Prediction of Self-Interacting Proteins from Protein Sequence Information Based on Random Projection Model and Fast Fourier Transform

 and
and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Performance Evaluation
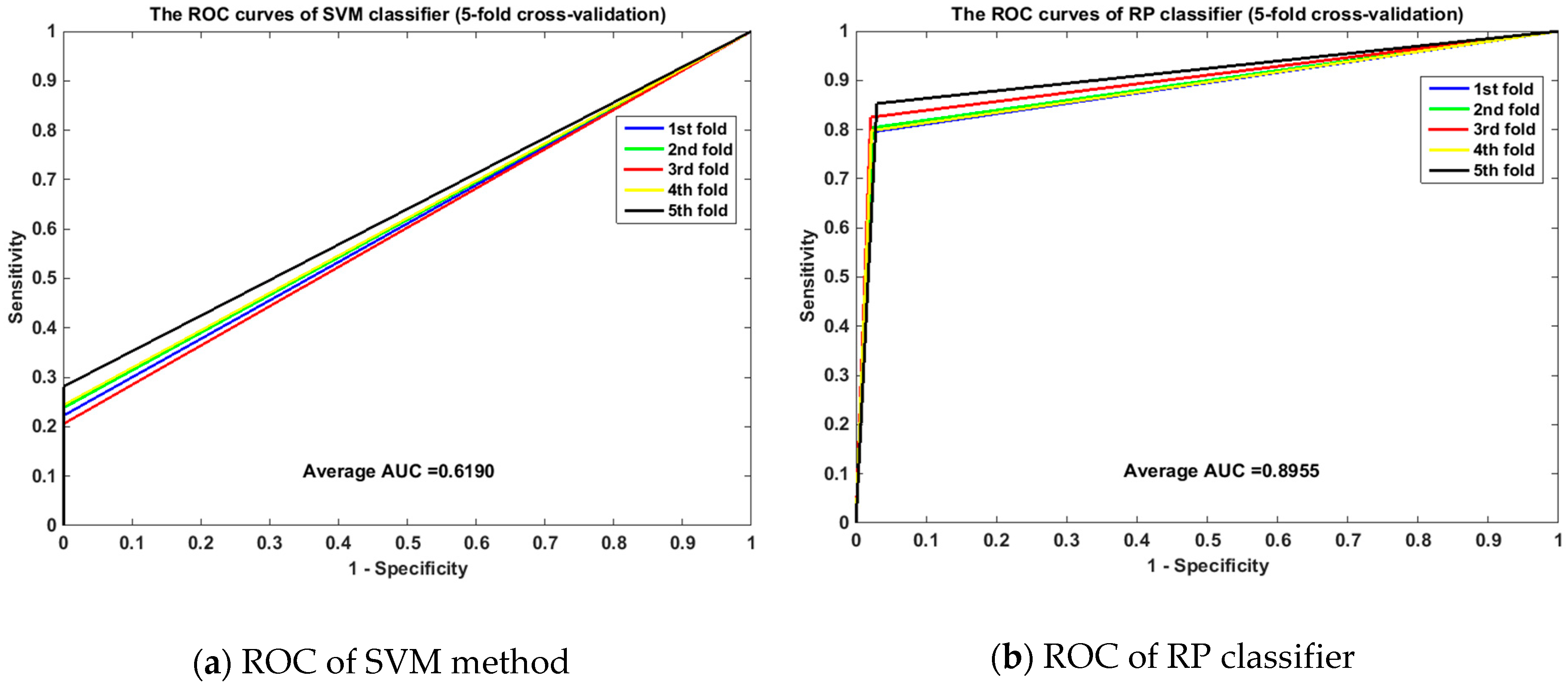
2.2. Performance of the Proposed Method
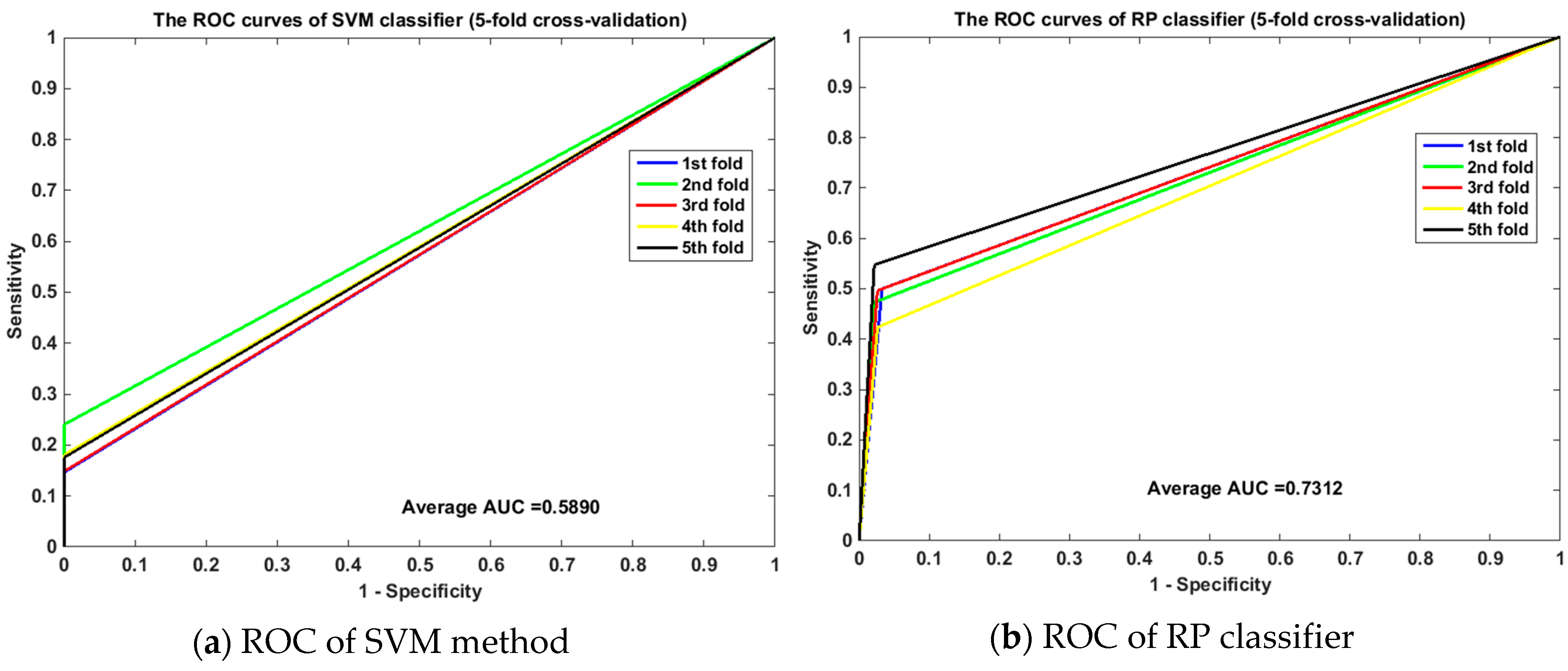
2.3. Comparison with Other Feature Extraction Methods
2.4. Comparison with the SVM-Based Method
2.5. Comparison with Other Existing Methods
3. Materials and Methodology
3.1. Datasets
3.2. Position-Specific Scoring Matrix
3.3. Fast Fourier Transform
3.4. Support Vector Machine
- (1)
- Linear: K(xi, xj) = xj.
- (2)
- Polynomial: K(xi, xj) = (γxj + r)d, γ > 0.
- (3)
- Radial basis function (RBF): K(xi, xj) = exp(−γ||xi − xj||2), γ > 0.
- (4)
- Sigmoid: K(xi, xj) = tan h(γxj + r).
3.5. Random Projection Classifier
- (1)
- The vectors are normally distributed over the q dimensional unit sphere.
- (2)
- The components of the vectors are selected Bernoulli +1/−1 distribution and the vectors are standardized so that ||ri||l2 = 1 for i = 1, …, n.
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Chen, Z.-H.; You, Z.-H.; Li, L.-P.; Wang, Y.-B.; Li, X. RP-FIRF: Prediction of Self-interacting Proteins Using Random Projection Classifier Combining with Finite Impulse Response Filter. In Proceedings of the International Conference on Intelligent Computing, Wuhan, China, 15–18 August 2018; pp. 232–234. [Google Scholar]
- Liu, Z.; Guo, F.; Zhang, J.; Wang, J.; Lu, L.; Li, D.; He, F. Proteome-wide prediction of self-interacting proteins based on multiple properties. Mol. Cell. Proteom. 2013. [Google Scholar] [CrossRef] [PubMed]
- Marianayagam, N.J.; Sunde, M.; Matthews, J.M. The power of two: Protein dimerization in biology. Trends Biochem. Sci. 2004, 29, 618–625. [Google Scholar] [CrossRef] [PubMed]
- Ispolatov, I.; Yuryev, A.; Mazo, I.; Maslov, S. Binding properties and evolution of homodimers in protein–protein interaction networks. Nucleic Acids Res. 2005, 33, 3629–3635. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-B.; You, Z.-H.; Li, L.-P.; Huang, Y.-A.; Yi, H.-C. Detection of interactions between proteins by using legendre moments descriptor to extract discriminatory information embedded in pssm. Molecules 2017, 22, 1366. [Google Scholar] [CrossRef] [PubMed]
- Woodcock, J.M.; Murphy, J.; Stomski, F.C.; Berndt, M.C.; Lopez, A.F. The dimeric versus monomeric status of 14-3-3ζ is controlled by phosphorylation of Ser58 at the dimer interface. J. Biol. Chem. 2003, 278, 36323–36327. [Google Scholar] [CrossRef]
- Baisamy, L.; Jurisch, N.; Diviani, D. Leucine zipper-mediated homo-oligomerization regulates the Rho-GEF activity of AKAP-Lbc. J. Biol. Chem. 2005, 280, 15405–15412. [Google Scholar] [CrossRef] [PubMed]
- Katsamba, P.; Carroll, K.; Ahlsen, G.; Bahna, F.; Vendome, J.; Posy, S.; Rajebhosale, M.; Price, S.; Jessell, T.; Ben-Shaul, A. Linking molecular affinity and cellular specificity in cadherin-mediated adhesion. Proc. Natl. Acad. Sci. USA 2009, 106, 11594–11599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koike, R.; Kidera, A.; Ota, M. Alteration of oligomeric state and domain architecture is essential for functional transformation between transferase and hydrolase with the same scaffold. Protein Sci. 2009, 18, 2060–2066. [Google Scholar] [CrossRef] [Green Version]
- Miller, S.; Lesk, A.M.; Janin, J.; Chothia, C. The accessible surface area and stability of oligomeric proteins. Nature 1987, 328, 834. [Google Scholar] [CrossRef]
- Zeng, X.; Liao, Y.; Liu, Y.; Zou, Q. Prediction and validation of disease genes using HeteSim Scores. IEEE/ACM Trans. Comput. Biol. Bioinform. (TCBB) 2017, 14, 687–695. [Google Scholar] [CrossRef]
- Zou, Q.; Wan, S.; Ju, Y.; Tang, J.; Zeng, X. Pretata: Predicting TATA binding proteins with novel features and dimensionality reduction strategy. BMC Syst. Biol. 2016, 10, 114. [Google Scholar] [CrossRef]
- Nanni, L.; Lumini, A.; Brahnam, S. A set of descriptors for identifying the protein–drug interaction in cellular networking. J. Theor. Biol. 2014, 359, 120–128. [Google Scholar] [CrossRef]
- Nanni, L.; Brahnam, S. Set of approaches based on 3D structure and Position Specific Scoring Matrix for predicting DNA-binding proteins. Bioinformatics 2018. [Google Scholar] [CrossRef]
- You, Z.-H.; Huang, Z.-A.; Zhu, Z.; Yan, G.-Y.; Li, Z.-W.; Wen, Z.; Chen, X. PBMDA: A novel and effective path-based computational model for miRNA-disease association prediction. PLoS Comput. Biol. 2017, 13, e1005455. [Google Scholar] [CrossRef]
- You, Z.-H.; Lei, Y.-K.; Gui, J.; Huang, D.-S.; Zhou, X. Using manifold embedding for assessing and predicting protein interactions from high-throughput experimental data. Bioinformatics 2010, 26, 2744–2751. [Google Scholar] [CrossRef] [Green Version]
- Zou, Q.; Li, J.; Song, L.; Zeng, X.; Wang, G. Similarity computation strategies in the microRNA-disease network: A survey. Brief. Funct. Genom. 2015, 15, 55–64. [Google Scholar] [CrossRef]
- Manavalan, B.; Shin, T.H.; Kim, M.O.; Lee, G. PIP-EL: A new ensemble learning method for improved proinflammatory peptide predictions. Front. Immunol. 2018, 9, 1783. [Google Scholar] [CrossRef]
- Wang, Y.-B.; You, Z.-H.; Li, X.; Jiang, T.-H.; Cheng, L.; Chen, Z.-H. Prediction of protein self-interactions using stacked long short-term memory from protein sequences information. BMC Syst. Biol. 2018, 12, 129. [Google Scholar] [CrossRef]
- Yi, H.-C.; You, Z.-H.; Huang, D.-S.; Li, X.; Jiang, T.-H.; Li, L.-P. A Deep Learning Framework for Robust and Accurate Prediction of ncRNA-Protein Interactions Using Evolutionary Information. Mol. Ther. Nucleic Acids 2018, 11, 337–344. [Google Scholar] [CrossRef]
- You, Z.-H.; Zhou, M.; Luo, X.; Li, S. Highly efficient framework for predicting interactions between proteins. IEEE Trans. Cybern. 2017, 47, 731–743. [Google Scholar] [CrossRef]
- Wang, L.; You, Z.-H.; Xia, S.-X.; Liu, F.; Chen, X.; Yan, X.; Zhou, Y. Advancing the prediction accuracy of protein-protein interactions by utilizing evolutionary information from position-specific scoring matrix and ensemble classifier. J. Theor. Biol. 2017, 418, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Pitre, S.; Dehne, F.; Chan, A.; Cheetham, J.; Duong, A.; Emili, A.; Gebbia, M.; Greenblatt, J.; Jessulat, M.; Krogan, N. PIPE: A protein-protein interaction prediction engine based on the re-occurring short polypeptide sequences between known interacting protein pairs. BMC Bioinform. 2006, 7, 365. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.-F.; Han, K.; Huang, D.-S. Sequence-based prediction of protein-protein interactions by means of rotation forest and autocorrelation descriptor. Protein Pept. Lett. 2010, 17, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-B.; You, Z.-H.; Li, X.; Jiang, T.-H.; Chen, X.; Zhou, X.; Wang, L. Predicting protein–protein interactions from protein sequences by a stacked sparse autoencoder deep neural network. Mol. BioSyst. 2017, 13, 1336–1344. [Google Scholar] [CrossRef] [PubMed]
- Basith, S.; Manavalan, B.; Shin, T.H.; Lee, G. iGHBP: Computational identification of growth hormone binding proteins from sequences using extremely randomised tree. Comput. Struct. Biotechnol. J. 2018, 16, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Manavalan, B.; Subramaniyam, S.; Shin, T.H.; Kim, M.O.; Lee, G. Machine-learning-based prediction of cell-penetrating peptides and their uptake efficiency with improved accuracy. J. Proteome Res. 2018, 17, 2715–2726. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Hu, J.; Li, F.; Song, J.; Su, R.; Zou, Q. Comparative analysis and prediction of quorum-sensing peptides using feature representation learning and machine learning algorithms. Brief. Bioinform. 2018. [Google Scholar] [CrossRef]
- Manavalan, B.; Shin, T.H.; Kim, M.O.; Lee, G. AIPpred: Sequence-Based Prediction of Anti-inflammatory Peptides Using Random Forest. Front. Pharmacol. 2018, 9, 276. [Google Scholar] [CrossRef]
- Wei, L.; Luan, S.; Nagai, L.A.E.; Su, R.; Zou, Q. Exploring sequence-based features for the improved prediction of DNA N4-methylcytosine sites in multiple species. Bioinformatics 2018. [Google Scholar] [CrossRef]
- Manavalan, B.; Govindaraj, R.G.; Shin, T.H.; Kim, M.O.; Lee, G. iBCE-EL: A new ensemble learning framework for improved linear B-cell epitope prediction. Front. Immunol. 2018, 9, 1695. [Google Scholar] [CrossRef]
- Wei, L.; Chen, H.; Su, R. M6APred-EL: A sequence-based predictor for identifying N6-methyladenosine sites using ensemble learning. Mol. Ther. Nucleic Acids 2018, 12, 635–644. [Google Scholar] [CrossRef]
- Gabere, M.N.; Noble, W.S. Empirical comparison of web-based antimicrobial peptide prediction tools. Bioinformatics 2017, 33, 1921–1929. [Google Scholar] [CrossRef]
- Manavalan, B.; Shin, T.H.; Lee, G. PVP-SVM: Sequence-based prediction of phage virion proteins using a support vector machine. Front. Microbiol. 2018, 9, 476. [Google Scholar] [CrossRef]
- Wei, L.; Zhou, C.; Chen, H.; Song, J.; Su, R. ACPred-FL: A sequence-based predictor based on effective feature representation to improve the prediction of anti-cancer peptides. Bioinformatics 2018, 34, 4007–4016. [Google Scholar] [CrossRef]
- Manavalan, B.; Shin, T.H.; Lee, G. DHSpred: Support-vector-machine-based human DNase I hypersensitive sites prediction using the optimal features selected by random forest. Oncotarget 2018, 9, 1944. [Google Scholar] [CrossRef]
- Wei, L.; Tang, J.; Zou, Q. SkipCPP-Pred: An improved and promising sequence-based predictor for predicting cell-penetrating peptides. BMC Genom. 2017, 18, 1. [Google Scholar] [CrossRef]
- Manavalan, B.; Basith, S.; Shin, T.H.; Choi, S.; Kim, M.O.; Lee, G. MLACP: Machine-learning-based prediction of anticancer peptides. Oncotarget 2017, 8, 77121. [Google Scholar] [CrossRef]
- Chou, K.-C. Some remarks on protein attribute prediction and pseudo amino acid composition. J. Theor. Biol. 2011, 273, 236–247. [Google Scholar] [CrossRef]
- Dao, F.-Y.; Lv, H.; Wang, F.; Feng, C.-Q.; Ding, H.; Chen, W.; Lin, H. Identify origin of replication in Saccharomyces cerevisiae using two-step feature selection technique. Bioinformatics 2018. [Google Scholar] [CrossRef]
- Manavalan, B.; Lee, J. SVMQA: Support–vector-machine-based protein single-model quality assessment. Bioinformatics 2017, 33, 2496–2503. [Google Scholar] [CrossRef]
- Nanni, L.; Lumini, A.; Brahnam, S. An empirical study of different approaches for protein classification. Sci. World J. 2014, 2014, 236717. [Google Scholar] [CrossRef] [PubMed]
- Nanni, L.; Brahnam, S.; Lumini, A. Wavelet images and Chou’s pseudo amino acid composition for protein classification. Amino Acids 2012, 43, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-C.; Lin, C.-J. LIBSVM: A library for support vector machines. ACM Trans. Intell. Syst. Technol. (TIST) 2011, 2, 27. [Google Scholar] [CrossRef]
- Du, X.; Cheng, J.; Zheng, T.; Duan, Z.; Qian, F. A novel feature extraction scheme with ensemble coding for protein–protein interaction prediction. Int. J. Mol. Sci. 2014, 15, 12731–12749. [Google Scholar] [CrossRef] [PubMed]
- Zahiri, J.; Yaghoubi, O.; Mohammad-Noori, M.; Ebrahimpour, R.; Masoudi-Nejad, A. PPIevo: Protein–protein interaction prediction from PSSM based evolutionary information. Genomics 2013, 102, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Zahiri, J.; Mohammad-Noori, M.; Ebrahimpour, R.; Saadat, S.; Bozorgmehr, J.H.; Goldberg, T.; Masoudi-Nejad, A. LocFuse: Human protein–protein interaction prediction via classifier fusion using protein localization information. Genomics 2014, 104, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yang, S.; Li, C.; Zhang, Z.; Song, J. SPAR: A random forest-based predictor for self-interacting proteins with fine-grained domain information. Amino Acids 2016, 48, 1655–1665. [Google Scholar] [CrossRef]
- Consortium, U. UniProt: A hub for protein information. Nucleic Acids Res. 2014, 43, D204–D212. [Google Scholar] [CrossRef]
- Salwinski, L.; Miller, C.S.; Smith, A.J.; Pettit, F.K.; Bowie, J.U.; Eisenberg, D. The database of interacting proteins: 2004 update. Nucleic Acids Res. 2004, 32, D449–D451. [Google Scholar] [CrossRef]
- Chatr-Aryamontri, A.; Oughtred, R.; Boucher, L.; Rust, J.; Chang, C.; Kolas, N.K.; O’Donnell, L.; Oster, S.; Theesfeld, C.; Sellam, A. The BioGRID interaction database: 2017 update. Nucleic Acids Res. 2017, 45, D369–D379. [Google Scholar] [CrossRef]
- Orchard, S.; Ammari, M.; Aranda, B.; Breuza, L.; Briganti, L.; Broackes-Carter, F.; Campbell, N.H.; Chavali, G.; Chen, C.; Del-Toro, N. The MIntAct project—IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 2013, 42, D358–D363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breuer, K.; Foroushani, A.K.; Laird, M.R.; Chen, C.; Sribnaia, A.; Lo, R.; Winsor, G.L.; Hancock, R.E.; Brinkman, F.S.; Lynn, D.J. InnateDB: Systems biology of innate immunity and beyond—recent updates and continuing curation. Nucleic Acids Res. 2012, 41, D1228–D1233. [Google Scholar] [CrossRef] [PubMed]
- Chautard, E.; Fatoux-Ardore, M.; Ballut, L.; Thierry-Mieg, N.; Ricard-Blum, S. MatrixDB, the extracellular matrix interaction database. Nucleic Acids Res. 2010, 39, D235–D240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gribskov, M.; McLachlan, A.D.; Eisenberg, D. Profile analysis: Detection of distantly related proteins. Proc. Natl. Acad. Sci. USA 1987, 84, 4355–4358. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; You, Z.; Li, X.; Chen, X.; Jiang, T.; Zhang, J. PCVMZM: Using the Probabilistic Classification Vector Machines Model Combined with a Zernike Moments Descriptor to Predict Protein–Protein Interactions from Protein Sequences. Int. J. Mol. Sci. 2017, 18, 1029. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-B.; You, Z.-H.; Li, L.-P.; Huang, D.-S.; Zhou, F.-F.; Yang, S. Improving Prediction of Self-interacting Proteins Using Stacked Sparse Auto-Encoder with PSSM profiles. Int. J. Biol. Sci. 2018, 14, 983–991. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Koonin, E.V. Iterated profile searches with PSI-BLAST—A tool for discovery in protein databases. Trends Biochem. Sci. 1998, 23, 444–447. [Google Scholar] [CrossRef]
- Ahmed, N.; Rao, K.R. Orthogonal Transforms for Digital Signal Processing; Springer Science & Business Media: Berlin, Germany, 2012. [Google Scholar]
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.-W.; Chang, C.-C.; Lin, C.-J. A Practical Guide to Support Vector Classification; National Taiwan University: Taipei, Taiwan, 2003. [Google Scholar]
- Schclar, A.; Rokach, L. Random projection ensemble classifiers. In Proceedings of the International Conference on Enterprise Information Systems, Milan, Italy, 6–10 May 2009; pp. 309–316. [Google Scholar]
- Candès, E.J.; Romberg, J.; Tao, T. Robust uncertainty principles: Exact signal reconstruction from highly incomplete frequency information. IEEE Trans. Inf. Theory 2006, 52, 489–509. [Google Scholar] [CrossRef]
- Donoho, D.L. Compressed sensing. IEEE Trans. Inf. Theory 2006, 52, 1289–1306. [Google Scholar] [CrossRef]
- Goel, N.; Bebis, G.; Nefian, A. Face recognition experiments with random projection. Proc. SPIE 2005, 5779, 426–438. [Google Scholar] [Green Version]
- Lumini, A.; Nanni, L.; Brahnam, S. Ensemble of texture descriptors and classifiers for face recognition. Appl. Comput. Inf. 2017, 13, 79–91. [Google Scholar] [CrossRef] [Green Version]
- Nanni, L.; Lumini, A.; Brahnam, S. Ensemble of texture descriptors for face recognition obtained by varying feature transforms and preprocessing approaches. Appl. Soft Comput. 2017, 61, 8–16. [Google Scholar] [CrossRef]
- Linial, M.; Linial, N.; Tishby, N.; Yona, G. Global self-organization of all known protein sequences reveals inherent biological signatures1. J. Mol. Biol. 1997, 268, 539–556. [Google Scholar] [CrossRef] [PubMed]
- Bingham, E.; Mannila, H. Random projection in dimensionality reduction: Applications to image and text data. In Proceedings of the Seventh ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 26–29 August 2001; pp. 245–250. [Google Scholar]



{kind=link}
{kind=link}
| Testing Set | Acc. (%) | Sen. (%) | Spe. (%) | MCC (%) |
|---|---|---|---|---|
| 1 | 94.44 | 88.28 | 100.00 | 89.36 |
| 2 | 92.53 | 85.37 | 100.00 | 86.07 |
| 3 | 92.19 | 85.48 | 100.00 | 85.51 |
| 4 | 93.75 | 86.76 | 100.00 | 88.08 |
| 5 | 94.81 | 89.73 | 100.00 | 90.12 |
| Average | 93.54 ± 1.15 | 87.12 ± 1.87 | 100.00 ± 0.00 | 87.83 ± 2.01 |
| Testing Set | Acc. (%) | Sen. (%) | Spe. (%) | MCC (%) |
|---|---|---|---|---|
| 1 | 80.99 | 97.12 | 65.52 | 65.71 |
| 2 | 83.45 | 92.14 | 75.00 | 68.03 |
| 3 | 82.04 | 97.89 | 66.20 | 67.57 |
| 4 | 84.86 | 95.14 | 74.29 | 71.13 |
| 5 | 83.45 | 92.41 | 74.10 | 67.83 |
| Average | 82.96 ± 1.48 | 94.94 ± 2.63 | 71.02 ± 4.73 | 68.05 ± 1.95 |
| Testing Set | Acc. (%) | Sen. (%) | Spe. (%) | MCC (%) | B_Acc. (%) |
|---|---|---|---|---|---|
| 1 | 96.23 | 79.51 | 97.74 | 75.72 | 88.63 |
| 2 | 96.20 | 80.34 | 97.65 | 75.89 | 89.00 |
| 3 | 96.58 | 82.49 | 97.89 | 78.61 | 90.19 |
| 4 | 96.40 | 79.78 | 97.79 | 75.40 | 88.79 |
| 5 | 96.00 | 85.28 | 97.01 | 76.68 | 91.15 |
| Average | 96.28 ± 0.22 | 81.48 ± 2.43 | 97.62 ± 0.35 | 76.46 ± 1.29 | 89.55 ± 1.08 |
| Testing Set | Acc. (%) | Sen. (%) | Spe. (%) | MCC (%) | B_Acc. (%) |
|---|---|---|---|---|---|
| 1 | 91.32 | 50.00 | 96.73 | 53.09 | 73.37 |
| 2 | 91.72 | 47.33 | 97.81 | 55.35 | 72.57 |
| 3 | 92.20 | 49.63 | 97.39 | 54.80 | 73.51 |
| 4 | 91.00 | 42.36 | 97.36 | 49.06 | 69.86 |
| 5 | 93.09 | 54.74 | 97.83 | 60.82 | 76.29 |
| Average | 91.87 ± 0.82 | 48.81 ± 4.50 | 97.42 ± 0.45 | 54.62 ± 4.25 | 73.12 ± 2.30 |
| Feature Extraction Methods | Acc. (%) | Sen. (%) | Spe. (%) | MCC (%) | B_Acc. (%) |
|---|---|---|---|---|---|
| SVD | 88.73 ± 0.75 | 10.25 ± 2.93 | 98.86 ± 0.43 | 19.76 ± 2.96 | 54.55 ± 1.31 |
| DCT | 90.35 ± 0.84 | 20.38 ± 2.62 | 99.36 ± 0.32 | 37.57 ± 1.74 | 59.87 ± 1.18 |
| COV | 91.93 ± 0.81 | 42.43 ± 4.82 | 98.31 ± 0.25 | 53.10 ± 4.91 | 70.37 ± 2.49 |
| FFT | 91.87 ± 0.82 | 48.81 ± 4.50 | 97.42 ± 0.45 | 54.62 ± 4.25 | 73.12 ± 2.30 |
| Model | Testing Set | Acc. (%) | Sen. (%) | Spe. (%) | MCC (%) | B_Acc. (%) |
|---|---|---|---|---|---|---|
| RP + FFT | 1 | 96.23 | 79.51 | 97.74 | 75.72 | 88.63 |
| 2 | 96.20 | 80.34 | 97.65 | 75.89 | 89.00 | |
| 3 | 96.58 | 82.49 | 97.89 | 78.61 | 90.19 | |
| 4 | 96.40 | 79.78 | 97.79 | 75.40 | 88.79 | |
| 5 | 96.00 | 85.28 | 97.01 | 76.68 | 91.15 | |
| Average | 96.28 ± 0.22 | 81.48 ± 2.43 | 97.62 ± 0.35 | 76.46 ± 1.29 | 89.55 ± 1.08 | |
| SVM + FFT | 1 | 93.55 | 22.22 | 100.00 | 45.57 | 61.11 |
| 2 | 93.64 | 23.79 | 100.00 | 47.17 | 61.90 | |
| 3 | 93.21 | 20.54 | 100.00 | 43.73 | 60.27 | |
| 4 | 94.19 | 24.34 | 100.00 | 47.86 | 62.17 | |
| 5 | 93.82 | 28.09 | 100.00 | 51.30 | 64.05 | |
| Average | 93.68 ± 0.36 | 23.80 ± 2.82 | 100.00 ± 0.00 | 47.13 ± 2.82 | 61.90 ± 1.41 |
| Model | Testing Set | Acc. (%) | Sen. (%) | Spe. (%) | MCC (%) | B_Acc. (%) |
|---|---|---|---|---|---|---|
| RP+FFT | 1 | 91.32 | 50.00 | 96.73 | 53.09 | 73.37 |
| 2 | 91.72 | 47.33 | 97.81 | 55.35 | 72.57 | |
| 3 | 92.20 | 49.63 | 97.39 | 54.80 | 73.51 | |
| 4 | 91.00 | 42.36 | 97.36 | 49.06 | 69.86 | |
| 5 | 93.09 | 54.74 | 97.83 | 60.82 | 76.29 | |
| Average | 91.87 ± 0.82 | 48.81 ± 4.50 | 97.42 ± 0.45 | 54.62 ± 4.25 | 73.12 ± 2.30 | |
| SVM+FFT | 1 | 90.11 | 14.58 | 100.00 | 36.22 | 57.29 |
| 2 | 90.84 | 24.00 | 100.00 | 46.62 | 62.00 | |
| 3 | 90.76 | 14.81 | 100.00 | 36.64 | 57.41 | |
| 4 | 90.51 | 18.06 | 100.00 | 40.38 | 59.03 | |
| 5 | 90.92 | 17.52 | 100.00 | 39.87 | 58.76 | |
| Average | 90.63 ± 0.33 | 17.79 ± 3.80 | 100.00 ± 0.00 | 39.95 ± 4.17 | 58.90 ± 1.90 |
| Model | Acc. (%) | Spe. (%) | Sen. (%) | MCC (%) | B_Acc. (%) |
|---|---|---|---|---|---|
| SLIPPER [2] | 71.90 | 72.18 | 69.72 | 28.42 | 70.95 |
| DXECPPI [45] | 87.46 | 94.93 | 29.44 | 28.25 | 62.19 |
| PPIevo [46] | 66.28 | 87.46 | 60.14 | 18.01 | 73.80 |
| LocFuse [47] | 66.66 | 68.10 | 55.49 | 15.77 | 61.80 |
| CRS [48] | 72.69 | 74.37 | 59.58 | 23.68 | 66.98 |
| SPAR [48] | 76.96 | 80.02 | 53.24 | 24.84 | 66.63 |
| Proposed method | 91.87 | 97.42 | 48.81 | 54.62 | 73.12 |
| Model | Acc. (%) | Spe. (%) | Sen. (%) | MCC (%) | B_Acc. (%) |
|---|---|---|---|---|---|
| SLIPPER [2] | 91.10 | 95.06 | 47.26 | 41.97 | 71.16 |
| DXECPPI [45] | 30.90 | 25.83 | 87.08 | 8.25 | 56.46 |
| PPIevo [46] | 78.04 | 25.82 | 87.83 | 20.82 | 56.83 |
| LocFuse [47] | 80.66 | 80.50 | 50.83 | 20.26 | 65.67 |
| CRS [48] | 91.54 | 96.72 | 34.17 | 36.33 | 65.45 |
| SPAR [48] | 92.09 | 97.40 | 33.33 | 38.36 | 65.37 |
| Proposed method | 96.28 | 97.62 | 81.48 | 76.46 | 89.55 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Z.-H.; You, Z.-H.; Li, L.-P.; Wang, Y.-B.; Wong, L.; Yi, H.-C. Prediction of Self-Interacting Proteins from Protein Sequence Information Based on Random Projection Model and Fast Fourier Transform. Int. J. Mol. Sci. 2019, 20, 930. https://doi.org/10.3390/ijms20040930
Chen Z-H, You Z-H, Li L-P, Wang Y-B, Wong L, Yi H-C. Prediction of Self-Interacting Proteins from Protein Sequence Information Based on Random Projection Model and Fast Fourier Transform. International Journal of Molecular Sciences. 2019; 20(4):930. https://doi.org/10.3390/ijms20040930
Chicago/Turabian StyleChen, Zhan-Heng, Zhu-Hong You, Li-Ping Li, Yan-Bin Wang, Leon Wong, and Hai-Cheng Yi. 2019. "Prediction of Self-Interacting Proteins from Protein Sequence Information Based on Random Projection Model and Fast Fourier Transform" International Journal of Molecular Sciences 20, no. 4: 930. https://doi.org/10.3390/ijms20040930