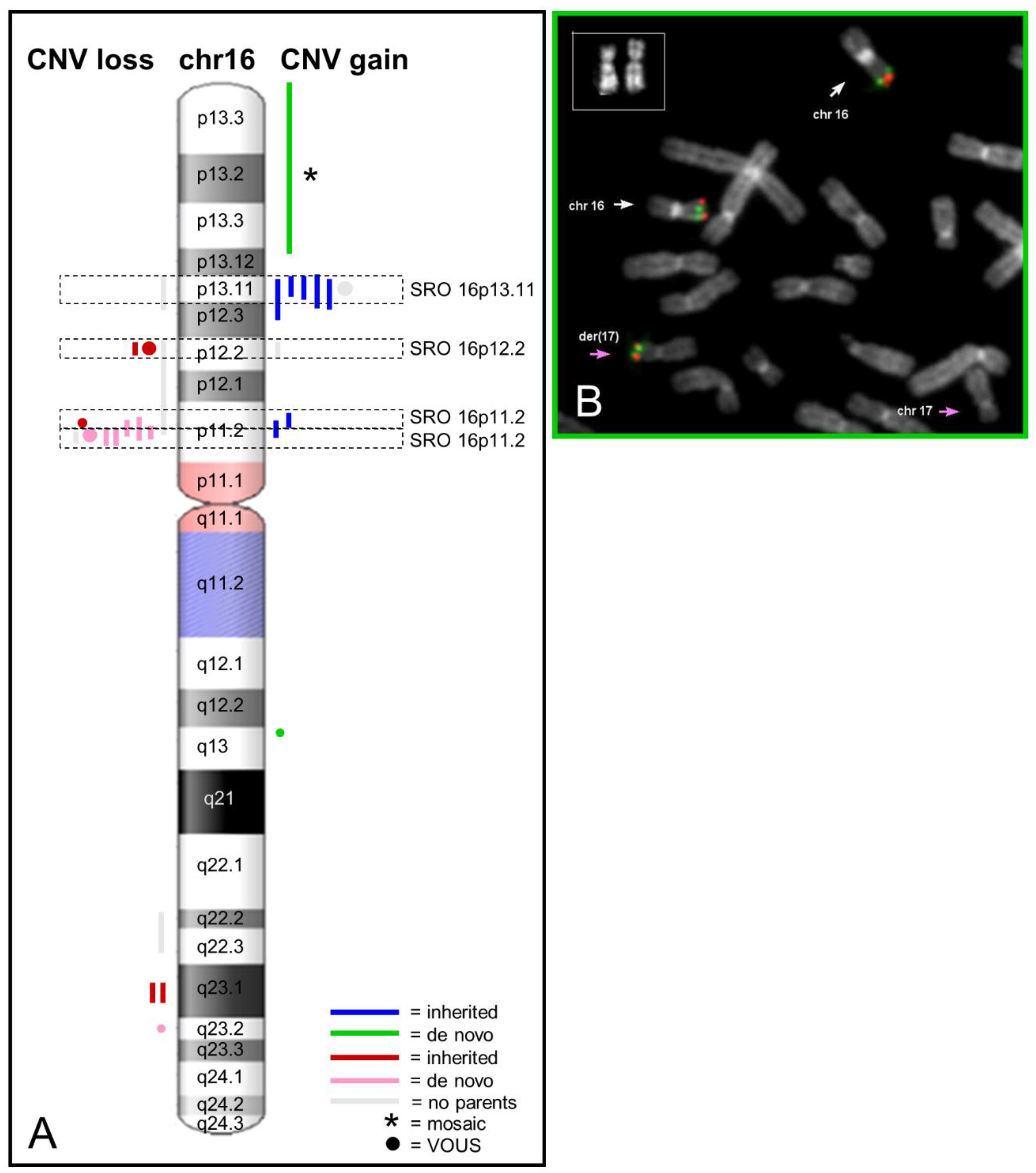
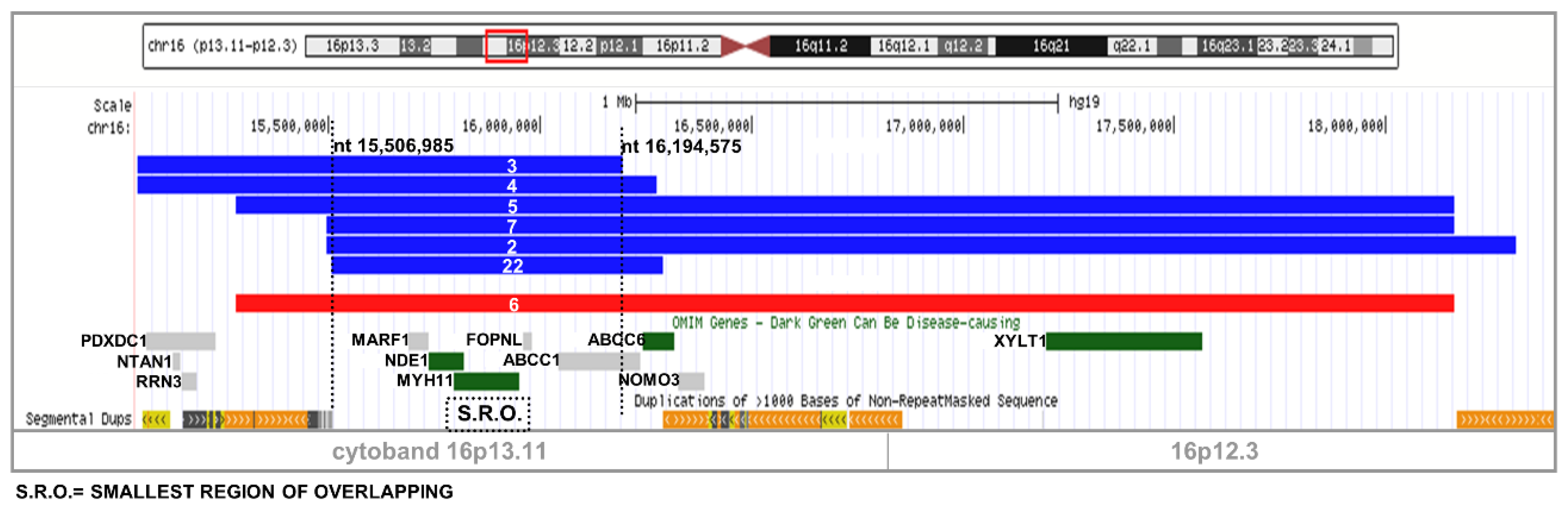
2.1. Recurrent Microdeletion/Duplication of 16p13.11
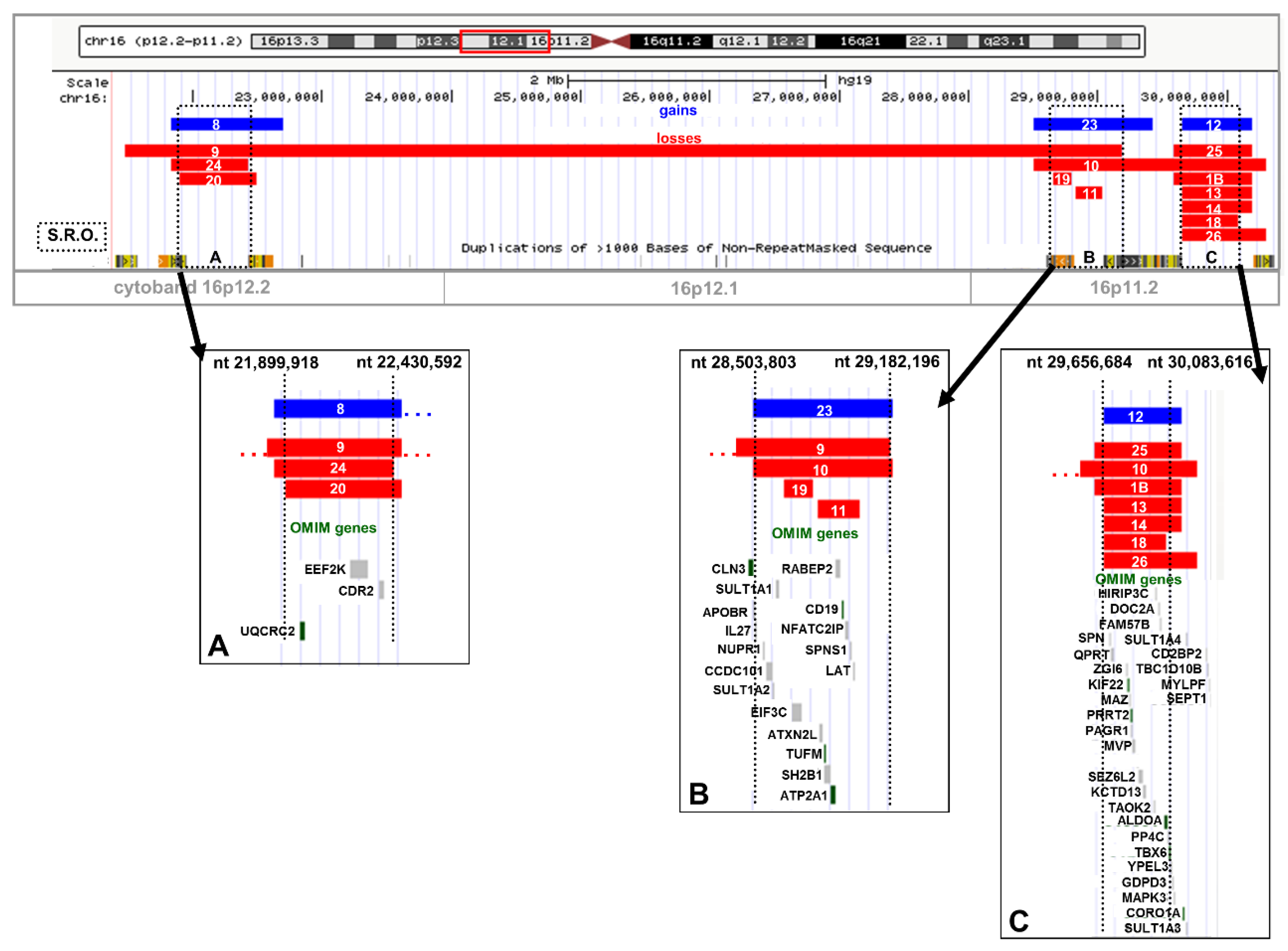
2.2. Recurrent Microdeletion/Duplication of 16p12.2
2.3. Recurrent Microdeletion/Duplication of 16p11.2
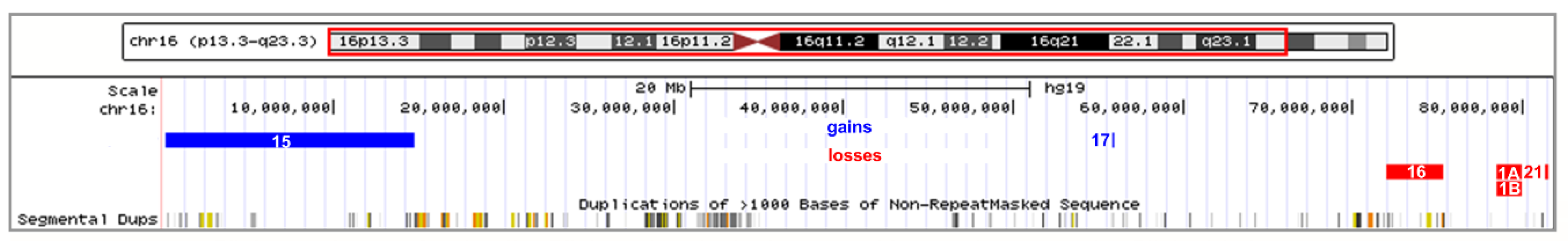
2.4. Atypical Rearrangements of Chromosome 16
2.5. Additional Variants
3. Discussion
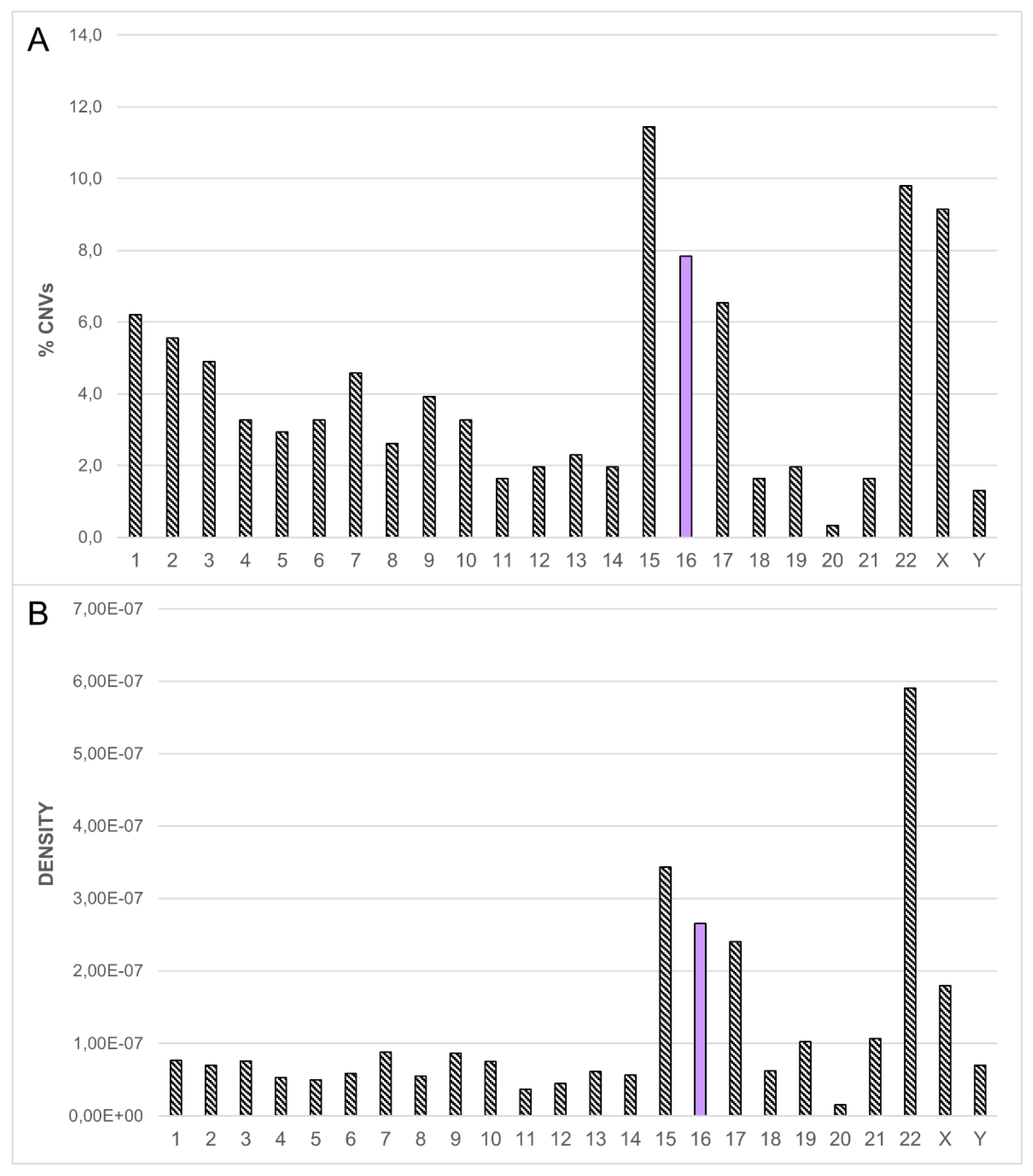
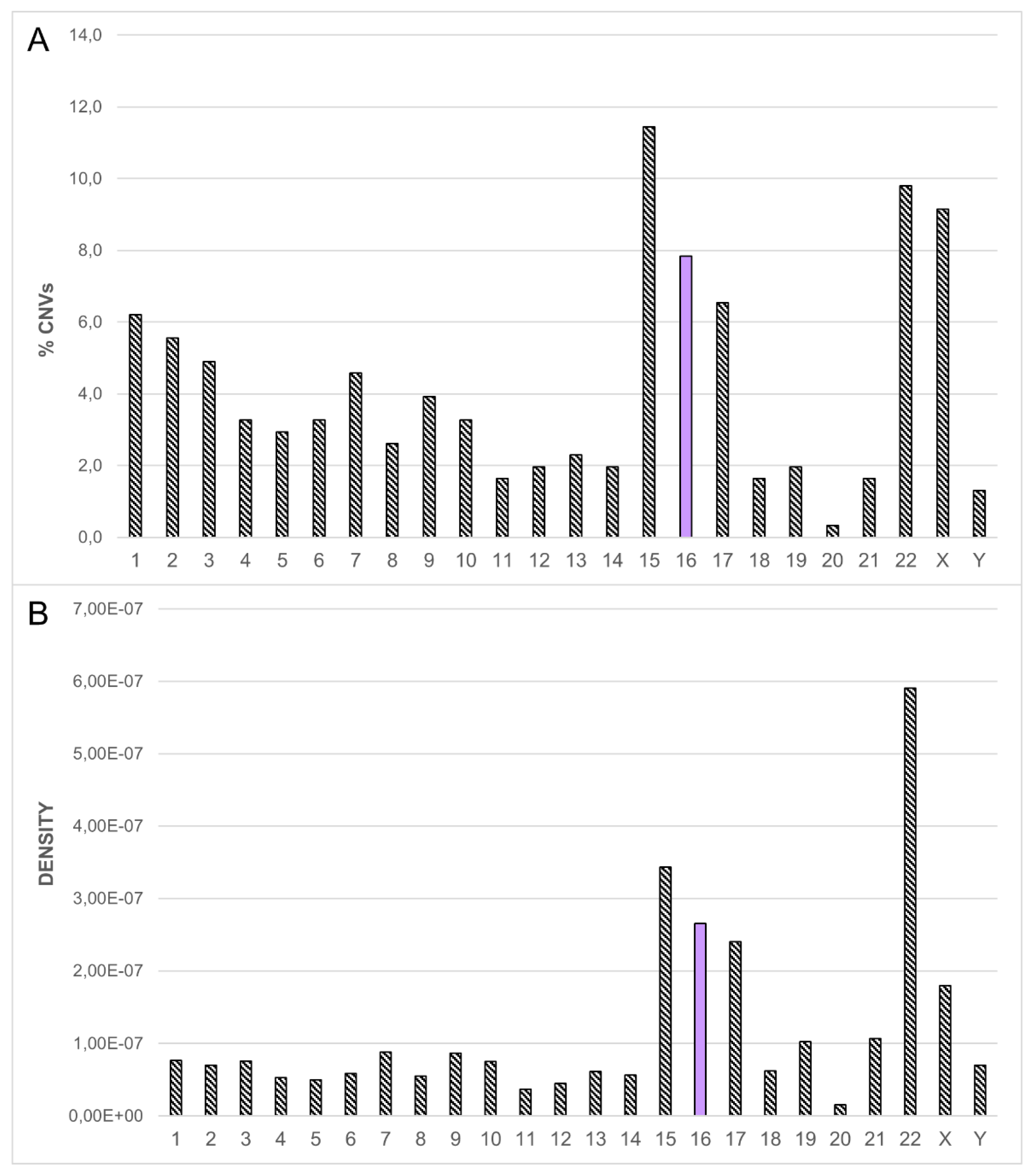
3.1. Dissection of the Chromosome 16 CNV-Morbidity Map
3.2. Supporting the Second-Site Genomic Alterations to Explain Incomplete Penetrance and Variable Expressivity
4. Materials and Methods
4.1. Participants
4.2. Cytogenetic and FISH Analysis
4.3. Array CGH Analysis
4.4. Evolutionary Genetic Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CAs | congenital abnormalities |
| CAS | childhood apraxia of speech |
| CGH | comparative genomic hybridization |
| CNVs | copy number variations |
| DLRS | derivative log ratio spread |
| EEG | electroencephalography |
| FISH | fluorescent in situ hybridization |
| ID | intellectual disability |
| ISCN | International System for Human Cytogenomic Nomenclature |
| IUGR | intrauterine growth restriction |
| MR/MRC | mental retardation and/or multiple congenital anomalies |
| NAHR | non-allelic homologous recombination |
| NDs | neurodevelopmental disorders |
| OMIM | online mendelian inheritance in man |
| SNP | single nucleotide polymorphism |
| SRO | smallest region of overlapping |
| WGD | whole genome duplication |
References
- Scherer, S. Guide to the Human Genome; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2010. [Google Scholar]
- Fatakia, S.N.; Mehta, I.S.; Rao, B.J. Systems-level chromosomal parameters represent a suprachromosomal basis for the non-random chromosomal arrangement in human interphase nuclei. Sci. Rep. 2016, 6, 36819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, J.; Han, C.; Gordon, L.A.; Terry, A.; Prabhakar, S.; She, X.; Xie, G.; Hellsten, U.; Chan, Y.M.; Altherr, M. The sequence and analysis of duplication-rich human chromosome 16. Nature 2004, 432, 988–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, M.E.; Cheng, Z.; Morrison, V.A.; Scherer, S.; Ventura, M.; Gibbs, R.A.; Green, E.D.; Eichler, E.E.; Program NIoHISCCS. Recurrent duplication-driven transposition of DNA during hominoid evolution. Proc. Natl. Acad. Sci. USA 2006, 103, 17626–17631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nuttle, X.; Giannuzzi, G.; Duyzend, M.H.; Schraiber, J.G.; Narvaiza, I.; Sudmant, P.H.; Penn, O.; Chiatante, G.; Malig, M.; Huddleston, J.; et al. Emergence of a Homo sapiens-specific gene family and chromosome 16p11.2 CNV susceptibility. Nature 2016, 536, 205–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, L.; Shang, X.; Yi, S.; Cai, R.; Li, Z.; Liu, C.; Liang, Y.; Cai, D.; Zhang, F.; Xu, X. Two novel copy number variations involving the α-globin gene cluster on chromosome 16 cause thalassemia in two Chinese families. Mol. Genet. Genomics 2016, 291, 1443–1450. [Google Scholar] [CrossRef] [PubMed]
- Torres, F.; Barbosa, M.; Maciel, P. Recurrent copy number variations as risk factors for neurodevelopmental disorders: Critical overview and analysis of clinical implications. J. Med. Genet. 2015, 53, 73–90. [Google Scholar] [CrossRef] [PubMed]
- Bijlsma, E.K.; Gijsbers, A.C.; Schuurs-Hoeijmakers, J.H.; van Haeringen, A.; Fransen van de Putte, D.E.; Anderlid, B.M.; Lundin, J.; Lapunzina, P.; Pérez Jurado, L.A.; Delle Chiaie, B.; et al. Extending the phenotype of recurrent rearrangements of 16p11.2: Deletions in mentally retarded patients without autism and in normal individuals. Eur. J. Med. Genet. 2009, 52, 77–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinman, K.J.; Spence, S.J.; Ramocki, M.B.; Proud, M.B.; Kessler, S.K.; Marco, E.J.; Green Snyder, L.; D’Angelo, D.; Chen, Q.; Chung, W.K.; et al. 16p11.2 deletion and duplication: Characterizing neurologic phenotypes in a large clinically ascertained cohort. Am. J. Med. Genet. A 2016, 170, 2943–2955. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, J.J.; Nelson, C.A. Deletion and duplication of 16p11.2 are associated with opposing effects on visual evoked potential amplitude. Mol. Autism 2016, 7, 30. [Google Scholar] [CrossRef] [PubMed]
- Bernier, R.; Hudac, C.M.; Chen, Q.; Zeng, C.; Wallace, A.S.; Gerdts, J.; Earl, R.; Peterson, J.; Wolken, A.; Peters, A.; et al. Developmental trajectories for young children with 16p11.2 copy number variation. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2017, 174, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Abdouh, M.; Facchino, S.; Chatoo, W.; Balasingam, V.; Ferreira, J.; Bernier, G. BMI1 sustains human glioblastoma multiforme stem cell renewal. J. Neurosci. 2009, 29, 8884–8896. [Google Scholar] [CrossRef] [PubMed]
- Jacquemont, S.; Reymond, A.; Zufferey, F.; Harewood, L.; Walters, R.G.; Kutalik, Z.; Martinet, D.; Shen, Y.; Valsesia, A.; Beckmann, N.D.; et al. Mirror extreme BMI phenotypes associated with gene dosage at the chromosome 16p11.2 locus. Nature 2011, 478, 97–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoppel, L.J.; Kazdoba, T.M.; Schaffler, M.D.; Preza, A.R.; Heynen, A.; Crawley, J.N.; Bear, M.F. R-Baclofen Reverses Cognitive Deficits and Improves Social Interactions in Two Lines of 16p11.2 Deletion Mice. Neuropsychopharmacology 2018, 43, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Walters, R.G.; Coin, L.J.; Ruokonen, A.; de Smith, A.J.; El-Sayed Moustafa, J.S.; Jacquemont, S.; Elliott, P.; Esko, T.; Hartikainen, A.L.; Laitinen, J.; et al. Rare genomic structural variants in complex disease: Lessons from the replication of associations with obesity. PLoS ONE 2013, 8, e58048. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, D.; Lebon, S.; Chen, Q.; Martin-Brevet, S.; Snyder, L.G.; Hippolyte, L.; Hanson, E.; Maillard, A.M.; Faucett, W.A.; Macé, A.; et al. Defining the Effect of the 16p11.2 Duplication on Cognition, Behavior, and Medical Comorbidities. JAMA Psychiatry 2016, 73, 20–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horev, G.; Ellegood, J.; Lerch, J.P.; Son, Y.E.; Muthuswamy, L.; Vogel, H.; Krieger, A.M.; Buja, A.; Henkelman, R.M.; Wigler, M.; et al. Dosage-dependent phenotypes in models of 16p11.2 lesions found in autism. Proc. Natl. Acad. Sci. USA 2011, 108, 17076–17081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giaroli, G.; Bass, N.; Strydom, A.; Rantell, K.; McQuillin, A. Does rare matter? Copy number variants at 16p11.2 and the risk of psychosis: A systematic review of literature and meta-analysis. Schizophr. Res. 2014, 159, 340–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raca, G.; Baas, B.S.; Kirmani, S.; Laffin, J.J.; Jackson, C.A.; Strand, E.A.; Jakielski, K.J.; Shriberg, L.D. Childhood Apraxia of Speech (CAS) in two patients with 16p11.2 microdeletion syndrome. Eur. J. Hum. Genet. 2013, 21, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Hannes, F.D.; Sharp, A.J.; Mefford, H.C.; de Ravel, T.; Ruivenkamp, C.A.; Breuning, M.H.; Fryns, J.P.; Devriendt, K.; Van Buggenhout, G.; Vogels, A.; et al. Recurrent reciprocal deletions and duplications of 16p13.11: The deletion is a risk factor for MR/MCA while the duplication may be a rare benign variant. J. Med. Genet. 2009, 46, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Kuang, S.Q.; Guo, D.C.; Prakash, S.K.; McDonald, M.L.; Johnson, R.J.; Wang, M.; Regalado, E.S.; Russell, L.; Cao, J.M.; Kwartler, C.; et al. Recurrent chromosome 16p13.1 duplications are a risk factor for aortic dissections. PLoS Genet. 2011, 7, e1002118. [Google Scholar] [CrossRef] [PubMed]
- Girirajan, S.; Rosenfeld, J.A.; Coe, B.P.; Parikh, S.; Friedman, N.; Goldstein, A.; Filipink, R.A.; McConnell, J.S.; Angle, B.; Meschino, W.S.; et al. Phenotypic heterogeneity of genomic disorders and rare copy-number variants. N. Engl. J. Med. 2012, 367, 1321–1331. [Google Scholar] [CrossRef] [PubMed]
- Tropeano, M.; Ahn, J.W.; Dobson, R.J.; Breen, G.; Rucker, J.; Dixit, A.; Pal, D.K.; McGuffin, P.; Farmer, A.; White, P.S.; et al. Male-biased autosomal effect of 16p13.11 copy number variation in neurodevelopmental disorders. PLoS ONE 2013, 8, e61365. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.; Deng, L.; Xia, Y.; Wei, X.; Cao, Y.; Guo, R.; Zhang, R.; Guo, J.; Liang, D.; Wu, L. A clear bias in parental origin of de novo pathogenic CNVs related to intellectual disability, developmental delay and multiple congenital anomalies. Sci. Rep. 2017, 7, 44446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ullmann, R.; Turner, G.; Kirchhoff, M.; Chen, W.; Tonge, B.; Rosenberg, C.; Field, M.; Vianna-Morgante, A.M.; Christie, L.; Krepischi-Santos, A.C.; et al. Array CGH identifies reciprocal 16p13.1 duplications and deletions that predispose to autism and/or mental retardation. Hum. Mutat. 2007, 28, 674–682. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, C.; Knijnenburg, J.; Bakker, E.; Vianna-Morgante, A.M.; Sloos, W.; Otto, P.A.; Kriek, M.; Hansson, K.; Krepischi-Santos, A.C.; Fiegler, H.; et al. Array-CGH detection of micro rearrangements in mentally retarded individuals: Clinical significance of imbalances present both in affected children and normal parents. J. Med. Genet. 2006, 43, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Kirchhoff, M.; Gerdes, T.; Brunebjerg, S.; Bryndorf, T. Investigation of patients with mental retardation and dysmorphic features using comparative genomic hybridization and subtelomeric multiplex ligation dependent probe amplification. Am. J. Med. Genet. A 2005, 139, 231–233. [Google Scholar] [CrossRef] [PubMed]
- Sanders, S.J.; Ercan-Sencicek, A.G.; Hus, V.; Luo, R.; Murtha, M.T.; Moreno-De-Luca, D.; Chu, S.H.; Moreau, M.P.; Gupta, A.R.; Thomson, S.A.; et al. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron 2011, 70, 863–885. [Google Scholar] [CrossRef] [PubMed]
- Hanson, E.; Bernier, R.; Porche, K.; Jackson, F.I.; Goin-Kochel, R.P.; Snyder, L.G.; Snow, A.V.; Wallace, A.S.; Campe, K.L.; Zhang, Y.; et al. The cognitive and behavioral phenotype of the 16p11.2 deletion in a clinically ascertained population. Biol. Psychiatry 2015, 77, 785–793. [Google Scholar] [CrossRef] [PubMed]
- Fedorenko, E.; Morgan, A.; Murray, E.; Cardinaux, A.; Mei, C.; Tager-Flusberg, H.; Fisher, S.E.; Kanwisher, N. A highly penetrant form of childhood apraxia of speech due to deletion of 16p11.2. Eur. J. Hum. Genet. 2016, 24, 302–306. [Google Scholar] [CrossRef] [PubMed]
- Laffin, J.J.; Raca, G.; Jackson, C.A.; Strand, E.A.; Jakielski, K.J.; Shriberg, L.D. Novel candidate genes and regions for childhood apraxia of speech identified by array comparative genomic hybridization. Genet. Med. 2012, 14, 928–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girirajan, S.; Rosenfeld, J.A.; Cooper, G.M.; Antonacci, F.; Siswara, P.; Itsara, A.; Vives, L.; Walsh, T.; McCarthy, S.E.; Baker, C.; et al. A recurrent 16p12.1 microdeletion supports a two-hit model for severe developmental delay. Nat. Genet. 2010, 42, 203–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duyzend, M.H.; Nuttle, X.; Coe, B.P.; Baker, C.; Nickerson, D.A.; Bernier, R.; Eichler, E.E. Maternal Modifiers and Parent-of-Origin Bias of the Autism-Associated 16p11.2 CNV. Am. J. Hum. Genet. 2016, 7, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Cheung, S.W.; Shaw, C.A.; Scott, D.A.; Patel, A.; Sahoo, T.; Bacino, C.A.; Pursley, A.; Li, J.; Erickson, R.; Gropman, A.L.; et al. Microarray-based CGH detects chromosomal mosaicism not revealed by conventional cytogenetics. Am. J. Med. Genet. A 2007, 143A, 1679–1686. [Google Scholar] [CrossRef] [PubMed]
- Vermeesch, J.R.; Brady, P.D.; Sanlaville, D.; Kok, K.; Hastings, R.J. Genome-wide arrays: Quality criteria and platforms to be used in routine diagnostics. Hum. Mutat. 2012, 33, 906–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, D.T.; Adam, M.P.; Aradhya, S.; Biesecker, L.G.; Brothman, A.R.; Carter, N.P.; Church, D.M.; Crolla, J.A.; Eichler, E.E.; Epstein, C.J.; et al. Consensus statement: Chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am. J. Hum. Genet. 2010, 86, 749–764. [Google Scholar] [CrossRef] [PubMed]
- Makino, T.; McLysaght, A. Ohnologs in the human genome are dosage balanced and frequently associated with disease. Proc. Natl. Acad. Sci. USA 2010, 107, 9270–9274. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case (n) | Sex/Age (y/m/w/d) | GD/PCTL | ID and/or LD | D | Major and Minor Malformations | Heart Defects | CNS Anomalies | Other | CNV on 16 Second Hit | Size (Mb) (SRO Figure 4) | Origin |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 22 AF | M, 22w+2d | - | - | + | - | - | - | Non−immune hydrops fetalis | 16p13.11 dup | 0.76 | ? |
| 3 | M, 11y | − | Moderate ID Severe LD | + | − | − | Syncopal episodes with spasticity NE−EEG abnormalities | Aggressiveness hypermetropy | 16p13.11dup | 1.15 | mat |
| 4 | F, 4y | − | − | − | − | − | Epilepsy | - | 16p13.11 dup | 1.3 | mat |
| 5 | F, 7y | − | Mild ID | + | MI Clinodactily Absence of the 12th rib | - | Abnormal signal intensity of frontal white matter | - | 16p13.11 dup | 2.88 | pat |
| 2 | M, 22y | − | ID | + | - | Bicuspid aorta | - | Cryptorchidism Aggressiveness Hypertrichosis | 16p13.11–p12.3 dup 15q11.1q11.2 del | 2.8 2.7 | mat mat |
| 7 | F, 16m | + | − | + | rMA | Tricuspid insufficiency | Hypertonia | Silver Russel like phenotype | 16p13.11–p12.3 dup | 2.7 | mat |
| 6 | M, 38y | − | ID | − | Hip dysplasia Pectus carinatum | − | − | - | 16p13.11–p12.3 del | 2.9 | ? |
| 24 AF | F, 21w+1d | IUGR | - | − | − | − | Oligohydramnios | 16p12.2 del 22q11.23 dup | 0.59 A 1.29 | pat mat | |
| 20 | F, 3m | − | - | − | Preaxial polydactyly of the hand | 2 VSD, PFO, SH | − | Ectopic kidney | 16p12.2 del | 0.59 A | pat |
| 8 | M, 8y | − | ID and LD | + | − | − | Hypotonia Areflexia | Conductive hearing loss | 16p12.2 dup | 0.86 A | pat |
| 9 | M, 24y | + | Absent speech | + | Bilateral clubfoot PUV Bifid uvula | - | Hypotonia | Behavioural problem Hypomobility of palate | 16p12.2–p11.2 del | 7.7 A−B | ? |
| 11 | M, 10y | 97th | MLD Executive dyspraxia | + | Brachydactyly, Mild flat feet Dolichocephaly | - | EEG abnormalities | Ascending testis | 16p11.2 del 3p26.3 dup | 0.21 B 0.36 | de novo pat |
| 19 | M, 10y | 3rd | Mild ID MLD Dyspraxia | + | Genu valgum Extra rotation feet | - | - | Strabism Gynecomasty Hypogenitalism | 16p11.2 del | 0.14 B | mat |
| 23 AF twins | F, 18w+1d | - | - | - | Left ventricular hypoplasia | - | - | 16p11.2 dup | 0.92 B | mat | |
| F, 18w+1d | - | - | - | - | - | - | 16p11.2 dup | 0.92 B | mat | ||
| 10 | M, 4y | − | Mild PDD Severe LD | − | Metatarsus adductus | ASD FPO | Hypertonia | Aggressiveness Hypermetropy | 16p11.2 del | 1.8 B–C | de novo |
| 13 | M, 13y | − | Mild ID Severe LD Verbal dyspraxia | + | Genu valgum Flat feet | − | Chiari malformation type 1 | Hepatic steatosis Scoliosis | 16p11.2 del | 0.53 C | de novo |
| 14 | F, 8y | 97th | Mild ID | + | − | − | Epilepsy Periventricular nodular heterotopia | Lumbar hyperlordosis Isolated premature thelarche | 16p11.2 del | 0.53 C | ? |
| 18 | M, 7y | − | LD Dyspraxia | + | − | − | − | Scoliosis | 16p11.2 del | 0.43 C | de novo |
| 1B* | F, 32y | 5th | PDD | − | Syndactyly | − | − | Bilateral cataract | 16p11.2 del 16q23.1–q23.2 del 5q34 del | 0.59 C 1.425 3.811 | de novo pat mat |
| 25 | M, 13y | − | Mild ID LD | + | MA Metatarsus adductus Feet overpronation Crossover toe | − | − | Conductive hypoacusia Scoliosis adenoid hypertrophy Hypermetropy Astigmatism Precocious puberty Obesity | 16p11.2 del 22q11.21–q11.22 dup | 0.59 C 1.42 | de novo mat |
| 26 | M, 2y | − | PDD | − | − | − | Hypotonia hyporeflexia | Hyperkeratosis | 16p11.2 del | 0.65 C | ? |
| 12 | M, 12y | + | Severe ID Severe LD | + | − | − | Cerebellar hypoplasia | Cornelia De Lange-like phenotype | 16p11.2 dup | 0.54 C | mat |
| 15 | F, 33y | + | Severe ID Severe LD | + | MI | − | Spasticity Epilepsy | Pervasive developmental disorder | 16p13.1–16p13.3 gain + | 14.6 | de novo |
| 1A | M, 2y | − | - | + | Clubfoot Hip delay maturation | − | Axial hypotonia Lower limb hypertonia Hyporeflexia | Bilateral cryptorchidism | 16q23.1–q23.2 del 5q34 del | 1.425 3.811 | pat pat |
| 16 | F, 15m | + | Mild PDD | + | Plagiocephaly | − | Axial hypotonia | Ptosis GE reflux | 16q22.2–23.1 del | 3.35 | ? |
| 17 | F, 23y | + | DD | + | − | − | Mood disorder | - | 16q12.2 dup 8q12.1 dup | 0.022 0.025 | de novo mat |
| 21 | M, 10y | − | Mild LD | − | − | − | − | − | 16q23.2 del | 0.206 | de novo |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Redaelli, S.; Maitz, S.; Crosti, F.; Sala, E.; Villa, N.; Spaccini, L.; Selicorni, A.; Rigoldi, M.; Conconi, D.; Dalprà, L.; et al. Refining the Phenotype of Recurrent Rearrangements of Chromosome 16. Int. J. Mol. Sci. 2019, 20, 1095. https://doi.org/10.3390/ijms20051095
Redaelli S, Maitz S, Crosti F, Sala E, Villa N, Spaccini L, Selicorni A, Rigoldi M, Conconi D, Dalprà L, et al. Refining the Phenotype of Recurrent Rearrangements of Chromosome 16. International Journal of Molecular Sciences. 2019; 20(5):1095. https://doi.org/10.3390/ijms20051095
Chicago/Turabian StyleRedaelli, Serena, Silvia Maitz, Francesca Crosti, Elena Sala, Nicoletta Villa, Luigina Spaccini, Angelo Selicorni, Miriam Rigoldi, Donatella Conconi, Leda Dalprà, and et al. 2019. "Refining the Phenotype of Recurrent Rearrangements of Chromosome 16" International Journal of Molecular Sciences 20, no. 5: 1095. https://doi.org/10.3390/ijms20051095