Hydrodynamic and Electrophoretic Properties of Trastuzumab/HER2 Extracellular Domain Complexes as Revealed by Experimental Techniques and Computational Simulations

 ,
, 
Abstract
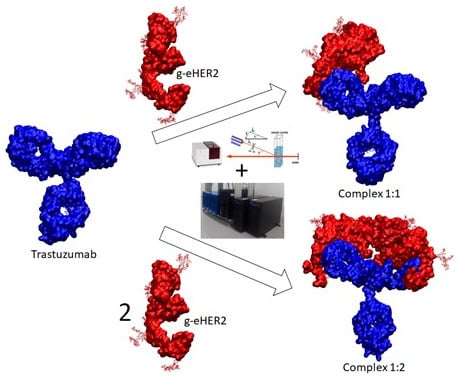
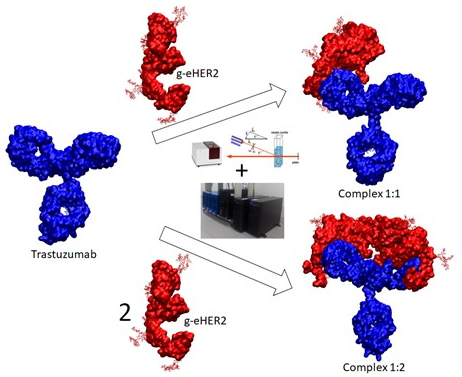
:
1. Introduction
2. Results
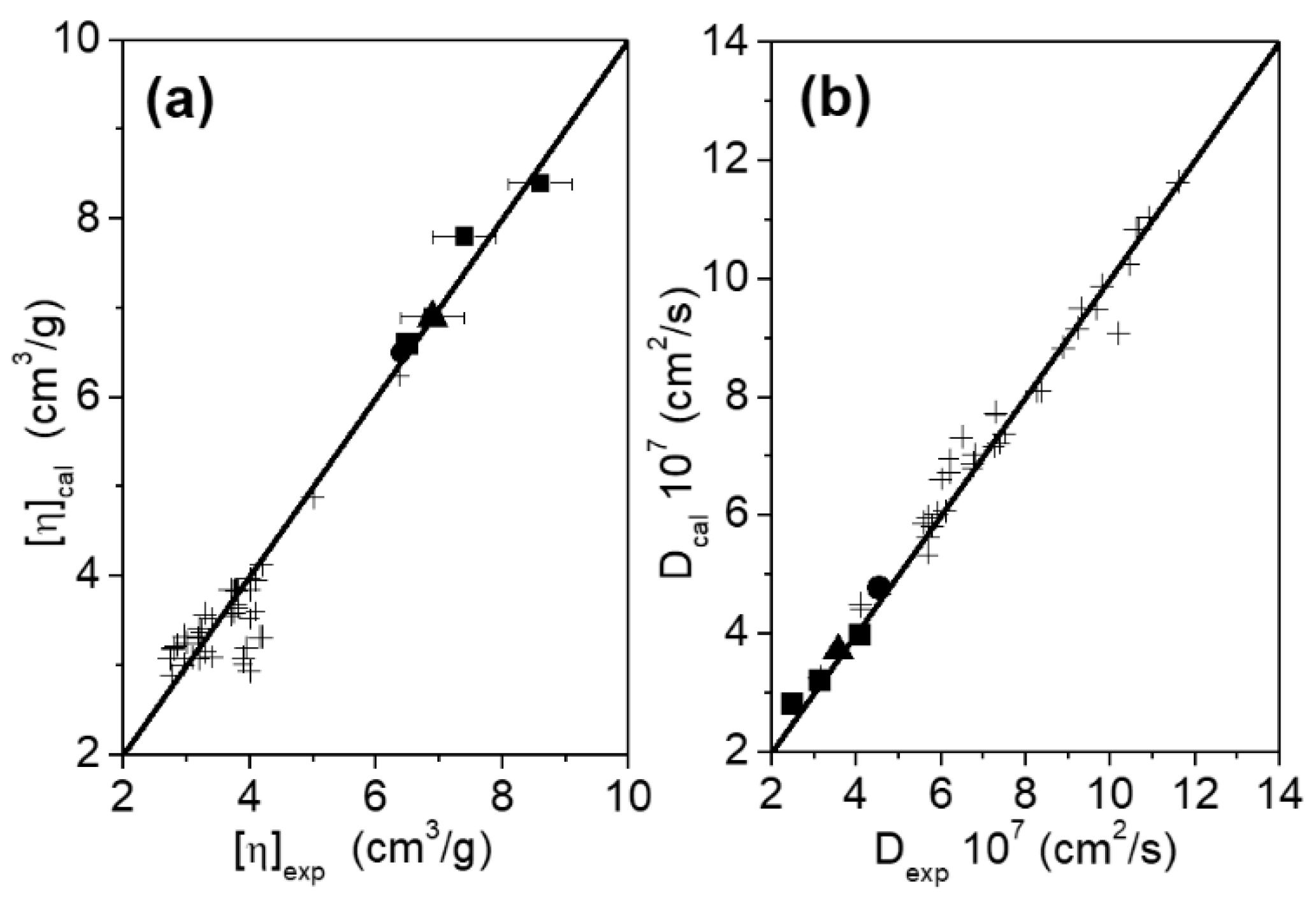
2.1. Molecular Sizes and Hydrodynamic Properties
2.1.1. SEC Experiments
2.1.2. DLS Experiments
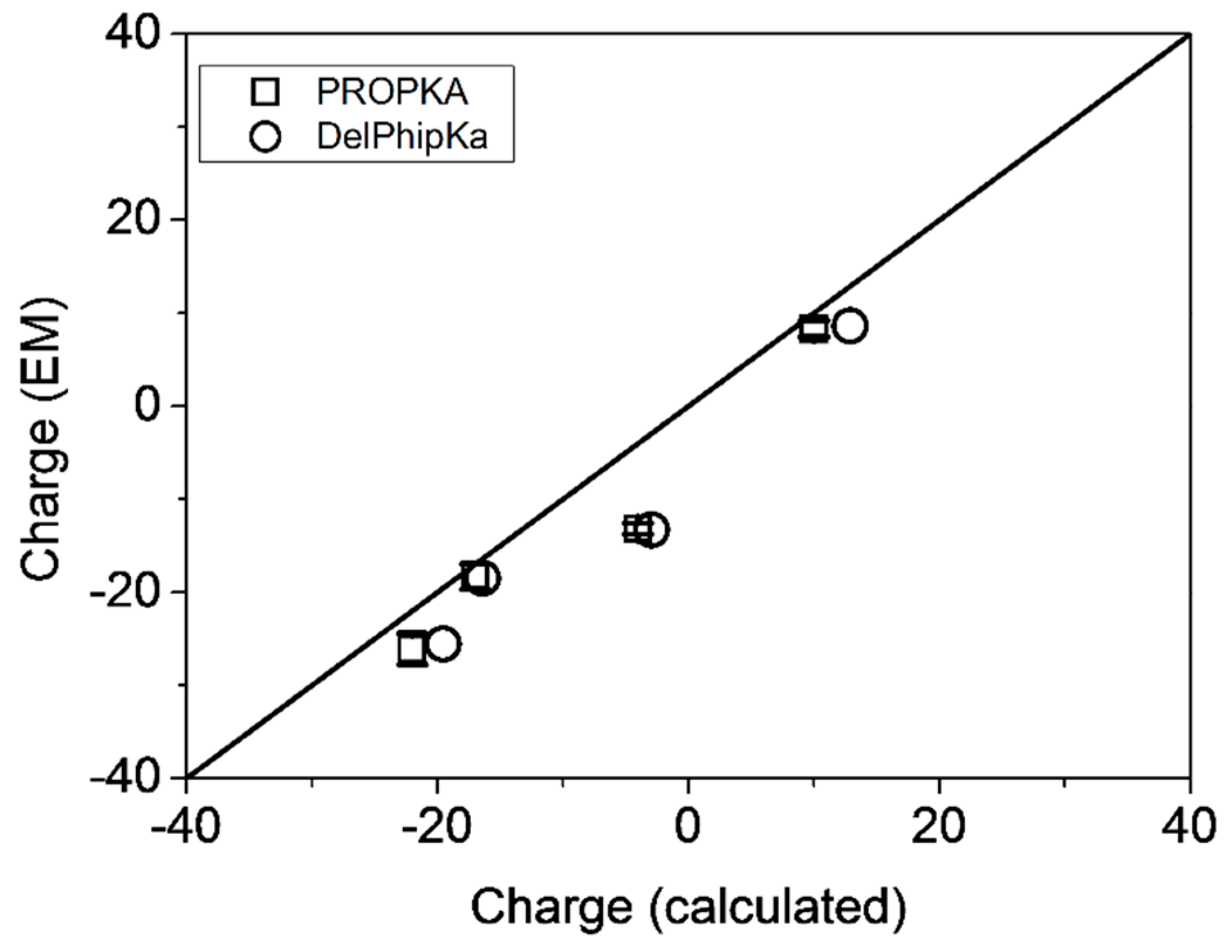
2.1.3. Electrostatic Properties
2.2. MD Simulations and Hydrodynamic Modeling
2.2.1. TZM:g-eHER2-his Interactions
2.2.2. Hydrodynamic Properties Calculation
3. Discussion
4. Materials and Methods
4.1. Experimental Details
4.2. Computational Simulations
4.2.1. Construction of the Models
4.2.2. Atomistic Molecular Dynamics (MD) Simulations
4.2.3. Hydrodynamic Calculations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| EGFR | Epidermal Growth Factor Receptors |
| HER2 | Human Epidermal Factor type 2 |
| TZM | Trastuzumab |
| PDB | Protein Data Bank |
| SOMO | SOlution MOdeller |
| BEST | Boundary Element numerical solution of the hydrodynamic problem with STick boundary conditions |
| DLS | Dynamic Light Scattering |
| SEC | Size Exclusion Chromatography |
| EM | Electrophoretic Mobility |
| MD | Molecular Dynamics |
| pI | Isoelectric point |
References
- Claret, F.; Vu, T. Trastuzumab: Updated Mechanisms of Action and Resistance in Breast Cancer. Front. Oncol. 2012, 2, 62. [Google Scholar]
- Pegram, M.D.; Lipton, A.; Hayes, D.F.; Weber, B.L.; Baselga, J.M.; Tripathy, D.; Baly, D.; Baughman, S.A.; Twaddell, T.; Glaspy, J.A.; et al. Phase II study of receptor-enhanced chemosensitivity using recombinant humanized anti-p185HER2/neu monoclonal antibody plus cisplatin in patients with HER2/neu-overexpressing metastatic breast cancer refractory to chemotherapy treatment. J. Clin. Oncol. 1998, 16, 2659–2671. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.D.; Sisk, J.; Ansari, R.; Gize, G.; Nattam, S.; Pennington, K.; Monaco, F.; Sledge, G.W., Jr. Gemcitabine, paclitaxel, and trastuzumab in metastatic breast cancer. Oncology 2001, 15 (Suppl. 3), 38–40. [Google Scholar] [PubMed]
- Pegram, M.D.; Pienkowski, T.; Northfelt, D.W.; Eiermann, W.; Patel, R.; Fumoleau, P.; Quan, E.; Crown, J.; Toppmeyer, D.; Smylie, M.; et al. Results of two open-label, multicenter phase II studies of docetaxel, platinum salts, and trastuzumab in HER2-positive advanced breast cancer. J. Natl. Cancer Inst. 2004, 96, 759–769. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J.; Tripathy, D.; Mendelsohn, J.; Baughman, S.; Benz, C.C.; Dantis, L.; Sklarin, N.T.; Seidman, A.D.; Hudis, C.A.; Moore, J.; et al. Phase II study of weekly intravenous recombinant humanized anti-p185HER2 monoclonal antibody in patients with HER2/neu-overexpressing metastatic breast cancer. J. Clin. Oncol. 1996, 14, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Cobleigh, M.A.; Vogel, C.L.; Tripathy, D.; Robert, N.J.; Scholl, S.; Fehrenbacher, L.; Wolter, J.M.; Paton, V.; Shak, S.; Lieberman, G.; et al. Multinational Study of the Efficacy and Safety of Humanized Anti-HER2 Monoclonal Antibody in Women Who Have HER2-Overexpressing Metastatic Breast Cancer That Has Progressed After Chemotherapy for Metastatic Disease. J. Clin. Oncol. 1999, 17, 2639. [Google Scholar] [CrossRef] [PubMed]
- Cortés, J.; Curigliano, G.; Diéras, V. Expert perspectives on biosimilar monoclonal antibodies in breast cancer. Breast Cancer Res. Treat. 2014, 144, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Maximiano, S.; Magalhães, P.; Guerreiro, M.P.; Morgado, M. Trastuzumab in the Treatment of Breast Cancer. BioDrugs 2016, 30, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.-S.; Mason, K.; Ramyar, K.X.; Stanley, A.M.; Gabelli, S.B.; Denney, D.W., Jr.; Leahy, D.J. Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature 2003, 421, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Franklin, M.C.; Carey, K.D.; Vajdos, F.F.; Leahy, D.J.; de Vos, A.M.; Sliwkowski, M.X. Insights into ErbB signaling from the structure of the ErbB2-pertuzumab complex. Cancer Cell 2004, 5, 317–328. [Google Scholar] [CrossRef]
- Li, S.; Schmitz, K.R.; Jeffrey, P.D.; Wiltzius, J.J.W.; Kussie, P.; Ferguson, K.M. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell 2005, 7, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Schmiedel, J.; Blaukat, A.; Li, S.; Knoechel, T.; Ferguson, K.M. Matuzumab binding to EGFR prevents the conformational rearrangement required for dimerization. Cancer Cell 2008, 13, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Stephen, E.H. Protein Hydrodynamics. In Protein: A Comprehensive Treatise; Science, E., Ed.; JAI Press: Greenwich, CT, USA, 1999; Volume 2, pp. 271–301. [Google Scholar]
- Erickson, H.P. Size and Shape of Protein Molecules at the Nanometer Level Determined by Sedimentation, Gel Filtration, and Electron Microscopy. Biol. Proced. Online 2009, 11, 32. [Google Scholar] [CrossRef] [PubMed]
- Harding, S.E.; Chowdhry, B.Z. Protein-Ligand Interactions: Hydrodynamics and Calorimetry: A Practical Approach; Oxford University Press: Oxford, UK, 2001. [Google Scholar]
- Al Kayal, T.; Russo, E.; Pieri, L.; Caminati, G.; Berti, D.; Bucciantini, M.; Stefani, M.; Baglioni, P. Interactions of lysozyme with phospholipid vesicles: Effects of vesicle biophysical features on protein misfolding and aggregation. Soft Matter 2012, 8, 9115–9126. [Google Scholar] [CrossRef]
- Kayitmazer, A.B.; Seeman, D.; Minsky, B.B.; Dubin, P.L.; Xu, Y. Protein–polyelectrolyte interactions. Soft Matter 2013, 9, 2553–2583. [Google Scholar] [CrossRef]
- Hewitt, D.; Marklund, E.; Scott, D.J.; Robinson, C.V.; Borysik, A.J. A Hydrodynamic Comparison of Solution and Gas Phase Proteins and Their Complexes. J. Phys. Chem. B 2014, 118, 8489–8495. [Google Scholar] [CrossRef] [PubMed]
- Patel, T.R.; Chojnowski, G.; Astha; Koul, A.; McKenna, S.A.; Bujnicki, J.M. Structural studies of RNA-protein complexes: A hybrid approach involving hydrodynamics, scattering, and computational methods. Methods 2017, 118–119, 146–162. [Google Scholar] [CrossRef] [PubMed]
- Vega, J.F.; Vicente-Alique, E.; Núñez-Ramírez, R.; Wang, Y.; Martínez-Salazar, J. Evidences of Changes in Surface Electrostatic Charge Distribution during Stabilization of HPV16 Virus-Like Particles. PLoS ONE 2016, 11, e0149009. [Google Scholar] [CrossRef] [PubMed]
- Gokarn, Y.R.; McLean, M.; Laue, T.M. Effect of PEGylation on Protein Hydrodynamics. Mol. Pharm. 2012, 9, 762–773. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.; Sreedhara, A.; Kanai, S.; Liu, J.; Lien, S.; Lowman, H.; Kalonia, D.S.; Shire, S.J. Establishing a Link Between Amino Acid Sequences and Self-Associating and Viscoelastic Behavior of Two Closely Related Monoclonal Antibodies. Pharm. Res. 2011, 28, 1750–1764. [Google Scholar] [CrossRef] [PubMed]
- Bethea, D.; Wu, S.-J.; Luo, J.; Hyun, L.; Lacy, E.R.; Teplyakov, A.; Jacobs, S.A.; O’Neil, K.T.; Gilliland, G.L.; Feng, Y. Mechanisms of self-association of a human monoclonal antibody CNTO607. Protein Eng. Des. Sel. 2012, 25, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Nicoud, L.; Lattuada, M.; Yates, A.; Morbidelli, M. Impact of aggregate formation on the viscosity of protein solutions. Soft Matter 2015, 11, 5513–5522. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, K.M.; Darling, P.J.; Mohan, M.J.; Macatee, T.L.; Lemmon, M.A. Extracellular domains drive homo- but not hetero-dimerization of erbB receptors. EMBO J. 2000, 19, 4632–4643. [Google Scholar] [CrossRef] [PubMed]
- García de la Torre, J.; Harding, S.E. Hydrodynamic modelling of protein conformation in solution: ELLIPS and HYDRO. Biophys. Rev. 2013, 5, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Amorós, D.; Ortega, A.; Harding, S.E.; García de la Torre, J. Multi-scale calculation and global-fit analysis of hydrodynamic properties of biological macromolecules: Determination of the overall conformation of antibody IgG molecules. Eur. Biophys. J. 2010, 39, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Ortega, A.; Amorós, D.; García de la Torre, J. Prediction of Hydrodynamic and Other Solution Properties of Rigid Proteins from Atomic- and Residue-Level Models. Biophys. J. 2011, 101, 892–898. [Google Scholar] [CrossRef] [PubMed]
- De la Torre, J.G. The HYDRO Software Suite for the Prediction of Solution Properties of Rigid and Flexible Macromolecules and Nanoparticles. In Analytical Ultracentrifugation: Instrumentation, Software, and Applications; Uchiyama, S., Arisaka, F., Stafford, W.F., Laue, T., Eds.; Springer: Tokyo, Japan, 2016; pp. 195–217. [Google Scholar]
- Rai, N.; Nöllmann, M.; Spotorno, B.; Tassara, G.; Byron, O.; Rocco, M. SOMO (SOlution MOdeler): Differences between X-ray- and NMR-Derived Bead Models Suggest a Role for Side Chain Flexibility in Protein Hydrodynamics. Structure 2005, 13, 723–734. [Google Scholar] [CrossRef] [PubMed]
- Brookes, E.; Demeler, B.; Rosano, C.; Rocco, M. The implementation of SOMO (SOlution MOdeller) in the UltraScan analytical ultracentrifugation data analysis suite: Enhanced capabilities allow the reliable hydrodynamic modeling of virtually any kind of biomacromolecule. Eur. Biophys. J. 2010, 39, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Aragon, S. A precise boundary element method for macromolecular transport properties. J. Comput. Chem. 2004, 9, 1191–1205. [Google Scholar] [CrossRef] [PubMed]
- Brandt, J.P.; Patapoff, T.W.; Aragon, S.R. Construction, MD Simulation, and Hydrodynamic Validation of an All-Atom Model of a Monoclonal IgG Antibody. Biophys. J. 2010, 99, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Vega, J.F.; Ramos, J.; Cruz, V.L.; Vicente-Alique, E.; Sánchez-Sánchez, E.; Sánchez-Fernández, A.; Wang, Y.; Hu, P.; Cortés, J.; Martínez-Salazar, J. Molecular and hydrodynamic properties of human epidermal growth factor receptor HER2 extracellular domain and its homodimer: Experiments and multi-scale simulations. Biochim. Biophys. Acta (BBA) Gen. Subj. 2017, 1861, 2406–2416. [Google Scholar] [CrossRef] [PubMed]
- Stanfield, R.L.; Wilson, I.A. Antibody Molecular Structure. In Therapeutic Monoclonal Antibodies—From Bench to Clinic; An, Z., Ed.; Wiley: Hoboken, NJ, USA, 2009; pp. 51–66. [Google Scholar]
- Harding, S.E. The intrinsic viscosity of biological macromolecules. Progress in measurement, interpretation and application to structure in dilute solution. Prog. Biophys. Mol. Biol. 1997, 68, 207–262. [Google Scholar] [CrossRef]
- Ahmad, F.; McPhie, P. The intrinsic viscosity of glycoproteins. Int. J. Biochem. 1980, 11, 91–96. [Google Scholar] [CrossRef]
- Franco-Gonzalez, J.F.; Cruz, V.L.; Ramos, J.; Martínez-Salazar, J. Conformational flexibility of the ErbB2 ectodomain and trastuzumab antibody complex as revealed by molecular dynamics and principal component analysis. J. Mol. Model. 2013, 19, 1227–1236. [Google Scholar] [CrossRef] [PubMed]
- Franco-Gonzalez, J.F.; Ramos, J.; Cruz, V.L.; Martínez-Salazar, J. Simulation of homology models for the extracellular domains (ECD) of ErbB3, ErbB4 and the ErbB2–ErbB3 complex in their active conformations. J. Mol. Model. 2013, 19, 931–941. [Google Scholar] [CrossRef] [PubMed]
- Franco-Gonzalez, J.F.; Ramos, J.; Cruz, V.L.; Martinez-Salazar, J. Exploring the dynamics and interaction of a full ErbB2 receptor and Trastuzumab-Fab antibody in a lipid bilayer model using Martini coarse-grained force field. J. Comput.-Aided Mol. Des. 2014, 28, 1093–1107. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.; Shire Steven, J.; Kalonia Devendra, S. Factors affecting the viscosity in high concentration solutions of different monoclonal antibodies. J. Pharm. Sci. 2010, 99, 4812–4829. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.; Liu, J.; Shire, S.J.; Kalonia, D.S. Specific interactions in high concentration antibody solutions resulting in high viscosity. J. Pharm. Sci. 2010, 99, 1152–1168. [Google Scholar] [CrossRef] [PubMed]
- Ortega, A.; García de la Torre, J. Equivalent Radii and Ratios of Radii from Solution Properties as Indicators of Macromolecular Conformation, Shape, and Flexibility. Biomacromolecules 2007, 8, 2464–2475. [Google Scholar] [CrossRef] [PubMed]
- Hahn, D.K.; Aragon, S.R. Intrinsic Viscosity of Proteins and Platonic Solids by Boundary Element Methods. J. Chem. Theory Comput. 2006, 2, 1416–1428. [Google Scholar] [CrossRef] [PubMed]
- Aragon, S.R. Recent advances in macromolecular hydrodynamic modeling. Methods 2011, 54, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Irina, G.; Carbeck, J.D.; Whitesides, G.M. Why Are Proteins Charged? Networks of Charge–Charge Interactions in Proteins Measured by Charge Ladders and Capillary Electrophoresis. Angew. Chem. Int. Ed. 2006, 45, 3022–3060. [Google Scholar]
- Swan, J.W.; Furst, E.M. A simpler expression for Henry’s function describing the electrophoretic mobility of spherical colloids. J. Colloid Interface Sci. 2012, 388, 92–94. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Azeim, S.; Chermak, E.; Vangone, A.; Oliva, R.; Cavallo, L. MDcons: Intermolecular contact maps as a tool to analyze the interface of protein complexes from molecular dynamics trajectories. BMC Bioinform. 2014, 15, S1. [Google Scholar] [CrossRef] [PubMed]
- Lehermayr, C.; Mahler, H.-C.; Mäder, K.; Fischer, S. Assessment of Net Charge and Protein–Protein Interactions of Different Monoclonal Antibodies. J. Pharm. Sci. 2011, 100, 2551–2562. [Google Scholar] [CrossRef] [PubMed]
- Roberts, D.; Keeling, R.; Tracka, M.; van der Walle, C.F.; Uddin, S.; Warwicker, J.; Curtis, R. The Role of Electrostatics in Protein–Protein Interactions of a Monoclonal Antibody. Mol. Pharm. 2014, 11, 2475–2489. [Google Scholar] [CrossRef] [PubMed]
- Lowry, G.V.; Hill, R.J.; Harper, S.; Rawle, A.F.; Hendren, C.O.; Klaessig, F.; Nobbmann, U.; Sayre, P.; Rumble, J. Guidance to improve the scientific value of zeta-potential measurements in nanoEHS. Environ. Sci. Nano 2016, 3, 953–965. [Google Scholar] [CrossRef]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An automated pipeline for the setup of Poisson–Boltzmann electrostatics calculations. Nucleic Acids Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef] [PubMed]
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, L.; Alexov, E. Predictions for Proteins, RNAs and DNAs with the Gaussian Dielectric Function Using DelPhiPKa. Proteins 2015, 83, 2186–2197. [Google Scholar] [CrossRef] [PubMed]
- Goto, Y.; Takahashi, N.; Fink, A.L. Mechanism of acid-induced folding of proteins. Biochemistry 1990, 29, 3480–3488. [Google Scholar] [CrossRef] [PubMed]
- Ebel, C.; Faou, P.; Kernel, B.; Zaccai, G. Relative Role of Anions and Cations in the Stabilization of Halophilic Malate Dehydrogenase. Biochemistry 1999, 38, 9039–9047. [Google Scholar] [CrossRef] [PubMed]
- Ramos, C.H.I.; Baldwin, R.L. Sulfate anion stabilization of native ribonuclease A both by anion binding and by the Hofmeister effect. Protein Sci. Publ. Protein Soc. 2002, 11, 1771–1778. [Google Scholar] [CrossRef] [PubMed]
- Sedlák, E.; Stagg, L.; Wittung-Stafshede, P. Effect of Hofmeister ions on protein thermal stability: Roles of ion hydration and peptide groups? Arch. Biochem. Biophys. 2008, 479, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Gokarn Yatin, R.; Fesinmeyer, R.M.; Saluja, A.; Razinkov, V.; Chase Susan, F.; Laue Thomas, M.; Brems David, N. Effective charge measurements reveal selective and preferential accumulation of anions, but not cations, at the protein surface in dilute salt solutions. Protein Sci. 2011, 20, 580–587. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Tan, H.; Fesinmeyer, R.M.; Li, C.; Catrone, D.; Le, D.; Remmele, R.L., Jr.; Zhang, J. Antibody solubility behavior in monovalent salt solutions reveals specific anion effects at low ionic strength. J. Pharm. Sci. 2012, 101, 965–977. [Google Scholar] [CrossRef] [PubMed]
- Medda, L.; Barse, B.; Cugia, F.; Boström, M.; Parsons, D.F.; Ninham, B.W.; Monduzzi, M.; Salis, A. Hofmeister Challenges: Ion Binding and Charge of the BSA Protein as Explicit Examples. Langmuir 2012, 28, 16355–16363. [Google Scholar] [CrossRef] [PubMed]
- Goyon, A.; Excoffier, M.; Janin-Bussat, M.-C.; Bobaly, B.; Fekete, S.; Guillarme, D.; Beck, A. Determination of isoelectric points and relative charge variants of 23 therapeutic monoclonal antibodies. J. Chromatogr. B 2017, 1065–1066, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, G.; Peitsch, M.C. SWISS-MODEL and the Swiss-Pdb Viewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar]
- Fair, M.C.; Anderson, J.L. Electrophoresis of nonuniformly charged ellipsoidal particles. J. Colloid Interface Sci. 1989, 127, 388–400. [Google Scholar] [CrossRef]
- Yoon, B.J. Electrophoretic motion of spherical particles with a nonuniform charge distribution. J. Colloid Interface Sci. 1991, 142, 575–581. [Google Scholar] [CrossRef]
- Kim, J.Y.; Ahn, S.H.; Kang, S.T.; Yoon, B.J. Electrophoretic mobility equation for protein with molecular shape and charge multipole effects. J. Colloid Interface Sci. 2006, 299, 486–492. [Google Scholar] [CrossRef] [PubMed]
- Duval, J.F.L.; Ohshima, H. Electrophoresis of Diffuse Soft Particles. Langmuir 2006, 22, 3533–3546. [Google Scholar] [CrossRef] [PubMed]
- Deiber, J.A.; Piaggio, M.V.; Peirotti, M.B. Determination of electrokinetic and hydrodynamic parameters of proteins by modeling their electrophoretic mobilities through the electrically charged spherical soft particle. Electrophoresis 2013, 34, 708–715. [Google Scholar] [CrossRef] [PubMed]
- Božič, A.L.; Podgornik, R. Symmetry effects in electrostatic interactions between two arbitrarily charged spherical shells in the Debye-Hückel approximation. J. Chem. Phys. 2013, 138, 074902. [Google Scholar] [CrossRef] [PubMed]
- Lošdorfer Božič, A.; Podgornik, R. pH Dependence of Charge Multipole Moments in Proteins. Biophys. J. 2017, 113, 1454–1465. [Google Scholar] [CrossRef] [PubMed]
- Sybyl-X Molecular Modeling Software Packages, Version 2.0; TRIPOS Associates, Inc.: St. Louis, MO, USA, 2012.
- Case, D.A.; Bavin, V.; Berryman, J.T.; Betz, R.M.; Cai, Q.; Cerutti, D.S.; Cheatham, I.T.E.; Darden, T.A.; Duke, R.E.; Gohlke, H.; et al. AMBER 14. AMBER 2014; University of California: San Francisco, CA, USA, 2014. [Google Scholar]
- Connolly, M.L. The molecular surface package. J. Mol. Gr. 1993, 11, 139–141. [Google Scholar] [CrossRef]
- De la Torre, J.G.; Ortega, Á.; Amorós, D.; Schmidt, R.R.; Cifre, J.G.H. Methods and Tools for the Prediction of Hydrodynamic Coefficients and Other Solution Properties of Flexible Macromolecules in Solution. A Tutorial Minireview. Macromol. Biosci. 2010, 10, 721–730. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Mw (kDa) SEC | [η] (cm3⋅g−1) SEC | rh (nm) SEC a | D × 107 (cm2⋅s−1) DLS | rh (nm) DLS |
|---|---|---|---|---|---|
| g-eHER2-his b | 88.7 ± 1.8 | 6.4 ± 0.2 | 4.5 ± 0.1 | 4.46 ± 0.02 | 4.7 ± 0.1 |
| TZM b | 149.0 ± 3.0 | 6.5 ± 0.1 | 5.4 ± 0.1 | 4.01 ± 0.02 | 5.2 ± 0.1 |
| Complex 1:1 | 245.0 ± 5.0 | 7.4 ± 0.3 | 6.8 ± 0.1 | 3.16 ± 0.02 | 6.6 ± 0.1 |
| Complex 1:2 | 349.0 ± 13.0 | 8.6 ± 0.3 | 7.7 ± 0.1 | 2.50 ± 0.02 | 8.4 ± 0.1 |
| Sample | μe (μm⋅cm⋅V−1⋅s−1) | ζ (mV) | Z (EM) | Z (PropKa/Delphi) a | ΔZ b | pI |
|---|---|---|---|---|---|---|
| g-eHER2 | −0.76 ± 0.04 | −9.1 ± 0.5 | −18.3 ± 1.4 | −17.0/−16.6 | 1.3/1.7 | 5.9/6.3 |
| TZM | +0.28 ± 0.02 | +3.3 ± 0.3 | +8.3 ± 0.9 | +10.0/+12.9 | 1.7/4.6 | 8.8/9.7 |
| Complex 1:2 | −0.45 ± 0.03 | −5.0 ± 0.3 | −26.1 ± 1.7 | −22.0/−19.9 | 4.1/6.4 | 6.7/6.8 |
| Complex 1:1 | −0.29 ± 0.01 | −3.3 ± 0.1 | −13.2 ± 0.6 | −4.0/−3.1 | 9.2 /10.1 | 7.3/7.2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramos, J.; Vega, J.F.; Cruz, V.; Sanchez-Sanchez, E.; Cortes, J.; Martinez-Salazar, J. Hydrodynamic and Electrophoretic Properties of Trastuzumab/HER2 Extracellular Domain Complexes as Revealed by Experimental Techniques and Computational Simulations. Int. J. Mol. Sci. 2019, 20, 1076. https://doi.org/10.3390/ijms20051076
Ramos J, Vega JF, Cruz V, Sanchez-Sanchez E, Cortes J, Martinez-Salazar J. Hydrodynamic and Electrophoretic Properties of Trastuzumab/HER2 Extracellular Domain Complexes as Revealed by Experimental Techniques and Computational Simulations. International Journal of Molecular Sciences. 2019; 20(5):1076. https://doi.org/10.3390/ijms20051076
Chicago/Turabian StyleRamos, Javier, Juan Francisco Vega, Victor Cruz, Eduardo Sanchez-Sanchez, Javier Cortes, and Javier Martinez-Salazar. 2019. "Hydrodynamic and Electrophoretic Properties of Trastuzumab/HER2 Extracellular Domain Complexes as Revealed by Experimental Techniques and Computational Simulations" International Journal of Molecular Sciences 20, no. 5: 1076. https://doi.org/10.3390/ijms20051076