Regulation of c-Raf Stability through the CTLH Complex


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
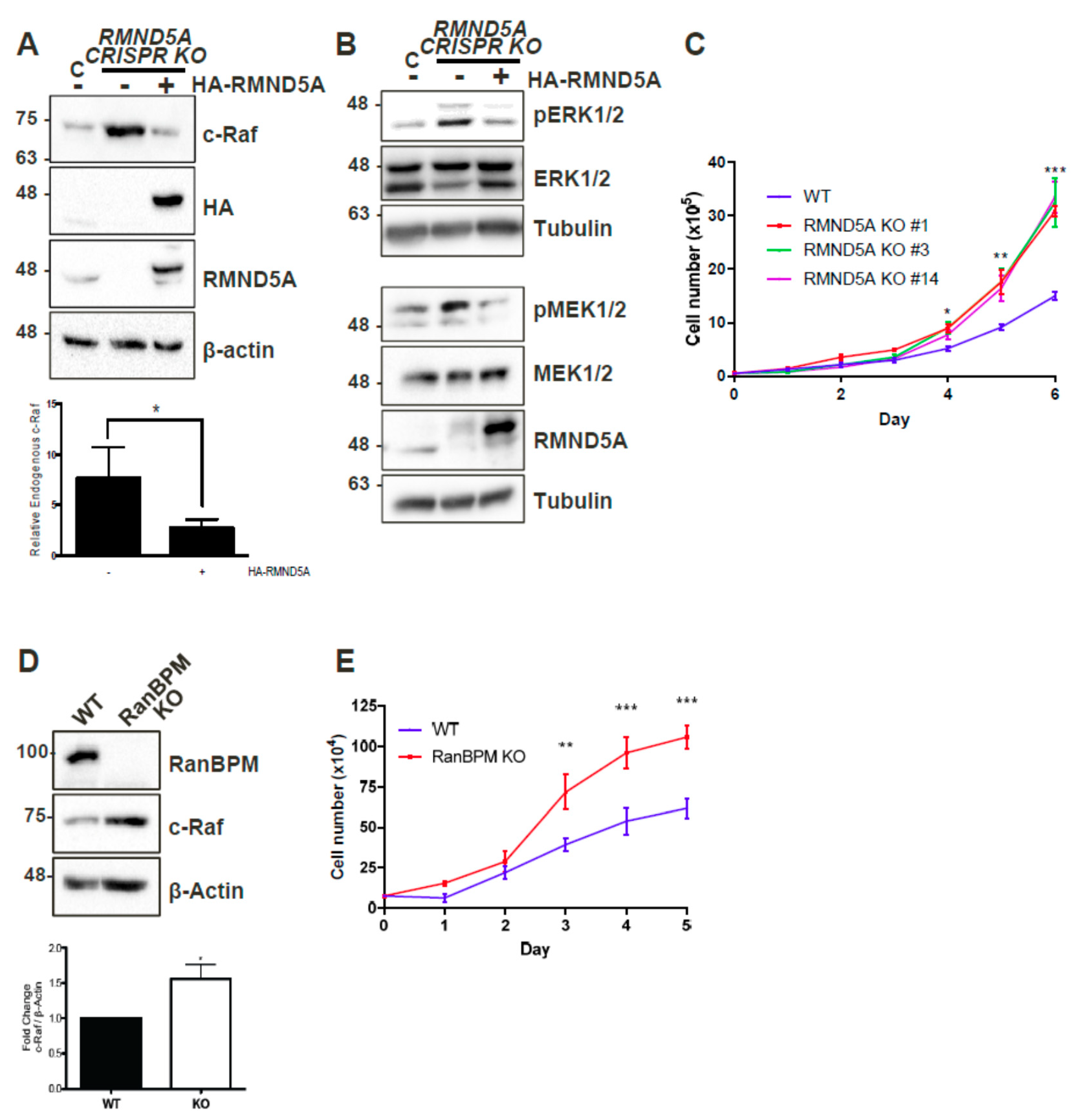
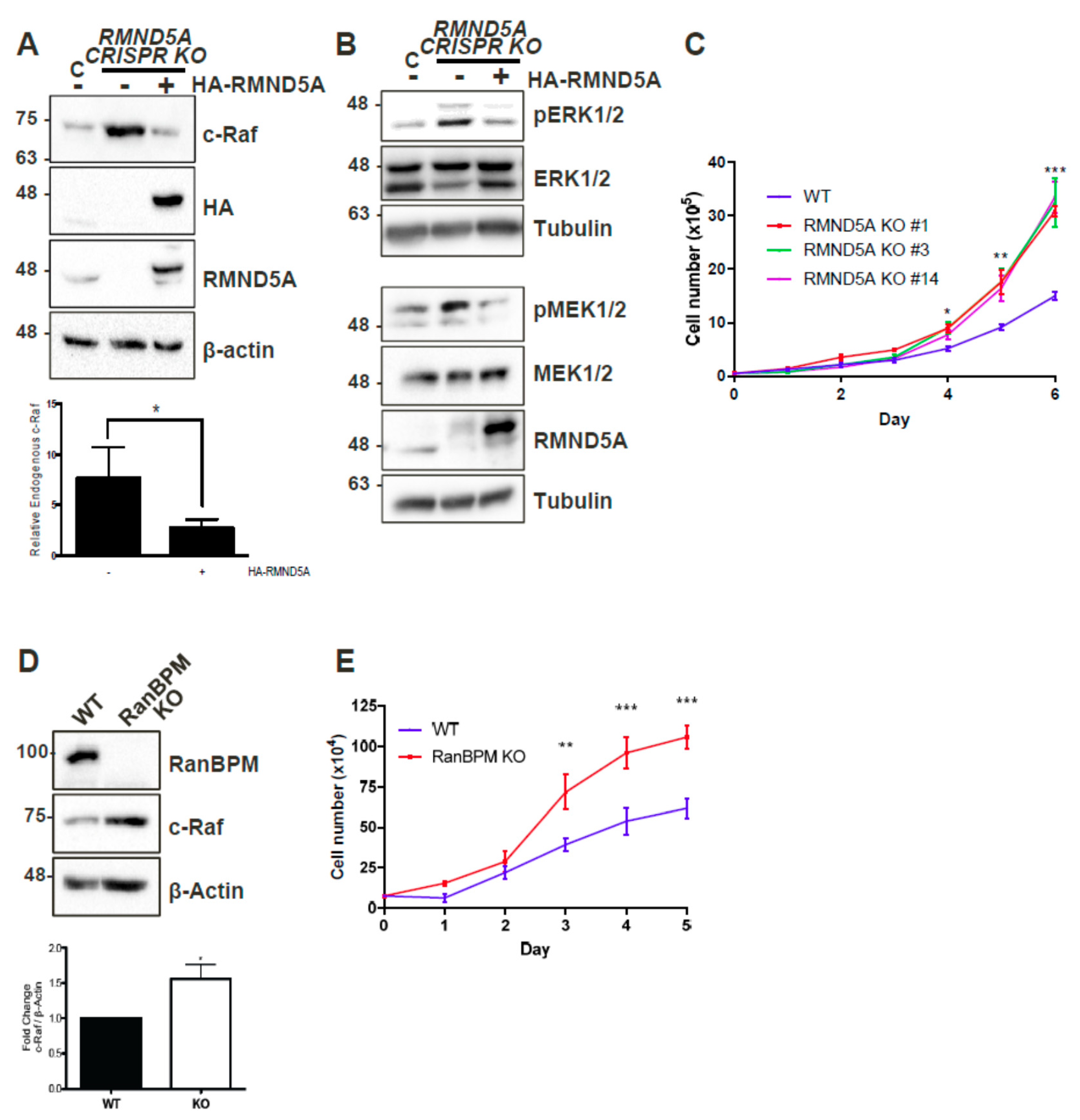
2.1. CTLH Complex Members Regulate c-Raf Levels and Cell Growth
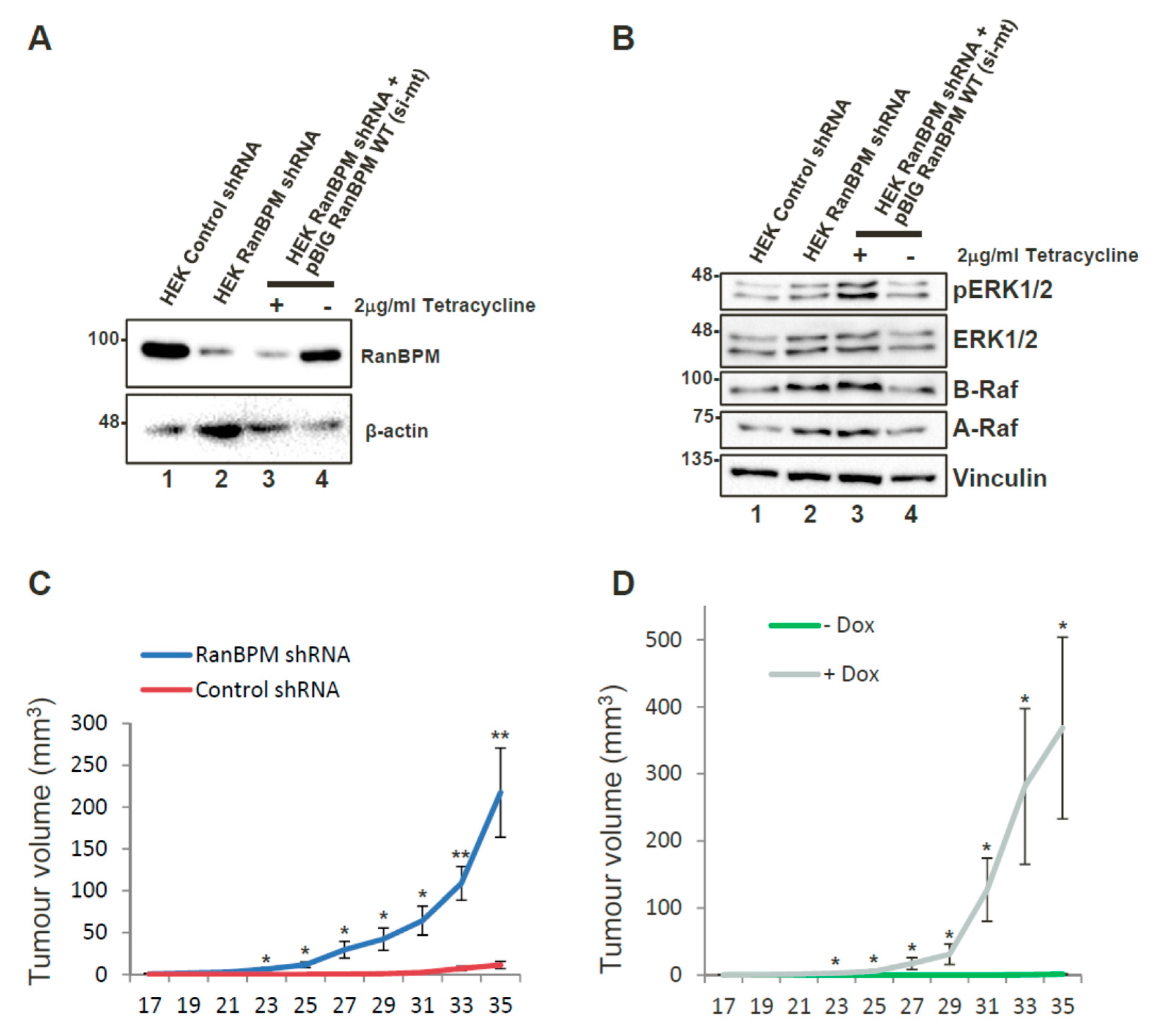
2.2. RanBPM Expression Prevents Tumour Formation in Mouse Models
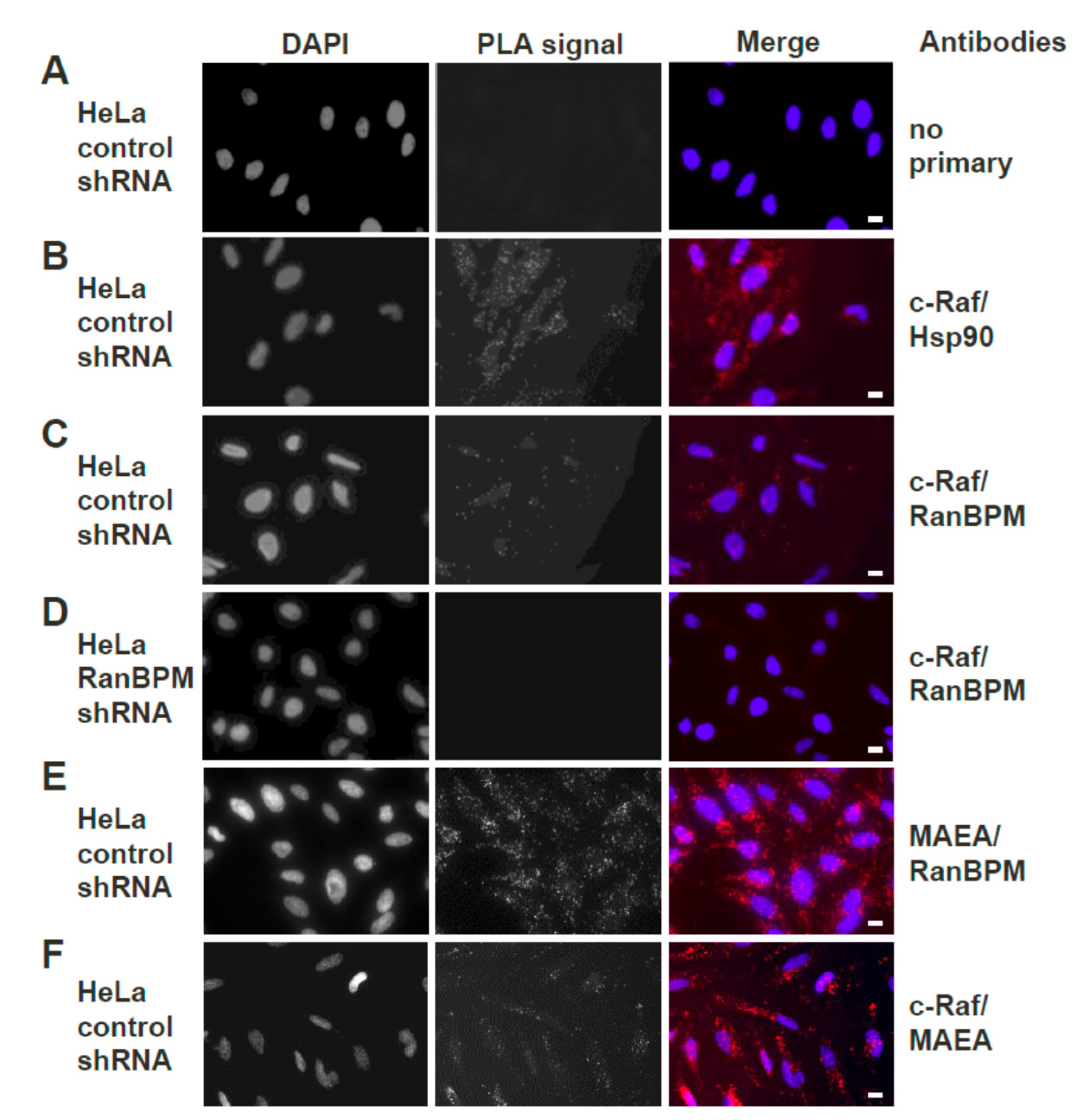
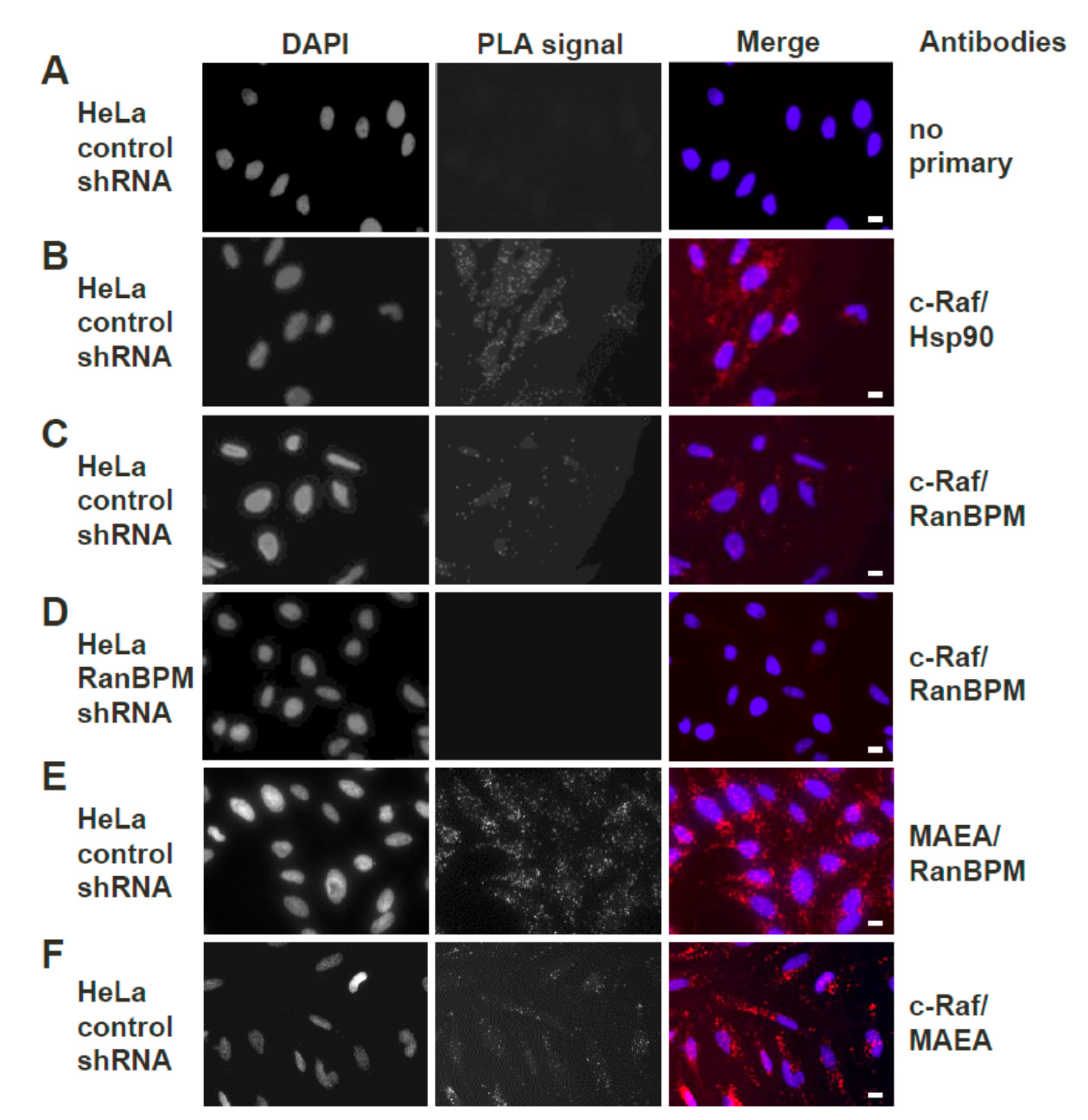
2.3. Both Endogenous RanBPM and MAEA Form a Complex with c-Raf In Situ
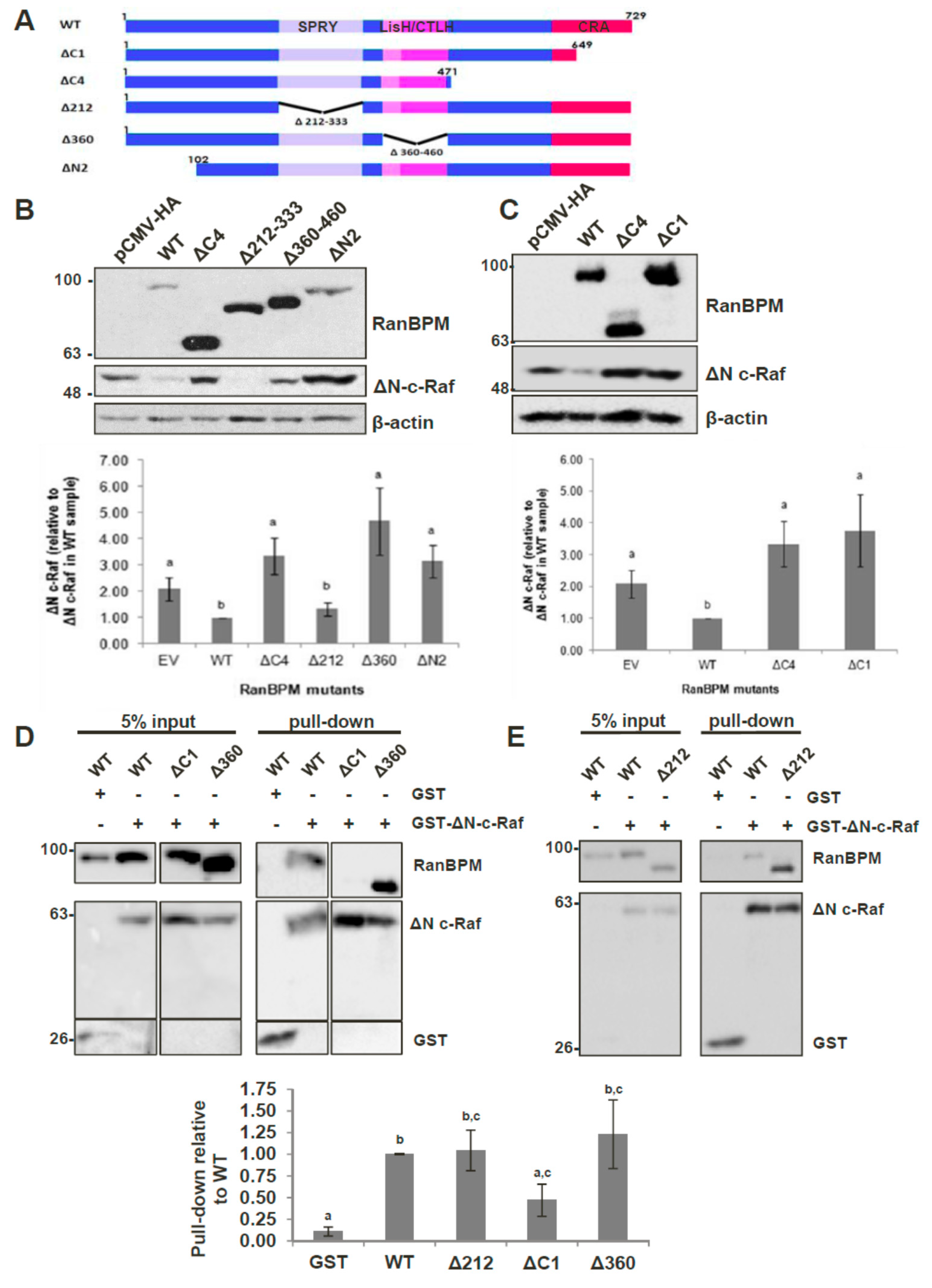
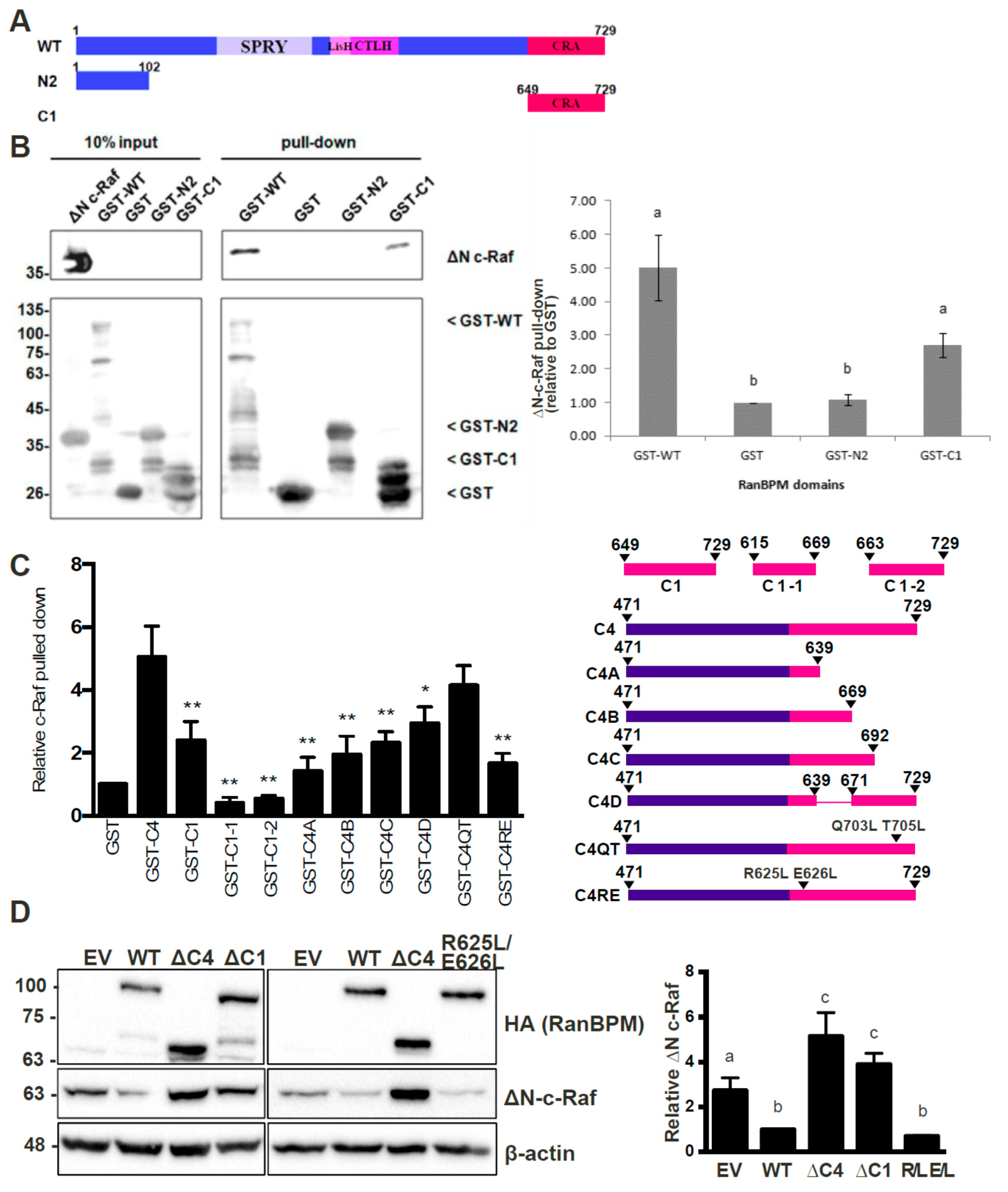
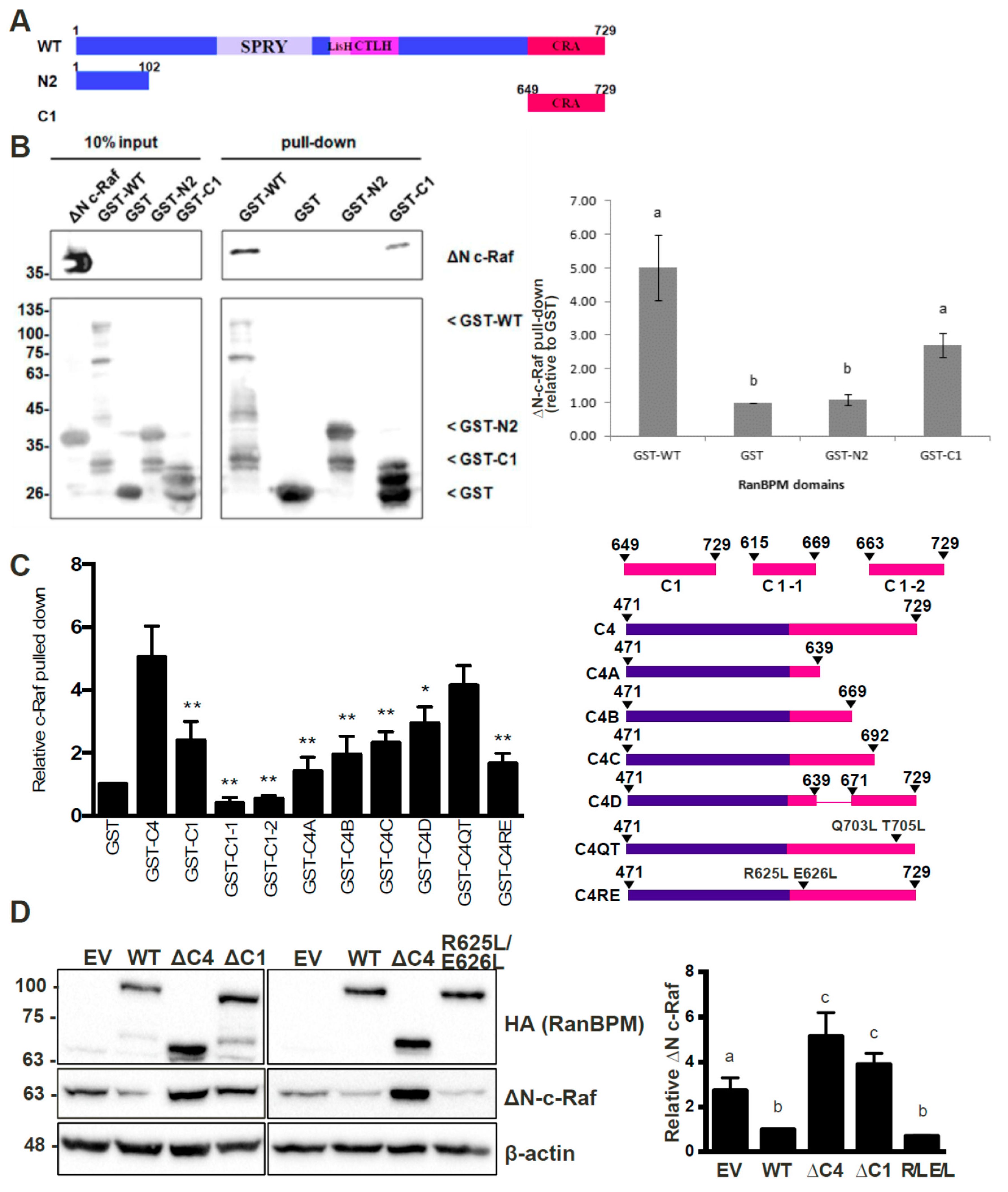
2.4. The N-Terminus, CRA and LisH/CTLH Domains of RanBPM Are Required for c-Raf Downregulation
2.5. The CRA Domain of RanBPM Is Required for Interaction with c-Raf
2.6. RanBPM Interacts Directly with c-Raf through the CRA Domain
2.7. Analysis of the RanBPM CRA Domain Interaction with c-Raf
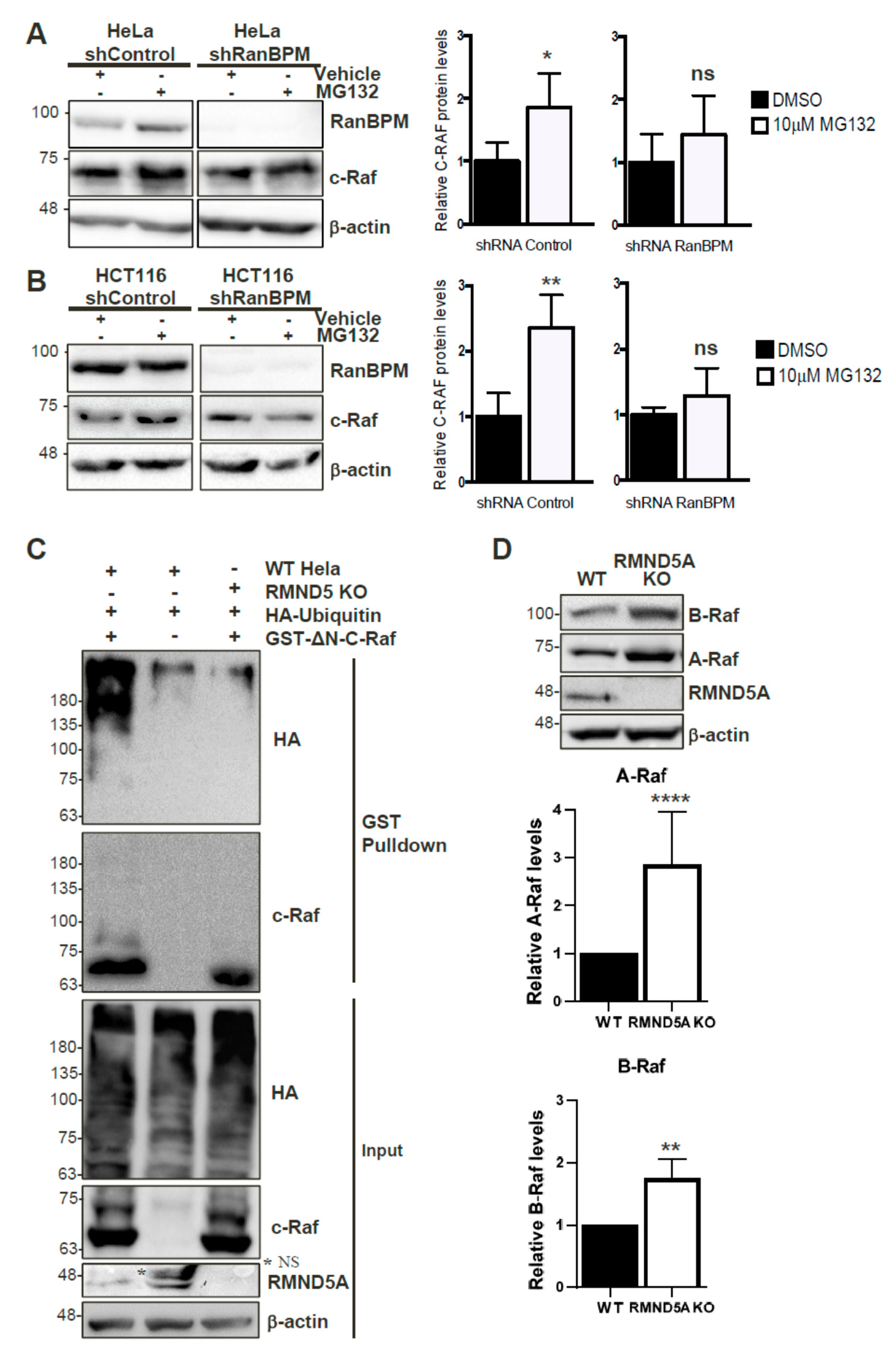
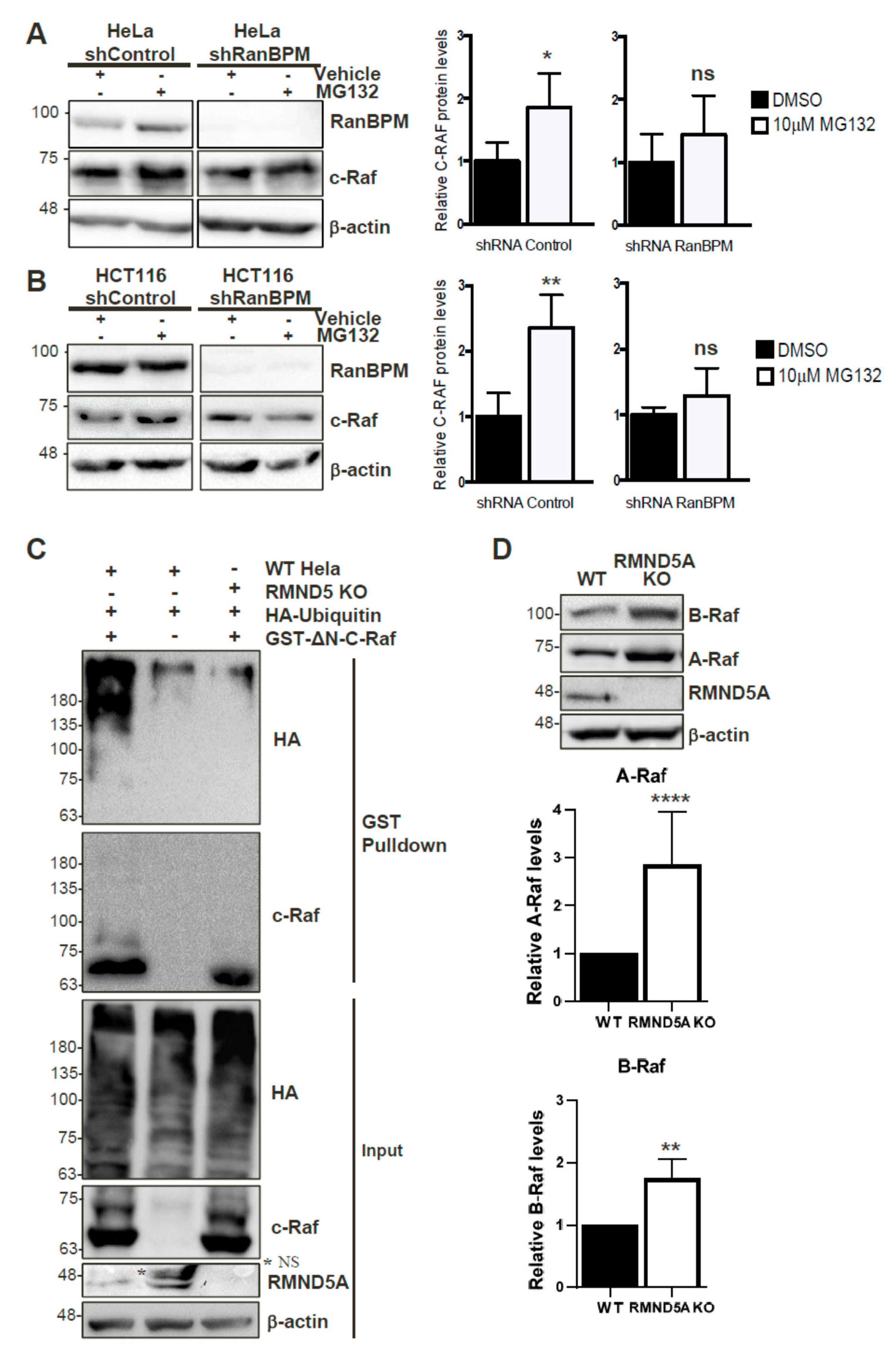
2.8. c-Raf Stability Is Regulated through the Proteasome and Its Ubiquitination Is Dependent on the CTLH Complex
2.9. The CTLH Complex Regulation Is Conserved for A- and B-Raf
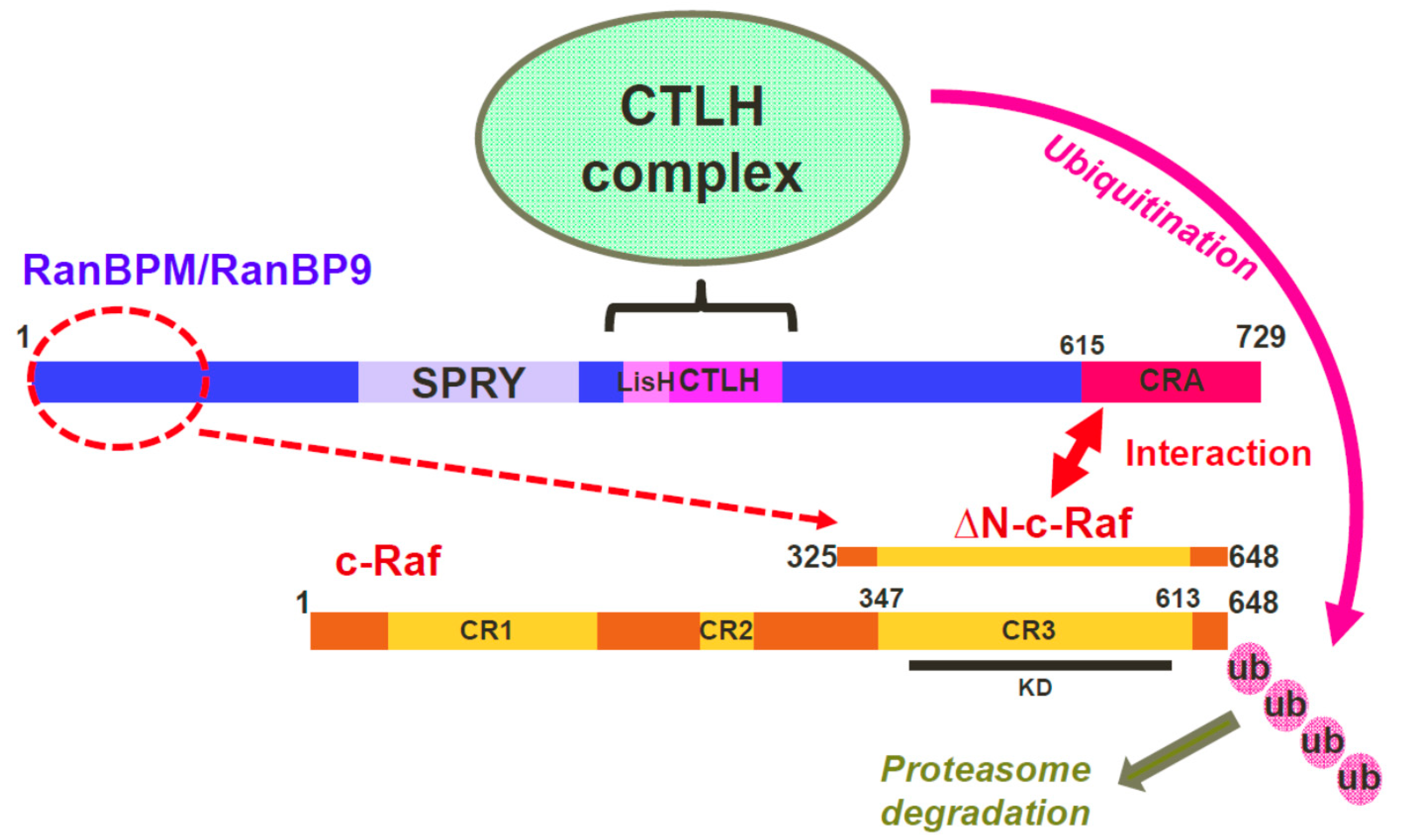
3. Discussion
4. Materials and Methods
4.1. Plasmid Constructs
4.2. Cell Lines and Cell Culture and Transfections
4.3. In Situ Proximity Ligation Assay (PLA)
4.4. Cell Extracts and Western Blot Analyses
4.5. GST Pull-Down Assays
4.6. Mouse Tumour Models
4.7. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CHIP | C-terminus of constitutive heat shock protein (Hsc) 70-interacting protein |
| CR3 | Conserved Region 3 |
| CRA | CT11-RanBPM |
| CTLH | C-terminal to LisH (LIS1-homology motif) |
| ELF3 | E74-like factor 3 |
| ERK | Extracellular signal-Regulated Kinase |
| Hsp90 | Heat Shock Protein 90 |
| KO | Knockout |
| L1CAM | L1 cell adhesion molecule |
| MAEA | Macrophage erythroblast attacher |
| RanBPM | Ran Binding Protein M |
| RMND5A | Required for Meiotic Nuclear Division 5A |
| RON | Recepteur d’origine nantais |
| TG2 | transglutaminase 2 |
| XIAP | X-linked inhibitor of apoptosis protein |
| WT | Wild-type |
References
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matallanas, D.; Birtwistle, M.; Romano, D.; Zebisch, A.; Rauch, J.; von Kriegsheim, A.; Kolch, W. Raf family kinases: Old dogs have learned new tricks. Genes Cancer 2011, 2, 232–260. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. RAF protein-serine/threonine kinases: Structure and regulation. Biochem. Biophys. Res. Commun. 2010, 399, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Chong, H.; Lee, J.; Guan, K.L. Positive and negative regulation of Raf kinase activity and function by phosphorylation. EMBO J. 2001, 20, 3716–3727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhillon, A.S.; Meikle, S.; Yazici, Z.; Eulitz, M.; Kolch, W. Regulation of Raf-1 activation and signalling by dephosphorylation. EMBO J. 2002, 21, 64–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, N.H.; Wu, X.; Frost, J.A. B-Raf and Raf-1 Are Regulated by Distinct Autoregulatory Mechanisms. J. Biol. Chem. 2005, 280, 16244–16253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubicek, M.; Pacher, M.; Abraham, D.; Podar, K.; Eulitz, M.; Baccarini, M. Dephosphorylation of Ser-259 Regulates Raf-1 Membrane Association. J. Biol. Chem. 2002, 277, 7913–7919. [Google Scholar] [CrossRef] [PubMed]
- Rushworth, L.K.; Hindley, A.D.; O’Neill, E.; Kolch, W. Regulation and Role of Raf-1/B-Raf Heterodimerization. Mol. Cell. Biol. 2006, 26, 2262–2272. [Google Scholar] [CrossRef] [Green Version]
- Xiang, X.; Zang, M.; Waelde, C.A.; Wen, R.; Luo, Z. Phosphorylation of 338SSYY341 Regulates Specific Interaction between Raf-1 and MEK1. J. Biol. Chem. 2002, 277, 44996–45003. [Google Scholar] [CrossRef] [Green Version]
- Freeman, A.K.; Ritt, D.A.; Morrison, D.K. Effects of Raf Dimerization and Its Inhibition on Normal and Disease-Associated Raf Signaling. Mol. Cell 2013, 49, 751–758. [Google Scholar] [CrossRef]
- Schulte, T.W.; Blagosklonny, M.V.; Romanova, L.; Mushinski, J.F.; Monia, B.P.; Johnston, J.F.; Nguyen, P.; Trepel, J.; Neckers, L.M. Destabilization of Raf-1 by geldanamycin leads to disruption of the Raf-1-MEK-mitogen-activated protein kinase signalling pathway. Mol. Cell. Biol. 1996, 16, 5839–5845. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.K.; Kolch, W.; Kholodenko, B.N. When ubiquitination meets phosphorylation: A systems biology perspective of EGFR/MAPK signalling. Cell Commun. Signal 2013, 11, 52. [Google Scholar] [CrossRef] [PubMed]
- Schulte, T.W.; An, W.G.; Neckers, L.M. Geldanamycin-induced destabilization of Raf-1 involves the proteasome. Biochem. Biophys. Res. Commun. 1997, 239, 655–659. [Google Scholar] [CrossRef] [PubMed]
- Paul, I.; Ghosh, M.K. The E3 ligase CHIP: Insights into its structure and regulation. Biomed. Res. Int. 2014, 2014, 918183. [Google Scholar] [CrossRef] [PubMed]
- Dogan, T.; Harms, G.S.; Hekman, M.; Karreman, C.; Oberoi, T.K.; Alnemri, E.S.; Rapp, U.R.; Rajalingam, K. X-linked and cellular IAPs modulate the stability of C-RAF kinase and cell motility. Nat. Cell Biol. 2008, 10, 1447–1455. [Google Scholar] [CrossRef] [PubMed]
- Tran, N.H.; Frost, J.A. Phosphorylation of Raf-1 by p21-activated kinase 1 and Src regulates Raf-1 autoinhibition. J. Biol. Chem. 2003, 278, 11221–11226. [Google Scholar] [CrossRef] [PubMed]
- Noble, C. CRAF autophosphorylation of serine 621 is required to prevent its proteasome-mediated degradation. Mol. Cell 2008, 31, 862–872. [Google Scholar] [CrossRef]
- Du, J.; Zeng, J.; Ou, X.; Ren, X.; Cai, S. Methylglyoxal downregulates Raf-1 protein through a ubiquitination-mediated mechanism. Int. J. Biochem. Cell Biol. 2006, 38, 1084–1091. [Google Scholar] [CrossRef]
- Manenti, S.; Delmas, C.; Darbon, J.M. Cell adhesion protects c-Raf-1 against ubiquitin-dependent degradation by the proteasome. Biochem. Biophys. Res. Commun. 2002, 294, 976–980. [Google Scholar] [CrossRef]
- Johnson, S.E.; Winner, D.G., Jr.; Wang, X. Ran binding protein 9 interacts with Raf kinase but does not contribute to downstream ERK1/2 activation in skeletal myoblasts. Biochem. Biophys. Res. Commun. 2006, 340, 409–416. [Google Scholar] [CrossRef]
- Atabakhsh, E.; Schild-Poulter, C. RanBPM Is an Inhibitor of ERK Signaling. PLoS ONE 2012, 7, e47803. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Suresh, B.; Kim, H.; Ramakrishna, S. RanBPM: A potential therapeutic target for modulating diverse physiological disorders. Drug Discov. Today 2017, 22, 1816–1824. [Google Scholar] [CrossRef]
- Salemi, L.M.; Maitland, M.E.R.; McTavish, C.J.; Schild-Poulter, C. Cell signalling pathway regulation by RanBPM: Molecular insights and disease implications. Open Biol. 2017, 7, 170081. [Google Scholar] [CrossRef] [PubMed]
- Lampert, F.; Stafa, D.; Goga, A.; Soste, M.V.; Gilberto, S.; Olieric, N.; Picotti, P.; Stoffel, M.; Peter, M. The multi-subunit GID/CTLH E3 ubiquitin ligase promotes cell proliferation and targets the transcription factor Hbp1 for degradation. Elife 2018, 7, e35528. [Google Scholar] [CrossRef] [PubMed]
- Francis, O.; Han, F.; Adams, J.C. Molecular phylogeny of a RING E3 ubiquitin ligase, conserved in eukaryotic cells and dominated by homologous components, the muskelin/RanBPM/CTLH complex. PLoS ONE 2013, 8, e75217. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Cheng, S.; Zabkiewicz, C.; Chen, J.; Zhang, L.; Ye, L.; Jiang, W.G. Reduced Expression of RanBPM Is Associated with Poorer Survival from Lung Cancer and Increased Proliferation and Invasion of Lung Cancer Cells In Vitro. Anticancer Res. 2017, 37, 4389–4397. [Google Scholar] [Green Version]
- Atabakhsh, E.; Wang, J.H.; Wang, X.; Carter, D.E.; Schild-Poulter, C. RanBPM expression regulates transcriptional pathways involved in development and tumorigenesis. Am. J. Cancer Res. 2012, 2, 549–565. [Google Scholar]
- Altevogt, P.; Doberstein, K.; Fogel, M. L1CAM in human cancer. Int. J. Cancer 2016, 138, 1565–1576. [Google Scholar] [CrossRef]
- Eckert, R.L.; Fisher, M.L.; Grun, D.; Adhikary, G.; Xu, W.; Kerr, C. Transglutaminase is a tumor cell and cancer stem cell survival factor. Mol. Carcinog. 2015, 54, 947–958. [Google Scholar] [CrossRef] [Green Version]
- Faham, N.; Welm, A.L. RON Signaling Is a Key Mediator of Tumor Progression in Many Human Cancers. Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 177–188. [Google Scholar] [CrossRef] [Green Version]
- Oliver, J.R.; Kushwah, R.; Hu, J. Multiple roles of the epithelium-specific ETS transcription factor, ESE-1, in development and disease. Lab. Investig. 2012, 92, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Canis, M.; Lechner, A.; Mack, B.; Zengel, P.; Laubender, R.P.; Koehler, U.; Heissmeyer, V.; Gires, O. CD133 induces tumour-initiating properties in HEK293 cells. Tumour Biol. 2013, 34, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Soderberg, O.; Gullberg, M.; Jarvius, M.; Ridderstrale, K.; Leuchowius, K.J.; Jarvius, J.; Wester, K.; Hydbring, P.; Bahram, F.; Larsson, L.G.; et al. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods 2006, 3, 995–1000. [Google Scholar] [CrossRef] [PubMed]
- Atabakhsh, E.; Bryce, D.M.; Lefebvre, K.J.; Schild-Poulter, C. RanBPM has pro-apoptotic activities that regulate cell death pathways in response to DNA damage. Mol. Cancer Res. 2009, 7, 1962–1972. [Google Scholar] [CrossRef] [PubMed]
- Stancato, L.F.; Chow, Y.H.; Hutchison, K.A.; Perdew, G.H.; Jove, R.; Pratt, W.B. Raf exists in a native heterocomplex with hsp90 and p50 that can be reconstituted in a cell-free system. J. Biol. Chem. 1993, 268, 21711–21716. [Google Scholar] [PubMed]
- Salemi, L.M.; Loureiro, S.O.; Schild-Poulter, C. Characterization of RanBPM determinants that control its subcellular localization. PLoS ONE 2015, 10, e0117655. [Google Scholar] [CrossRef] [PubMed]
- Lakshmana, M.K.; Chung, J.Y.; Wickramarachchi, S.; Tak, E.; Bianchi, E.; Koo, E.H.; Kang, D.E. A fragment of the scaffolding protein RanBP9 is increased in Alzheimer’s disease brains and strongly potentiates amyloid-{beta} peptide generation. FASEB J. 2010, 24, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Kallberg, M.; Wang, H.; Wang, S.; Peng, J.; Wang, Z.; Lu, H.; Xu, J. Template-based protein structure modeling using the RaptorX web server. Nat. Protoc. 2012, 7, 1511–1522. [Google Scholar] [CrossRef] [Green Version]
- Menon, R.P.; Gibson, T.J.; Pastore, A. The C terminus of fragile X mental retardation protein interacts with the multi-domain Ran-binding protein in the microtubule-organising centre. J. Mol. Biol. 2004, 343, 43–53. [Google Scholar] [CrossRef]
- Menssen, R.; Schweiggert, J.; Schreiner, J.; Kusevic, D.; Reuther, J.; Braun, B.; Wolf, D.H. Exploring the topology of the Gid complex, the E3 ubiquitin ligase involved in catabolite-induced degradation of gluconeogenic enzymes. J. Biol. Chem. 2012, 287, 25602–25614. [Google Scholar] [CrossRef]
- Salemi, L.M.; Almawi, A.W.; Lefebvre, K.J.; Schild-Poulter, C. Aggresome formation is regulated by RanBPM through an interaction with HDAC6. Biol. Open 2014, 3, 418–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, T.; Martinez-Martinez, A.; Cubillos-Rojas, M.; Bartrons, R.; Ventura, F.; Rosa, J.L. The E3 ubiquitin ligase HERC1 controls the ERK signaling pathway targeting C-RAF for degradation. Oncotarget 2018, 9, 31531–31548. [Google Scholar] [CrossRef] [PubMed]
- Jang, E.R.; Shi, P.; Bryant, J.; Chen, J.; Dukhande, V.; Gentry, M.S.; Jang, H.; Jeoung, M.; Galperin, E. HUWE1 is a molecular link controlling RAF-1 activity supported by the Shoc2 scaffold. Mol. Cell. Biol. 2014, 34, 3579–3593. [Google Scholar] [CrossRef] [PubMed]
- Salemi, L.M.; Maitland, M.E.R.; Yefet, E.R.; Schild-Poulter, C. Inhibition of HDAC6 activity through interaction with RanBPM and its associated CTLH complex. BMC Cancer 2017, 17, 460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batchu, S.N.; Brijmohan, A.S.; Advani, A. The therapeutic hope for HDAC6 inhibitors in malignancy and chronic disease. Clin. Sci. 2016, 130, 987–1003. [Google Scholar] [CrossRef]
- Dai, H.; Lv, Y.-F.; Yan, G.-N.; Meng, G.; Zhang, X.; Guo, Q.-N. RanBP9/TSSC3 complex cooperates to suppress anoikis resistance and metastasis via inhibiting Src-mediated Akt signaling in osteosarcoma. Cell Death Dis. 2016, 7, e2572. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ma, W.; Tian, S.; Fan, Z.; Ma, X.; Yang, X.; Zhao, Q.; Tan, K.; Chen, H.; Chen, D.; et al. RanBPM interacts with TbetaRI, TRAF6 and curbs TGF induced nuclear accumulation of TbetaRI. Cell Signal. 2014, 26, 162–172. [Google Scholar] [CrossRef]
- Fell, V.L.; Walden, E.A.; Hoffer, S.M.; Rogers, S.R.; Aitken, A.S.; Salemi, L.M.; Schild-Poulter, C. Ku70 phosphorylation mediates Aurora B inhibition and activation of the DNA damage response. Sci. Rep. 2016, 6, 37194. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McTavish, C.J.; Bérubé-Janzen, W.; Wang, X.; Maitland, M.E.R.; Salemi, L.M.; Hess, D.A.; Schild-Poulter, C. Regulation of c-Raf Stability through the CTLH Complex. Int. J. Mol. Sci. 2019, 20, 934. https://doi.org/10.3390/ijms20040934
McTavish CJ, Bérubé-Janzen W, Wang X, Maitland MER, Salemi LM, Hess DA, Schild-Poulter C. Regulation of c-Raf Stability through the CTLH Complex. International Journal of Molecular Sciences. 2019; 20(4):934. https://doi.org/10.3390/ijms20040934
Chicago/Turabian StyleMcTavish, Christina J., Wesley Bérubé-Janzen, Xu Wang, Matthew E. R. Maitland, Louisa M. Salemi, David A. Hess, and Caroline Schild-Poulter. 2019. "Regulation of c-Raf Stability through the CTLH Complex" International Journal of Molecular Sciences 20, no. 4: 934. https://doi.org/10.3390/ijms20040934