Gene Selection and Evolutionary Modeling Affect Phylogenomic Inference of Neuropterida Based on Transcriptome Data

 , , ,
, , , 
Abstract
:1. Introduction
2. Results
2.1. Illumina Sequencing, Sequence Assembly, and Data Matrix Construction
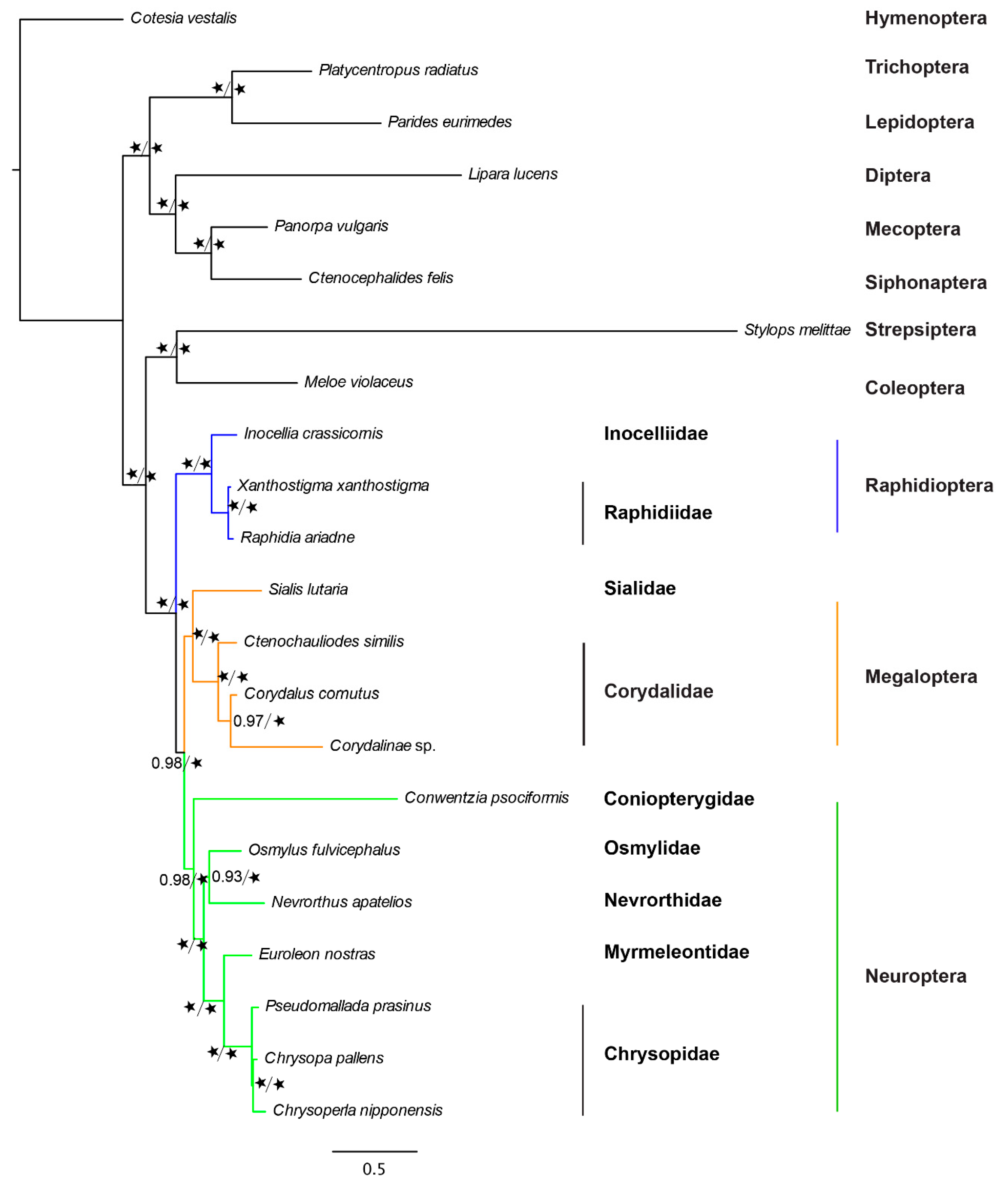
2.2. Phylogenetic Analysis Under a Site-Homogeneous Model
2.3. Phylogenetic Analysis Using Genes with Strong Signals
2.4. Heterogeneous Sequence Divergence Test
2.5. Phylogenetic Analysis Using Site-Heterogeneous Model
3. Discussion
4. Materials and Methods
4.1. Insect Samples and RNA Extraction
4.2. cDNA Library Construction and Sequencing
4.3. Transcriptome Analysis and Assembly
4.4. Data Matrix Construction
4.5. Gene Alignment
4.6. Phylogenetic Inference Under Site-Homogeneous Model
4.7. Phylogenetic Inference Under Site-Heterogeneous Model
4.8. Evaluation of Incongruence
4.9. Data Availability
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ABS | Average bootstrap support |
| nt | Nucleotide |
| aa | Amino acid |
| BS | Bootstrap support |
| UFBS | Ultrafast bootstrap support |
| ML | Maximum likelihood |
| IC | Internode certainty |
| ICA | Internode certainty including all conflicting bipartitions |
| TC | Tree certainty; |
| TCA | Tree certainty including all conflicting bipartitions |
| pp | Posterior probability |
| RTCA | Relative tree certainty including all conflicting bipartitions; |
| EB | Ethidium bromide |
| SRA | Short Read Archive |
| TSA | Transcriptome Shotgun Assembly |
References
- Wiegmann, B.M.; Trautwein, M.D.; Kim, J.W.; Cassel, B.K.; Bertone, M.A.; Winterton, S.L.; Yeates, D.K. Single-copy nuclear genes resolve the phylogeny of the holometabolous insects. BMC Biol. 2009, 7, 34. [Google Scholar] [CrossRef] [PubMed]
- Misof, B.; Liu, S.; Meusemann, K.; Peters, R.S.; Donath, A.; Mayer, C.; Frandsen, P.B.; Ware, J.; Flouri, T.; Beutel, R.G. Phylogenomics resolves the timing and pattern of insect evolution. Science 2014, 346, 763–767. [Google Scholar] [CrossRef] [PubMed]
- Oswald, J.D. Lacewing Digital Library. Available online: http://lacewing.tamu.edu/ (accessed on 12 September 2015).
- Aspöck, U.; Plant, J.D.; Nemeschkal, H.L. Cladistic analysis of Neuroptera and their systematic position within Neuropterida (Insecta: Holometabola: Neuropterida: Neuroptera). Syst. Entomol. 2001, 26, 73–86. [Google Scholar] [CrossRef]
- Beutel, R.G.; Friedrich, F.; Aspöck, U. The larval head of Nevrorthidae and the phylogeny of Neuroptera (Insecta). Zool. J. Linn. Soc. 2010, 158, 533–562. [Google Scholar] [CrossRef]
- Zhao, C.; Liu, X.; Yang, D. Wing base structural data support the sister relationship of megaloptera and neuroptera (insecta: Neuropterida). PLoS ONE 2014, 9, e114695. [Google Scholar] [CrossRef] [PubMed]
- Haring, E.; Aspöck, U. Phylogeny of the Neuropterida: A first molecular approach. Syst. Entomol. 2004, 29, 415–430. [Google Scholar] [CrossRef]
- Kjer, K.M.; Carle, F.L.; Litman, J.; Ware, J. A Molecular Phylogeny of Hexapoda. Arthropod Syst. Phylogeny 2006, 64, 35–44. [Google Scholar]
- Cameron, S.; Sullivan, J.; Song, H.; Miller, K.; Whiting, M. A mitochondrial genome phylogeny of the Neuropterida (lace-wings, alderflies and snakeflies) and their relationship to the other holometabolous insect orders. Zool. Scr. 2009, 38, 575–590. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, X.; Winterton, S.L.; Yang, D. The first mitochondrial genome for the fishfly subfamily Chauliodinae and implications for the higher phylogeny of Megaloptera. PLoS ONE 2012, 7, e47302. [Google Scholar] [CrossRef] [PubMed]
- Winterton, S.L.; Lemmon, A.R.; Gillung, J.P.; Garzon, I.J.; Bakkes, D.K.; Breitkreuz, L.C.V.; Duelli, P.; Engel, M.S.; Lemmon, E.M.; Liu, X.Y.; et al. Phylogenomics of lacewings using anchored hybrid enrichment data (Insecta: Neuroptera). Syst. Entomol. 2018, 43, 330–354. [Google Scholar] [CrossRef]
- Winterton, S.L.; Hardy, N.B.; Wiegmann, B.M. On wings of lace: Phylogeny and Bayesian divergence time estimates of Neuropterida (Insecta) based on morphological and molecular data. Syst. Entomol. 2010, 35, 349–378. [Google Scholar] [CrossRef]
- Withycombe, C. Some aspects of the biology and morphology of the Neuroptera. With special reference to the immature stages and their possible phylogenetic significance. Trans. R. Entomol. Soc. Lond. 1924, 72, 303–411. [Google Scholar] [CrossRef]
- Winterton, S. Molecular Phylogeny of Neuropterida with Emphasis on the Lacewings (Neuroptera). Entomol. Abh. 2003, 61, 158–160. [Google Scholar]
- Aspöck, U.; Aspöck, H. Phylogenetic relevance of the genital sclerites of Neuropterida (Insecta: Holometabola). Syst. Entomol. 2008, 33, 97–127. [Google Scholar] [CrossRef]
- Zimmermann, D.; Klepal, W.; Aspöck, U. The first holistic SEM study of Coniopterygidae (Neuroptera)—Structural evidence and phylogenetic implications. Eur. J. Entomol. 2009, 106, 651–662. [Google Scholar] [CrossRef]
- Randolf, S.; Zimmermann, D.; Aspöck, U. Head anatomy of adult Sisyra terminalis (Insecta: Neuroptera: Sisyridae)—Functional adaptations and phylogenetic implications. Arthropod Struct. Dev. 2013, 42, 565–582. [Google Scholar] [CrossRef] [PubMed]
- Randolf, S.; Zimmermann, D.; Aspöck, U. Head anatomy of adult Nevrorthus apatelios and basal splitting events in Neuroptera (Neuroptera: Nevrorthidae). Arthropod Syst. Phylogeny 2014, 111, 111–136. [Google Scholar]
- Hittinger, C.T.; Johnston, M.; Tossberg, J.T.; Rokas, A. Leveraging skewed transcript abundance by RNA-Seq to increase the genomic depth of the tree of life. Proc. Natl. Acad. Sci. USA 2010, 107, 1476–1481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiménez-Guri, E.; Huerta-Cepas, J.; Cozzuto, L.; Wotton, K.R.; Kang, H.; Himmelbauer, H.; Roma, G.; Gabaldón, T.; Jaeger, J. Comparative transcriptomics of early dipteran development. BMC Genomics 2013, 14, 123. [Google Scholar] [CrossRef] [PubMed]
- Peters, R.S.; Meusemann, K.; Petersen, M.; Mayer, C.; Wilbrandt, J.; Ziesmann, T.; Donath, A.; Kjer, K.M.; Aspöck, U.; Aspöck, H.; et al. The evolutionary history of holometabolous insects inferred from transcriptome-based phylogeny and comprehensive morphological data. BMC Evol. Biol. 2014, 14, 380–393. [Google Scholar] [CrossRef] [PubMed]
- Zhong, B.; Liu, L.; Yan, Z.; Penny, D. Origin of land plants using the multispecies coalescent model. Trends Plant Sci. 2013, 18, 492–495. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Liu, L.; Edwards, S.V.; Wu, S. Resolving conflict in eutherian mammal phylogeny using phylogenomics and the multispecies coalescent model. Proc. Natl. Acad. Sci. USA 2012, 109, 14942–14947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirarab, S. ASTRAL-II: Coalescent-based species tree estimation with many hundreds of taxa and thousands of genes. Bioinformatics 2015, 31, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Lartillot, N.; Philippe, H. A Bayesian mixture model for across-site heterogeneities in the amino-acid replacement process. Mol. Biol. Evol. 2004, 21, 1095–1109. [Google Scholar] [CrossRef] [PubMed]
- Lartillot, N.; Philippe, H. Computing Bayes factors using thermodynamic integration. Syst. Biol. 2006, 55, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Salichos, L.; Rokas, A. Inferring ancient divergences requires genes with strong phylogenetic signals. Nature 2013, 497, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Salichos, L.; Stamatakis, A.; Rokas, A. Novel information theory-based measures for quantifying incongruence among phylogenetic trees. Mol. Biol. Evol. 2014, 31, 1261–1271. [Google Scholar] [CrossRef] [PubMed]
- Simão, A.F.O.; Waterhouse, M.R.; Ioannidis, P.; Kriventseva, V.E.; Zdobnov, M.E. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Lartillot, N.; Brinkmann, H.; Philippe, H. Suppression of long-branch attraction artefacts in the animal phylogeny using a site-heterogeneous model. BMC Evol. Biol. 2007, 7, S4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Shao, R.; Song, N.; Song, F.; Jiang, P.; Li, Z.; Cai, W. Higher-level phylogeny of paraneopteran insects inferred from mitochondrial genome sequences. Sci. Rep. 2015, 5, 8527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, F.; Li, H.; Jiang, P. Capturing the Phylogeny of Holometabola with Mitochondrial Genome Data and Bayesian Site-Heterogeneous Mixture Models. Gen. Biol. Evol. 2016, 8, 1411–1426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheffield, N.C.; Song, H.; Cameron, S.L.; Whiting, M.F. Nonstationary Evolution and Compositional Heterogeneity in Beetle Mitochondrial Phylogenomics. Syst. Biol. 2009, 58, 381–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kück, P.; Meid, S.A.; Groß, C.; Wägele, J.W.; Misof, B. AliGROOVE—Visualization of heterogeneous sequence divergence within multiple sequence alignments and detection of inflated branch support. BMC Bioinform. 2014, 15, 294. [Google Scholar] [CrossRef] [PubMed]
- Lartillot, N.; Lepage, T.; Blanquart, S. PhyloBayes 3: A Bayesian software package for phylogenetic reconstruction and molecular dating. Bioinformatics 2009, 25, 2286–2288. [Google Scholar] [CrossRef] [PubMed]
- Quang, L.S.; Gascuel, O.N. Empirical profile mixture models for phylogenetic reconstruction. Bioinformatics 2008, 24, 2317–2323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam-Tung, N.; Schmidt, H.A.; Arndt, V.H.; Bui Quang, M. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar]
- Yin, C.; Shen, G.; Guo, D.; Wang, S.; Ma, X.; Xiao, H.; Liu, J.; Zhang, Z.; Liu, Y.; Zhang, Y. InsectBase: A resource for insect genomes and transcriptomes. Nucleic Acids Res. 2015, 44, D801–D807. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, A.Y.; Breinholt, J.W. Phylogenomics provides strong evidence for relationships of butterflies and moths. Proc. Roy. Soc. B Biol. Sci. 2014, 281, 738–743. [Google Scholar] [CrossRef] [PubMed]
- Dell’Ampio, E.; Meusemann, K.; Szucsich, N.U.; Peters, R.S.; Meyer, B.; Borner, J.; Petersen, M.; Aberer, A.J.; Stamatakis, A.; Walzl, M.G. Decisive data sets in phylogenomics: Lessons from studies on the phylogenetic relationships of primarily wingless insects. Mol. Biol. Evol. 2014, 31, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Neafsey, D.E.; Waterhouse, R.M.; Abai, M.R.; Aganezov, S.S.; Alekseyev, M.A.; Allen, J.E.; Amon, J.; Arcà, B.; Arensburger, P.; Artemov, G. Highly evolvable malaria vectors: The genomes of 16 Anopheles mosquitoes. Science 2015, 347, 1258522. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, E.D.; Mirarab, S.; Aberer, A.J.; Li, B.; Houde, P.; Li, C.; Ho, S.Y.W.; Faircloth, B.C.; Nabholz, B.; Howard, J.T. Whole-genome analyses resolve early branches in the tree of life of modern birds. Science 2014, 346, 1320–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rokas, A.; Carroll, S.B. Bushes in the tree of life. PLoS Biol. 2006, 4, e352. [Google Scholar] [CrossRef] [PubMed]
- Hess, J.; Goldman, N. Addressing Inter-Gene Heterogeneity in Maximum Likelihood Phylogenomic Analysis: Yeasts Revisited. PLoS ONE 2011, 6, 685. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, G.; Vincent, D. Dealing with incongruence in phylogenomic analyses. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2008, 363, 4023–4029. [Google Scholar] [Green Version]
- Degnan, J.H.; Rosenberg, N.A. Gene tree discordance, phylogenetic inference and the multispecies coalescent. Trends Ecol. Evol. 2009, 24, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, M.C.; Pease, J.B.; Aaron, S.; Waterhouse, R.M.; Neafsey, D.E.; Sharakhov, I.V.; Xiaofang, J.; Hall, A.B.; Flaminia, C.; Evdoxia, K. Mosquito genomics. Extensive introgression in a malaria vector species complex revealed by phylogenomics. Science 2015, 347, 1258524. [Google Scholar] [CrossRef] [PubMed]
- Nater, A.; Burri, R.; Kawakami, T.; Smeds, L.; Ellegren, H. Resolving Evolutionary Relationships in Closely Related Species with Whole-Genome Sequencing Data. Syst. Biol. 2015, 64, 1000–1017. [Google Scholar] [CrossRef] [PubMed]
- Ricardo, B.R.; Naylor, G.J.P.; Guillermo, O. Conserved genes, sampling error, and phylogenomic inference. Syst. Biol. 2014, 63, 257–262. [Google Scholar]
- Jonathan, R.; Vincent, R.; Frédéric, D.; Nicolas, G.; Douzery, E.J.P. Less is more in mammalian phylogenomics: AT-rich genes minimize tree conflicts and unravel the root of placental mammals. Mol. Biol. Evol. 2013, 30, 2134–2144. [Google Scholar]
- Wang, Y.; Liu, X.; Garzón-Orduña, I.J.; Winterton, S.L.; Yan, Y.; Aspöck, U.; Aspöck, H.; Yang, D. Mitochondrial phylogenomics illuminates the evolutionary history of Neuropterida. Cladistics 2017, 33, 617–636. [Google Scholar] [CrossRef]
- Gillung, J.P.; Winterton, S.L.; Bayless, K.M.; Khouri, Z.; Borowiec, M.L.; Yeates, D.; Kimsey, L.S.; Misof, B.; Shin, S.; Zhou, X.; et al. Anchored phylogenomics unravels the evolution of spider flies (Diptera, Acroceridae) and reveals discordance between nucleotides and amino acids. Mol. Phylogen. Evol. 2018, 128, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Wu, G.; Tang, J.; Luo, R.; Patterson, J.; Liu, S.; Huang, W.; He, G.; Gu, S.; Li, S. SOAPdenovo-Trans: Transcriptome assembly with short RNA-Seq reads. Bioinformatics 2014, 30, 1660–1666. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.-X.; Zhou, X.; Kominek, J.; Kurtzman, C.P.; Hittinger, C.T.; Rokas, A. Reconstructing the backbone of the Saccharomycotina yeast phylogeny using genome-scale data. G3 Genes Genomes Genet. 2016, 6, 3927–3939. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Toh, H. Recent developments in the MAFFT multiple sequence alignment program. Brief. Bioinform. 2008, 9, 286–298. [Google Scholar] [CrossRef] [PubMed]
- Suyama, M.; Torrents, D.; Bork, P. PAL2NAL: Robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006, 34, W609–W612. [Google Scholar] [CrossRef] [PubMed]
- Capellagutiérrez, S.; Sillamartínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML Version 8: A tool for Phylogenetic Analysis and Post-Analysis of Large Phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Hoang, D.T.; Chernomor, O.; Von, H.A.; Minh, B.Q.; Le, S.V. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2017, 35, 518–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Number |
|---|---|
| Raw Data | 26,988,698 |
| Q20 of string one of raw data | 91.70% |
| Q20 of string two of raw data | 89.91% |
| GC content of string one raw data | 40.64% |
| GC content of string two raw data | 40.62% |
| Total number of clean reads | 25,017,948 |
| Q20 of string one of clean reads | 99.98% |
| Q20 of string two of clean reads | 99.95% |
| GC content of string one of clean reads | 40.26% |
| GC content of string two of clean reads | 40.21% |
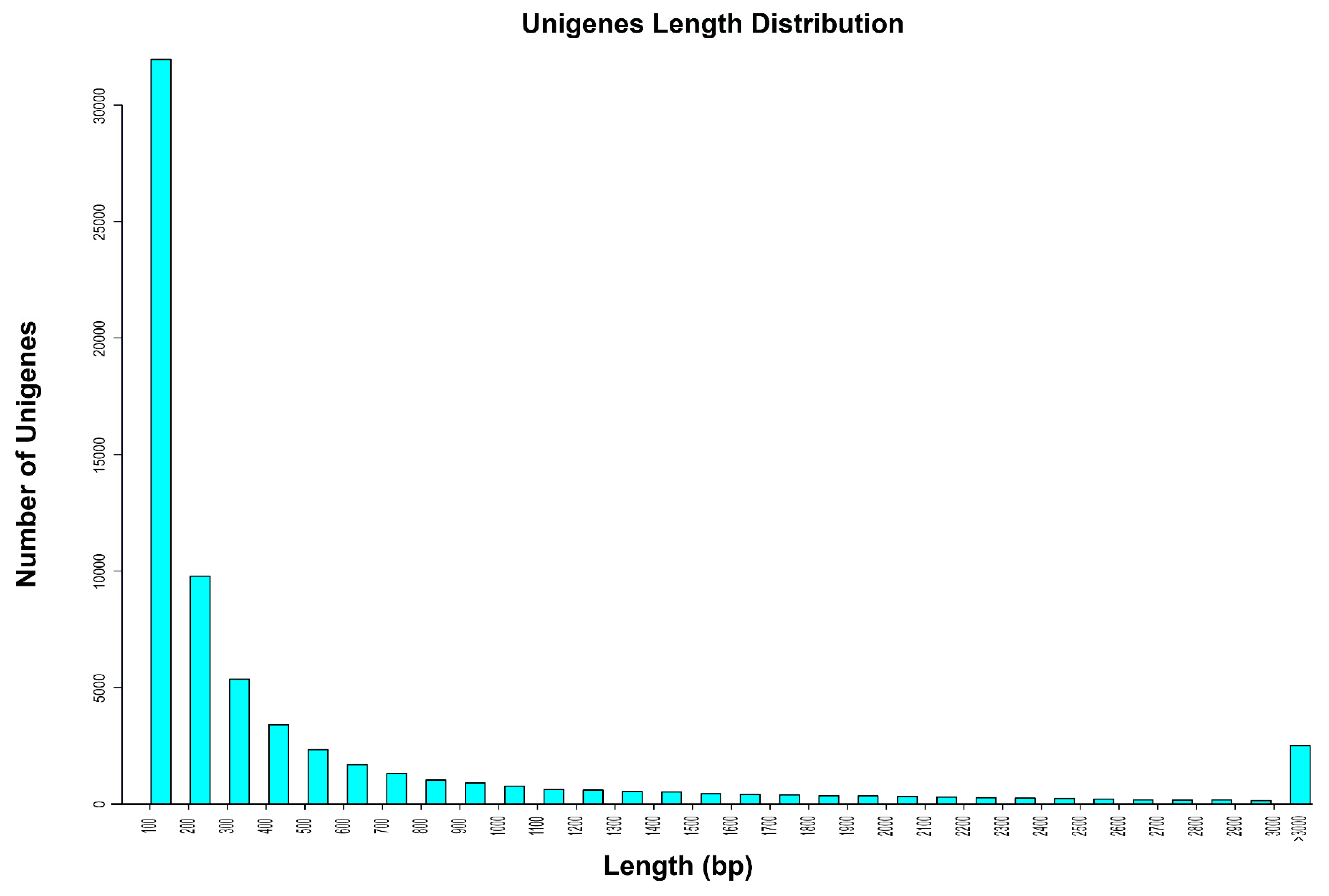
| Total number of unigenes | 67,683 |
| Minimum length of unigenes | 100 |
| Maximum length of unigenes | 50,138 |
| Mean length of unigenes | 585.26 |
| N50 of unigenes (nt) | 1675 |
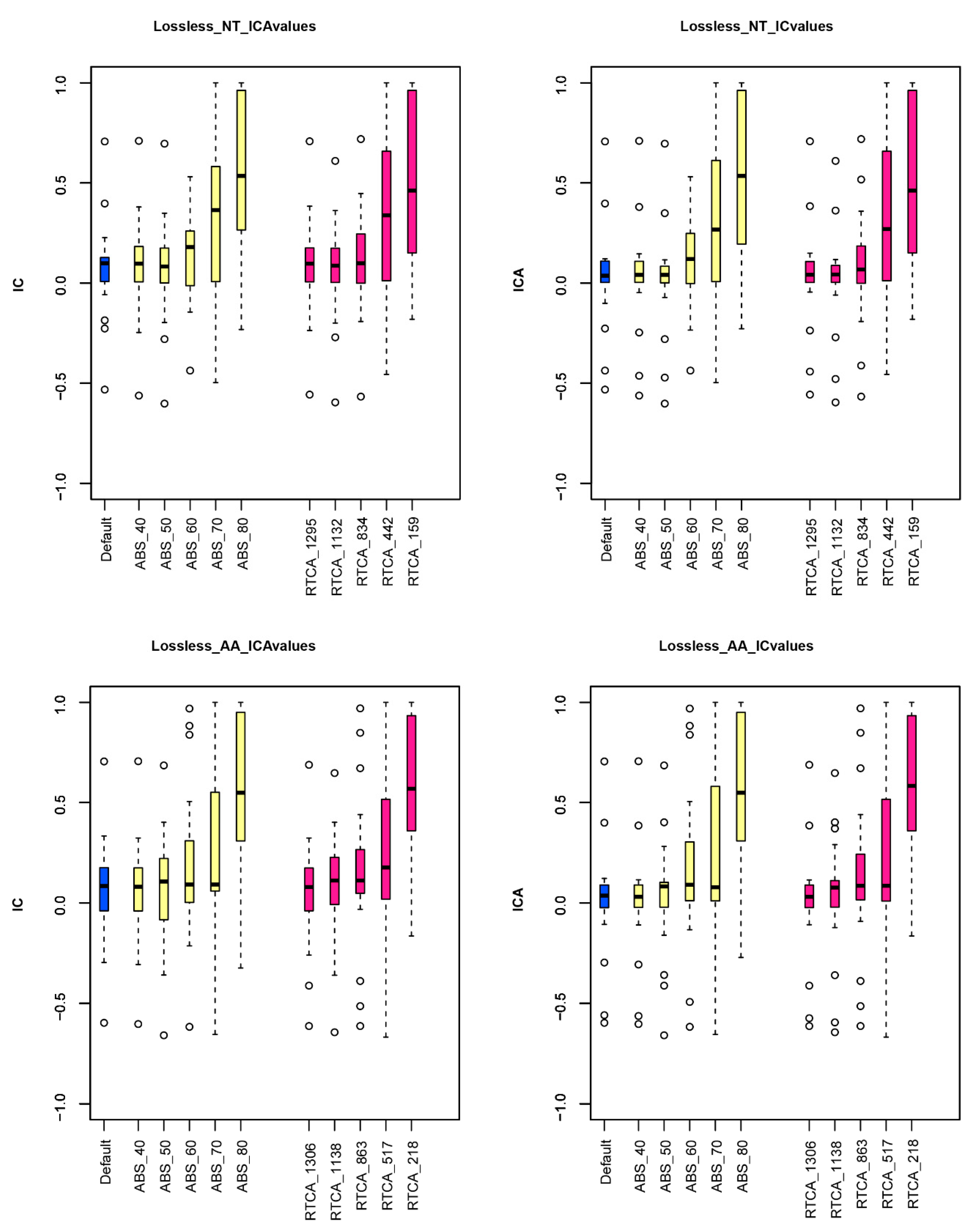
| Treatment (NT) | Treatment Details | TCA | RTCA | ICA Increases | ICA Decreases |
|---|---|---|---|---|---|
| Default analysis | 1392 genes | 1.41 | 0.07 | / | / |
| Selection of genes whose ML trees have high ABS | Genes with ABS ≥ 40% (1295 genes) | 1.45 | 0.08 | 8 | 11 |
| Genes with ABS ≥ 50% (1132 genes) | 1.23 | 0.07 | 5 | 14 | |
| Genes with ABS ≥ 60% (834 genes) | 2.51 | 0.13 | 14 | 5 | |
| Genes with ABS ≥ 70% (442 genes) | 6.41 | 0.34 | 18 | 1 | |
| Genes with ABS ≥ 80% (159 genes) | 10.42 | 0.55 | 16 | 3 | |
| Selection of genes whose ML trees have high RTC | Using only 1295 genes with the highest RTC | 1.47 | 0.08 | 5 | 10 |
| Using only 1132 genes with the highest RTC | 1.20 | 0.06 | 9 | 10 | |
| Using only 834 genes with the highest RTC | 2.05 | 0.11 | 11 | 7 | |
| Using only 442 genes with the highest RTC | 6.20 | 0.33 | 16 | 3 | |
| Using only 159 genes with the highest RTC | 10.11 | 0.53 | 17 | 2 |
| Treatment (AA) | Treatment Details | TCA | RTCA | ICA Increases | ICA Decreases |
|---|---|---|---|---|---|
| Default analysis | 1392 genes | 1.13 | 0.06 | / | / |
| Selection of genes whose ML trees have high ABS | Genes with ABS ≥ 40% (1306 genes) | 1.08 | 0.06 | 5 | 14 |
| Genes with ABS ≥ 50% (1138 genes) | 1.33 | 0.07 | 11 | 7 | |
| Genes with ABS ≥ 60% (863 genes) | 3.48 | 0.18 | 13 | 6 | |
| Genes with ABS ≥ 70% (517 genes) | 4.65 | 0.24 | 12 | 7 | |
| Genes with ABS ≥ 80% (218 genes) | 10.38 | 0.55 | 17 | 2 | |
| Selection of genes whose ML trees have high RTC | Using only 1306 genes with the highest RTC | 0.94 | 0.05 | 4 | 15 |
| Using only 1138 genes with the highest RTC | 1.55 | 0.08 | 11 | 8 | |
| Using only 863 genes with the highest RTC | 2.85 | 0.15 | 11 | 8 | |
| Using only 517 genes with the highest RTC | 4.73 | 0.25 | 14 | 5 | |
| Using only 218 genes with the highest RTC | 11.09 | 0.58 | 17 | 2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Zhou, X.; Wang, L.; Liu, X.; Yang, D.; Rokas, A. Gene Selection and Evolutionary Modeling Affect Phylogenomic Inference of Neuropterida Based on Transcriptome Data. Int. J. Mol. Sci. 2019, 20, 1072. https://doi.org/10.3390/ijms20051072
Wang Y, Zhou X, Wang L, Liu X, Yang D, Rokas A. Gene Selection and Evolutionary Modeling Affect Phylogenomic Inference of Neuropterida Based on Transcriptome Data. International Journal of Molecular Sciences. 2019; 20(5):1072. https://doi.org/10.3390/ijms20051072
Chicago/Turabian StyleWang, Yuyu, Xiaofan Zhou, Liming Wang, Xingyue Liu, Ding Yang, and Antonis Rokas. 2019. "Gene Selection and Evolutionary Modeling Affect Phylogenomic Inference of Neuropterida Based on Transcriptome Data" International Journal of Molecular Sciences 20, no. 5: 1072. https://doi.org/10.3390/ijms20051072