Participation of NADPH Oxidase-Related Reactive Oxygen Species in Leptin-Promoted Pulmonary Inflammation: Regulation of cPLA2α and COX-2 Expression

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
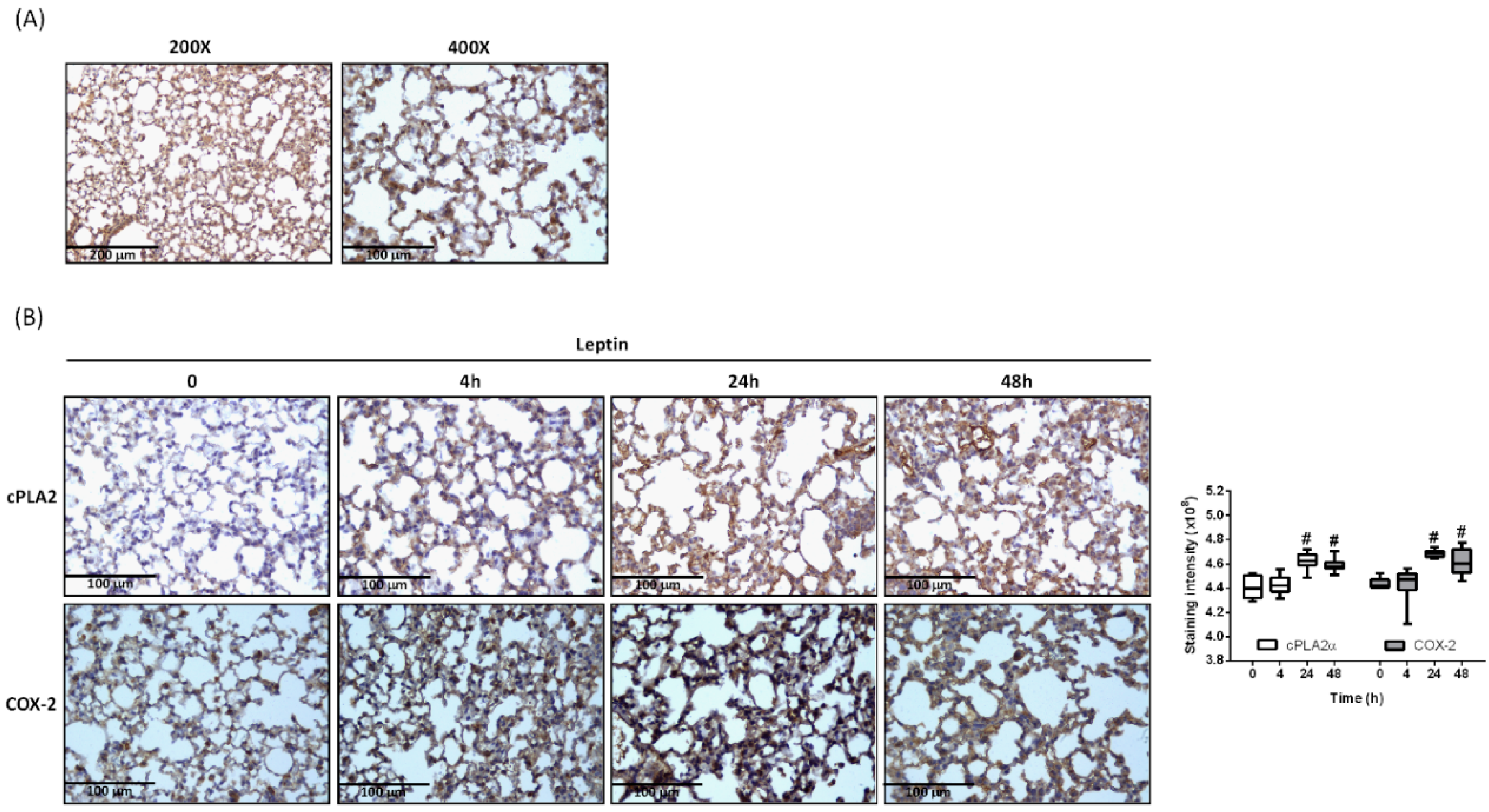
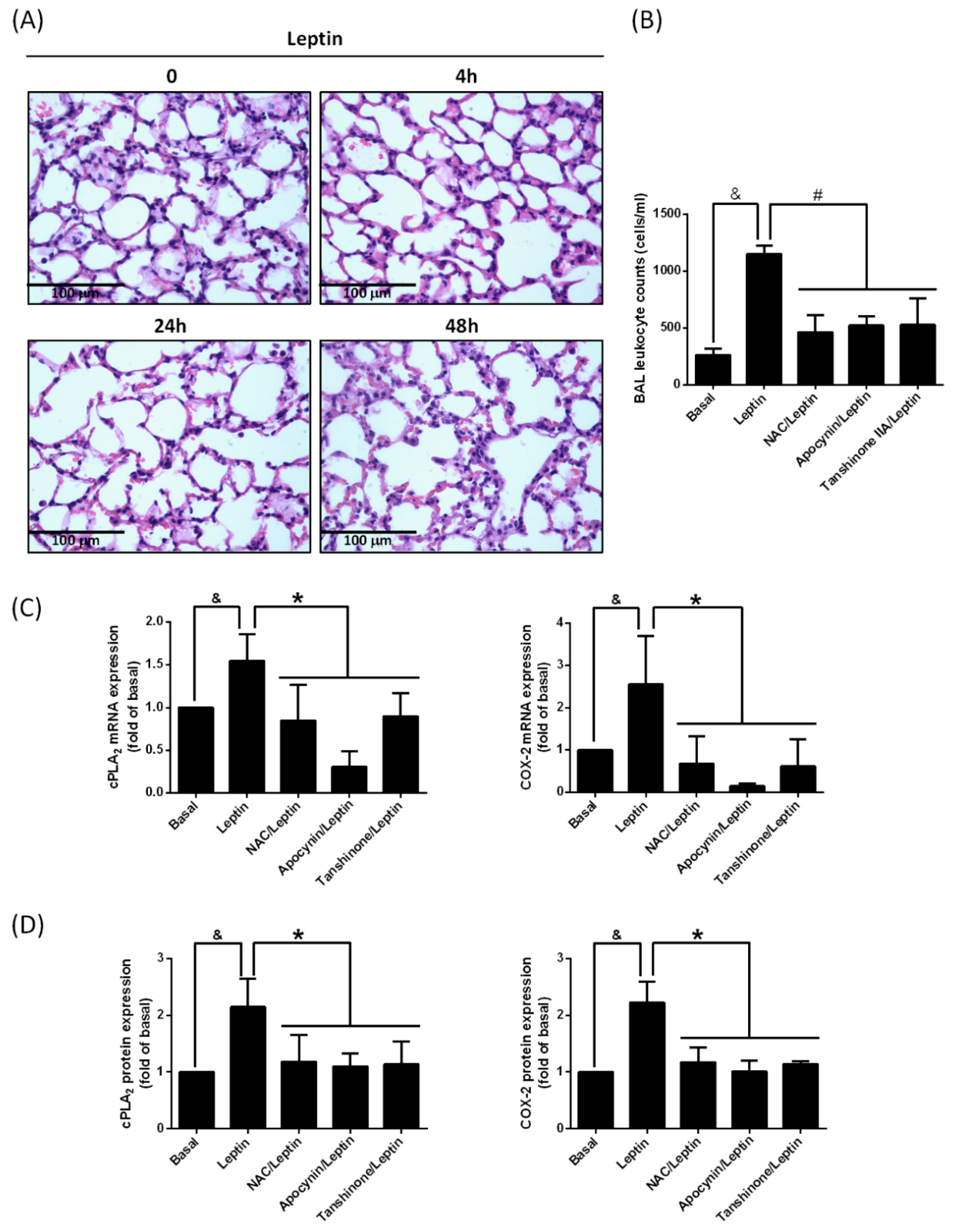
2.1. Leptin Promotes cPLA2α and COX-2 Expression in Lung Tissue
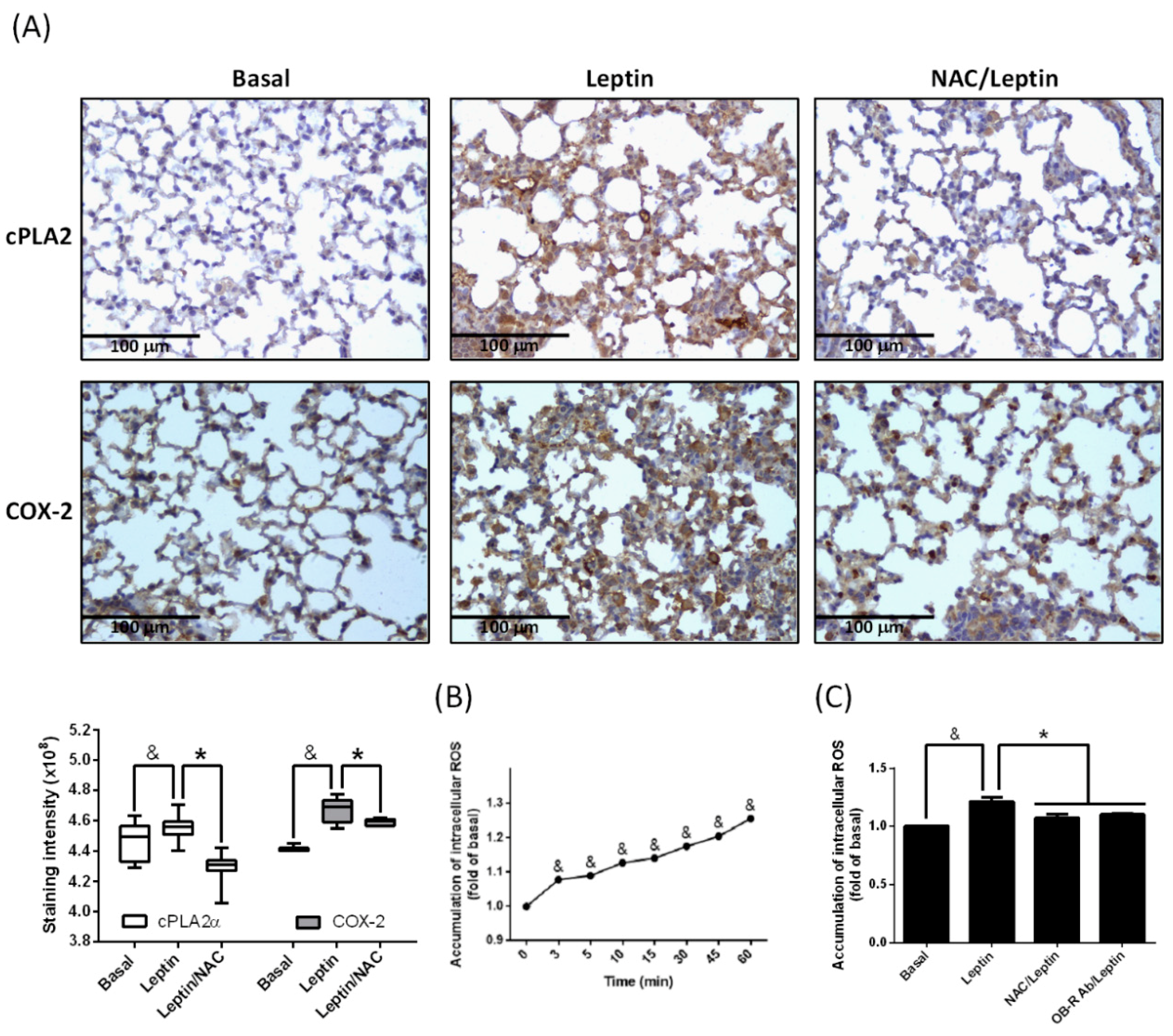
2.2. Participation of ROS in Leptin-Promoted cPLA2α and COX-2 Expression
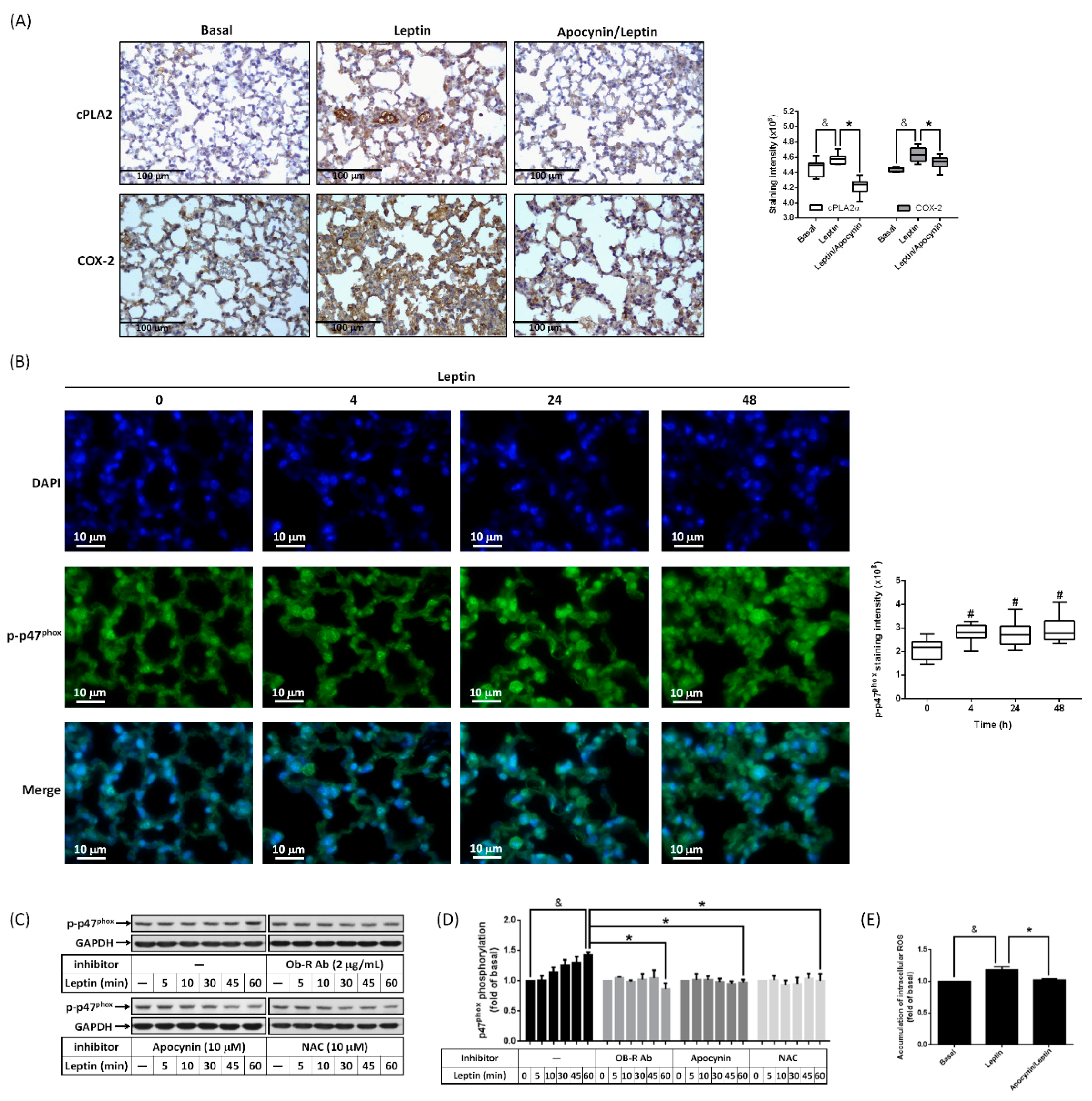
2.3. Activation of NADPH Oxidase Contributes to Leptin-Regulated cPLA2α and COX-2 Expression
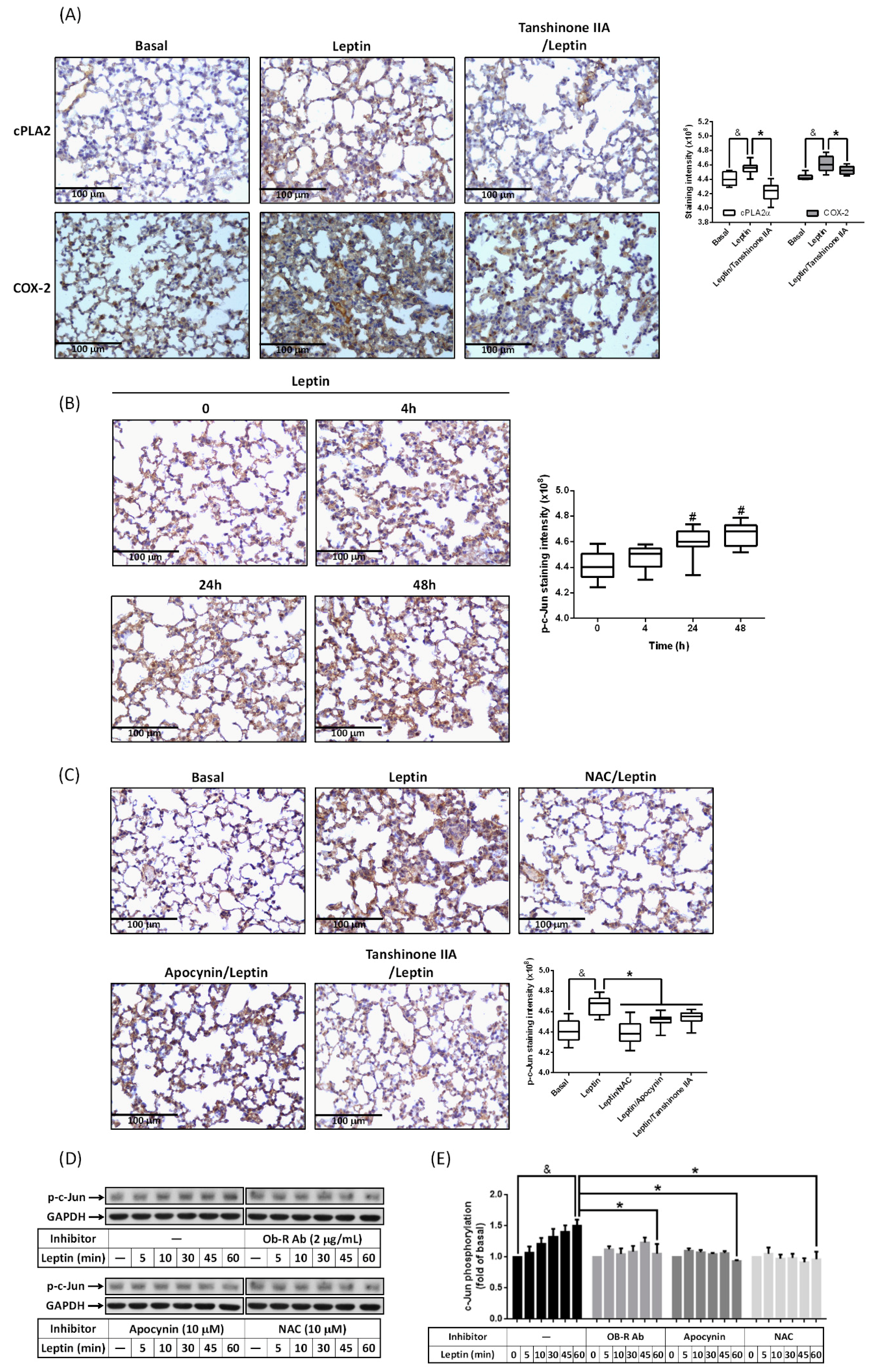
2.4. Involvement of AP-1 in Leptin-Mediated cPLA2α and COX-2 Expression
2.5. Leptin Enhances Leukocyte Recruitment in Lung Space
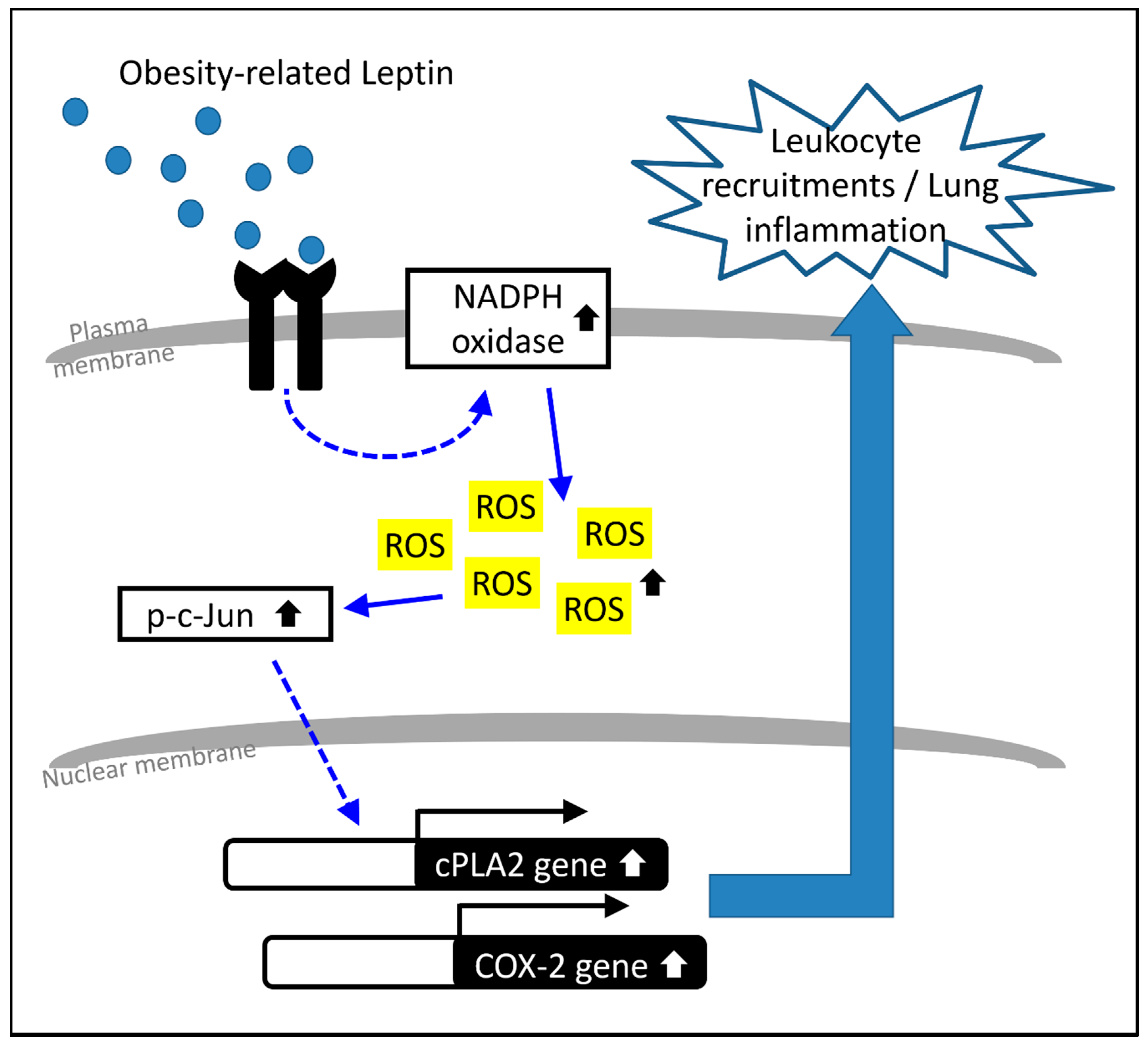
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Animal Treatment
4.3. Isolation of Bronchoalveolar Lavage (BAL)
4.4. Cell Culture of Human Alveolar Epithelial Cell Carcinoma (A549)
4.5. Protein Extraction and Western Blot
4.6. Total RNA Extraction and Gene Expression Analysis
4.7. ROS Measurement by CM-H2DCFDA Fluorescence
4.8. Immunohistochemical Staining and Immunofluorescence Assay
4.9. Statistical Analysis of Data
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Pabon, M.A.; Ma, K.C.; Choi, A.M. Autophagy and Obesity-Related Lung Disease. Am. J. Respir. Cell Mol. Biol. 2016, 54, 636–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhi, G.; Xin, W.; Ying, W.; Guohong, X.; Shuying, L. “Obesity Paradox” in Acute Respiratory Distress Syndrome: Asystematic Review and Meta-Analysis. PLoS ONE 2016, 11, e0163677. [Google Scholar]
- Thais Fantozzi, E.; Rodrigues-Garbin, S.; Yamamoto Ricardo-da-Silva, F.; Oliveira-Filho, R.M.; Spina, D.; Tavares-de-Lima, W.; Riffo-Vasquez, Y. Acute lung injury induced by intestinal ischemia and reperfusion is altered in obese female mice. Pulm. Pharmacol. Ther. 2018, 49, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Shah, D.; Romero, F.; Duong, M.; Wang, N.; Paudyal, B.; Suratt, B.T.; Kallen, C.B.; Sun, J.; Zhu, Y.; Walsh, K.; et al. Obesity-induced adipokine imbalance impairs mouse pulmonary vascular endothelial function and primes the lung for injury. Sci. Rep. 2015, 5, 11362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manicone, A.M.; Gong, K.; Johnston, L.K.; Giannandrea, M. Diet-induced obesity alters myeloid cell populations in naive and injured lung. Respir. Res. 2016, 17, 24. [Google Scholar] [CrossRef] [PubMed]
- Lambert, A.A.; Putcha, N.; Drummond, M.B.; Boriek, A.M.; Hanania, N.A.; Kim, V.; Kinney, G.L.; McDonald, M.N.; Brigham, E.P.; Wise, R.A.; et al. Obesity Is Associated with Increased Morbidity in Moderate to Severe COPD. Chest 2017, 151, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.N.; Lin, C.C.; Cheng, H.Y.; Yang, C.M. Regulation of cyclooxygenase-2 and cytosolic phospholipase A2 gene expression by lipopolysaccharide through the RNA-binding protein HuR: Involvement of NADPH oxidase, reactive oxygen species and mitogen-activated protein kinases. Br. J. Pharmacol. 2011, 163, 1691–1706. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.T.; Lin, C.C.; Lin, W.N.; Wu, W.L.; Hsiao, L.D.; Yang, C.M. Lung inflammation caused by adenosine-5′-triphosphate is mediated via Ca2+/PKCs-dependent COX-2/PGE2 induction. Int. J. Biochem. Cell Biol. 2013, 45, 1657–1668. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.Y.; Shao, D.; Liu, H.; Feng, J.; Feng, B.; Song, X.; Zhao, Q.; Chu, M.; Jiang, C.; Huang, W.; et al. Metabolomics analysis reveals that benzo[a]pyrene, a component of PM2.5, promotes pulmonary injury by modifying lipid metabolism in a phospholipase A2-dependent manner in vivo and in vitro. Redox Biol. 2017, 13, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.; Zhang, Y.; Do, D.C.; Ke, X.; Zhang, S.; Lambert, K.; Kumar, S.; Hu, C.; Zhou, Y.; Ishmael, F.T.; et al. miR-155 Modulates Cockroach Allergen- and Oxidative Stress-Induced Cyclooxygenase-2 in Asthma. J. Immunol. 2018, 201, 916–929. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.C.; Hsiao, F.C.; Chang, H.M.; Wabitsch, M.; Hsieh, P.S. Importance of adipocyte cyclooxygenase-2 and prostaglandin E2-prostaglandin E receptor 3 signaling in the development of obesity-induced adipose tissue inflammation and insulin resistance. FASEB J. 2016, 30, 2282–2297. [Google Scholar] [CrossRef] [PubMed]
- Yonker, L.M.; Pazos, M.A.; Lanter, B.B.; Mou, H.; Chu, K.K.; Eaton, A.D.; Bonventre, J.V.; Tearney, G.J.; Rajagopal, J.; Hurley, B.P. Neutrophil-Derived Cytosolic PLA2alpha Contributes to Bacterial-Induced Neutrophil Transepithelial Migration. J. Immunol. 2017, 199, 2873–2884. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Cifarelli, V.; Sun, S.; Kuda, O.; Abumrad, N.A.; Su, X. Major role of adipocyte prostaglandin E2 in lipolysis-induced macrophage recruitment. J. Lipid Res. 2016, 57, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Margetic, S.; Gazzola, C.; Pegg, G.G.; Hill, R.A. Leptin: A review of its peripheral actions and interactions. Int. J. Obes. Relat. Metab. Disord. 2002, 26, 1407–1433. [Google Scholar] [CrossRef] [PubMed]
- Ubags, N.D.; Stapleton, R.D.; Vernooy, J.H.; Burg, E.; Bement, J.; Hayes, C.M.; Ventrone, S.; Zabeau, L.; Tavernier, J.; Poynter, M.E.; et al. Hyperleptinemia is associated with impaired pulmonary host defense. JCI Insight 2016, 1. [Google Scholar] [CrossRef] [PubMed]
- Bodini, A.; Tenero, L.; Sandri, M.; Maffeis, C.; Piazza, M.; Zanoni, L.; Peroni, D.; Boner, A.; Piacentini, G. Serum and exhaled breath condensate leptin levels in asthmatic and obesity children: A pilot study. J. Breath Res. 2017, 11, 046005. [Google Scholar] [CrossRef] [PubMed]
- Suzukawa, M.; Koketsu, R.; Baba, S.; Igarashi, S.; Nagase, H.; Yamaguchi, M.; Matsutani, N.; Kawamura, M.; Shoji, S.; Hebisawa, A.; et al. Leptin enhances ICAM-1 expression, induces migration and cytokine synthesis, and prolongs survival of human airway epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L801–L811. [Google Scholar] [CrossRef] [PubMed]
- Hao, W.; Wang, J.; Zhang, Y.; Wang, Y.; Sun, L.; Han, W. Leptin positively regulates MUC5AC production and secretion induced by interleukin-13 in human bronchial epithelial cells. Biochem. Biophys. Res. Commun. 2017, 493, 979–984. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.S.; Wu, C.S.; Chang, J.F.; Lin, W.N. Leptin Promotes cPLA(2) Gene Expression through Activation of the MAPK/NF-kappaB/p300 Cascade. Int. J. Mol. Sci. 2015, 16, 27640–27658. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, K.; Shindo, S.; Movila, A.; Kayal, R.; Abdullah, A.; Savitri, I.J.; Ikeda, A.; Yamaguchi, T.; Howait, M.; Al-Dharrab, A.; et al. TRAP-positive osteoclast precursors mediate ROS/NO-dependent bactericidal activity via TLR4. Free Radic. Biol. Med. 2016, 97, 330–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, Y.S.; Kang, K.A.; Piao, M.J.; Ahn, M.J.; Yi, J.M.; Hyun, Y.M.; Kim, S.H.; Ko, M.K.; Park, C.O.; Hyun, J.W. Particulate matter induces inflammatory cytokine production via activation of NFkappaB by TLR5-NOX4-ROS signaling in human skin keratinocyte and mouse skin. Redox Biol. 2018, 21, 101080. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.F.; Liang, S.S.; Thanasekaran, P.; Chang, H.W.; Wen, L.L.; Chen, C.H.; Liou, J.C.; Yeh, J.C.; Liu, S.H.; Dai, H.M.; et al. Translational Medicine in Pulmonary-Renal Crosstalk: Therapeutic Targeting of p-Cresyl Sulfate Triggered Nonspecific ROS and Chemoattractants in Dyspneic Patients with Uremic Lung Injury. J. Clin. Med. 2018, 7, 266. [Google Scholar] [CrossRef] [PubMed]
- Xiang, L.; Lu, S.; Mittwede, P.N.; Clemmer, J.S.; Hester, R.L. Inhibition of NADPH oxidase prevents acute lung injury in obese rats following severe trauma. Am. J. Physiol.-Heart Circul. Physiol. 2014, 306, H684–H689. [Google Scholar] [CrossRef] [PubMed]
- Cho, R.L.; Yang, C.C.; Lee, I.T.; Lin, C.C.; Chi, P.L.; Hsiao, L.D.; Yang, C.M. Lipopolysaccharide induces ICAM-1 expression via a c-Src/NADPH oxidase/ROS-dependent NF-kappaB pathway in human pulmonary alveolar epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.K.; Lee, I.T.; Lin, C.C.; Hsiao, L.D.; Yang, C.M. Sphingosine-1-phosphate mediates COX-2 expression and PGE2 /IL-6 secretion via c-Src-dependent AP-1 activation. J. Cell. Physiol. 2015, 230, 702–715. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.T.; Lin, C.C.; Cheng, S.E.; Hsiao, L.D.; Hsiao, Y.C.; Yang, C.M. TNF-alpha induces cytosolic phospholipase A2 expression in human lung epithelial cells via JNK1/2- and p38 MAPK-dependent AP-1 activation. PLoS ONE 2013, 8, e72783. [Google Scholar] [CrossRef]
- Min, D.Y.; Jung, E.; Kim, J.; Lee, Y.H.; Shin, S.Y. Leptin stimulates IGF-1 transcription by activating AP-1 in human breast cancer cells. BMB Rep. 2018, 8, 4313. [Google Scholar]
- Rehman Khan, A.; Awan, F.R. Leptin Resistance: A Possible Interface Between Obesity and Pulmonary-Related Disorders. Int. J. Endocrinol. Metab. 2016, 14, e32586. [Google Scholar] [CrossRef] [PubMed]
- Dal Farra, C.; Zsurger, N.; Vincent, J.P.; Cupo, A. Binding of a pure 125I-monoiodoleptin analog to mouse tissues: A developmental study. Peptides 2000, 21, 577–587. [Google Scholar] [CrossRef]
- Blaser, H.; Dostert, C.; Mak, T.W.; Brenner, D. TNF and ROS Crosstalk in Inflammation. Trends Cell Biol. 2016, 26, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Baffi, C.W.; Wood, L.; Winnica, D.; Strollo, P.J., Jr.; Gladwin, M.T.; Que, L.G.; Holguin, F. Metabolic Syndrome and the Lung. Chest 2016, 149, 1525–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, U.; Suratt, B.T.; Bates, J.H.T.; Dixon, A.E. Beyond BMI: Obesity and Lung Disease. Chest 2018, 153, 702–709. [Google Scholar] [CrossRef] [PubMed]
- Sideleva, O.; Suratt, B.T.; Black, K.E.; Tharp, W.G.; Pratley, R.E.; Forgione, P.; Dienz, O.; Irvin, C.G.; Dixon, A.E. Obesity and asthma: An inflammatory disease of adipose tissue not the airway. Am. J. Respir. Crit. Care Med. 2012, 186, 598–605. [Google Scholar] [CrossRef] [PubMed]
- Holguin, F.; Rojas, M.; Hart, C.M. The peroxisome proliferator activated receptor gamma (PPARgamma) ligand rosiglitazone modulates bronchoalveolar lavage levels of leptin, adiponectin, and inflammatory cytokines in lean and obese mice. Lung 2007, 185, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, R.; Clark, S.; Bonventre, J.V.; Leong, J.M.; McCormick, B.A. Cytosolic Phospholipase A2alpha Promotes Pulmonary Inflammation and Systemic Disease during Streptococcus pneumoniae Infection. Infect. Immun. 2017, 85. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Liu, B.; Guan, H.; Mao, W.; Wang, L.; Zhang, C.; Hai, L.; Liu, K.; Cao, J. PGE2 increases inflammatory damage in Escherichia coli-infected bovine endometrial tissue in vitro via the EP4-PKA signaling pathway. Biol. Reprod. 2018. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buckley, C.D.; Pilling, D.; Lord, J.M.; Akbar, A.N.; Scheel-Toellner, D.; Salmon, M. Fibroblasts regulate the switch from acute resolving to chronic persistent inflammation. Trends Immunol. 2001, 22, 199–204. [Google Scholar] [CrossRef]
- Gui, X.; Chen, H.; Cai, H.; Sun, L.; Gu, L. Leptin promotes pulmonary fibrosis development by inhibiting autophagy via PI3K/Akt/mTOR pathway. Biochem. Biophys. Res. Commun. 2018, 498, 660–666. [Google Scholar] [CrossRef] [PubMed]
- Amatore, D.; Sgarbanti, R.; Aquilano, K.; Baldelli, S.; Limongi, D.; Civitelli, L.; Nencioni, L.; Garaci, E.; Ciriolo, M.R.; Palamara, A.T. Influenza virus replication in lung epithelial cells depends on redox-sensitive pathways activated by NOX4-derived ROS. Cell. Microbiol. 2015, 17, 131–145. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Kou, X.; Xie, L.; Cheng, F.; Geng, H. Effects of ambient PM2.5 on pathological injury, inflammation, oxidative stress, metabolic enzyme activity, and expression of c-fos and c-jun in lungs of rats. Environ. Sci Pollut. Res. Int. 2015, 22, 20167–20176. [Google Scholar] [CrossRef] [PubMed]
- Cho, R.L.; Yang, C.C.; Tseng, H.C.; Hsiao, L.D.; Lin, C.C.; Yang, C.M. Haem oxygenase-1 up-regulation by rosiglitazone via ROS-dependent Nrf2-antioxidant response elements axis or PPARgamma attenuates LPS-mediated lung inflammation. Br. J. Pharmacol. 2018, 175, 3928–3946. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.C.; Lin, W.N.; Cho, R.L.; Wang, C.Y.; Hsiao, L.D.; Yang, C.M. TNF-alpha-Induced cPLA2 Expression via NADPH Oxidase/Reactive Oxygen Species-Dependent NF-kappaB Cascade on Human Pulmonary Alveolar Epithelial Cells. Front. Pharmacol. 2016, 7, 447. [Google Scholar] [PubMed]
- Hsu, H.T.; Tseng, Y.T.; Wong, W.J.; Liu, C.M.; Lo, Y.C. Resveratrol prevents nanoparticles-induced inflammation and oxidative stress via downregulation of PKC-alpha and NADPH oxidase in lung epithelial A549 cells. BMC Complement. Altern. Med. 2018, 18, 211. [Google Scholar] [CrossRef] [PubMed]
- Grandvaux, N.; Mariani, M.; Fink, K. Lung epithelial NOX/DUOX and respiratory virus infections. Clin. Sci. 2015, 128, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Moyano, A.J.; Racca, A.C.; Soria, G.; Saka, H.A.; Andreoli, V.; Smania, A.M.; Sola, C.; Bocco, J.L. c-Jun Proto-Oncoprotein Plays a Protective Role in Lung Epithelial Cells Exposed to Staphylococcal alpha-Toxin. Front. Cell. Infect. Microbiol. 2018, 8, 170. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.R.; Chida, A.S.; Bauter, M.R.; Shafiq, N.; Seweryniak, K.; Maggirwar, S.B.; Kilty, I.; Rahman, I. Cigarette smoke induces proinflammatory cytokine release by activation of NF-kappaB and posttranslational modifications of histone deacetylase in macrophages. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 291, L46–L57. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.F.; Dasari, H.; Van Keulen, V.P.; Carmona, E.M. Canonical Stimulation of the NLRP3 Inflammasome by Fungal Antigens Links Innate and Adaptive B-Lymphocyte Responses by Modulating IL-1beta and IgM Production. Front. Immunol. 2017, 8, 1504. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Cai, L.; Zheng, L.; Hu, Y.; Yuan, W.; Guo, Z.; Li, W. Gefitinib Inhibits Bleomycin-Induced Pulmonary Fibrosis via Alleviating the Oxidative Damage in Mice. Oxidative Med. Cell. Longev. 2018, 2018, 8249693. [Google Scholar] [CrossRef] [PubMed]
- Foster, K.A.; Oster, C.G.; Mayer, M.M.; Avery, M.L.; Audus, K.L. Characterization of the A549 cell line as a type II pulmonary epithelial cell model for drug metabolism. Exp. Cell Res. 1998, 243, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.M.; Yang, C.M.; Chang, J.F.; Wu, C.S.; Sia, K.C.; Lin, W.N. AdipoR-increased intracellular ROS promotes cPLA2 and COX-2 expressions via activation of PKC and p300 in adiponectin-stimulated human alveolar type II cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 311, L255–L269. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsu, P.-S.; Lin, C.-M.; Chang, J.-F.; Wu, C.-S.; Sia, K.-C.; Lee, I.-T.; Huang, K.-Y.; Lin, W.-N. Participation of NADPH Oxidase-Related Reactive Oxygen Species in Leptin-Promoted Pulmonary Inflammation: Regulation of cPLA2α and COX-2 Expression. Int. J. Mol. Sci. 2019, 20, 1078. https://doi.org/10.3390/ijms20051078
Hsu P-S, Lin C-M, Chang J-F, Wu C-S, Sia K-C, Lee I-T, Huang K-Y, Lin W-N. Participation of NADPH Oxidase-Related Reactive Oxygen Species in Leptin-Promoted Pulmonary Inflammation: Regulation of cPLA2α and COX-2 Expression. International Journal of Molecular Sciences. 2019; 20(5):1078. https://doi.org/10.3390/ijms20051078
Chicago/Turabian StyleHsu, Pei-Sung, Chia-Mo Lin, Jia-Feng Chang, Chi-Sheng Wu, Kee-Chin Sia, I-Ta Lee, Kuo-Yang Huang, and Wei-Ning Lin. 2019. "Participation of NADPH Oxidase-Related Reactive Oxygen Species in Leptin-Promoted Pulmonary Inflammation: Regulation of cPLA2α and COX-2 Expression" International Journal of Molecular Sciences 20, no. 5: 1078. https://doi.org/10.3390/ijms20051078