The Anti-Tumor Agent Sodium Selenate Decreases Methylated PP2A, Increases GSK3βY216 Phosphorylation, Including Tau Disease Epitopes and Reduces Neuronal Excitability in SHSY-5Y Neurons


Abstract
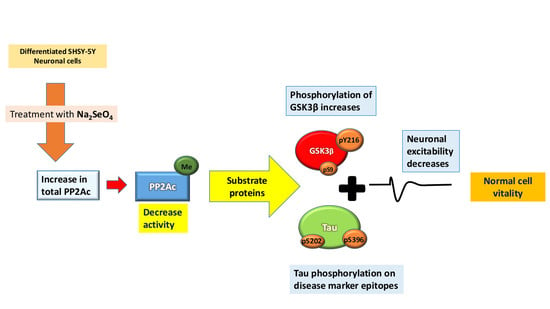
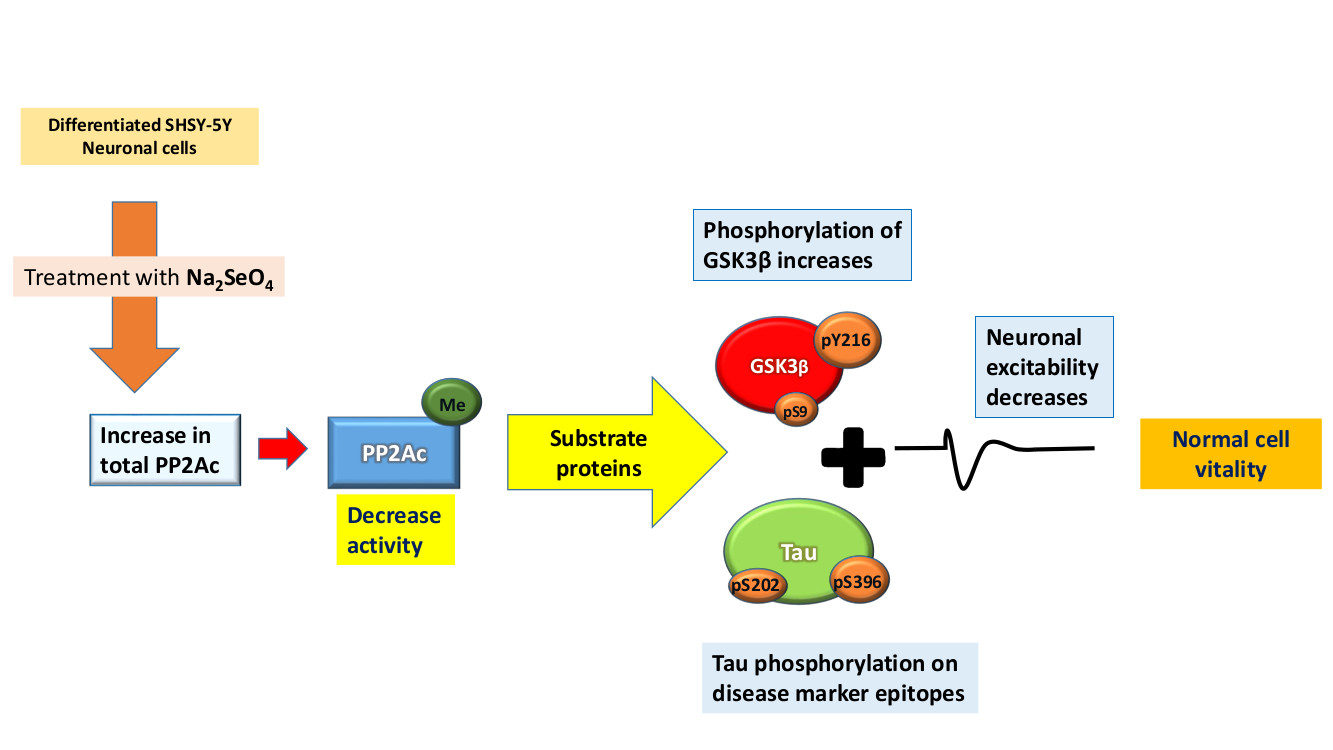
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
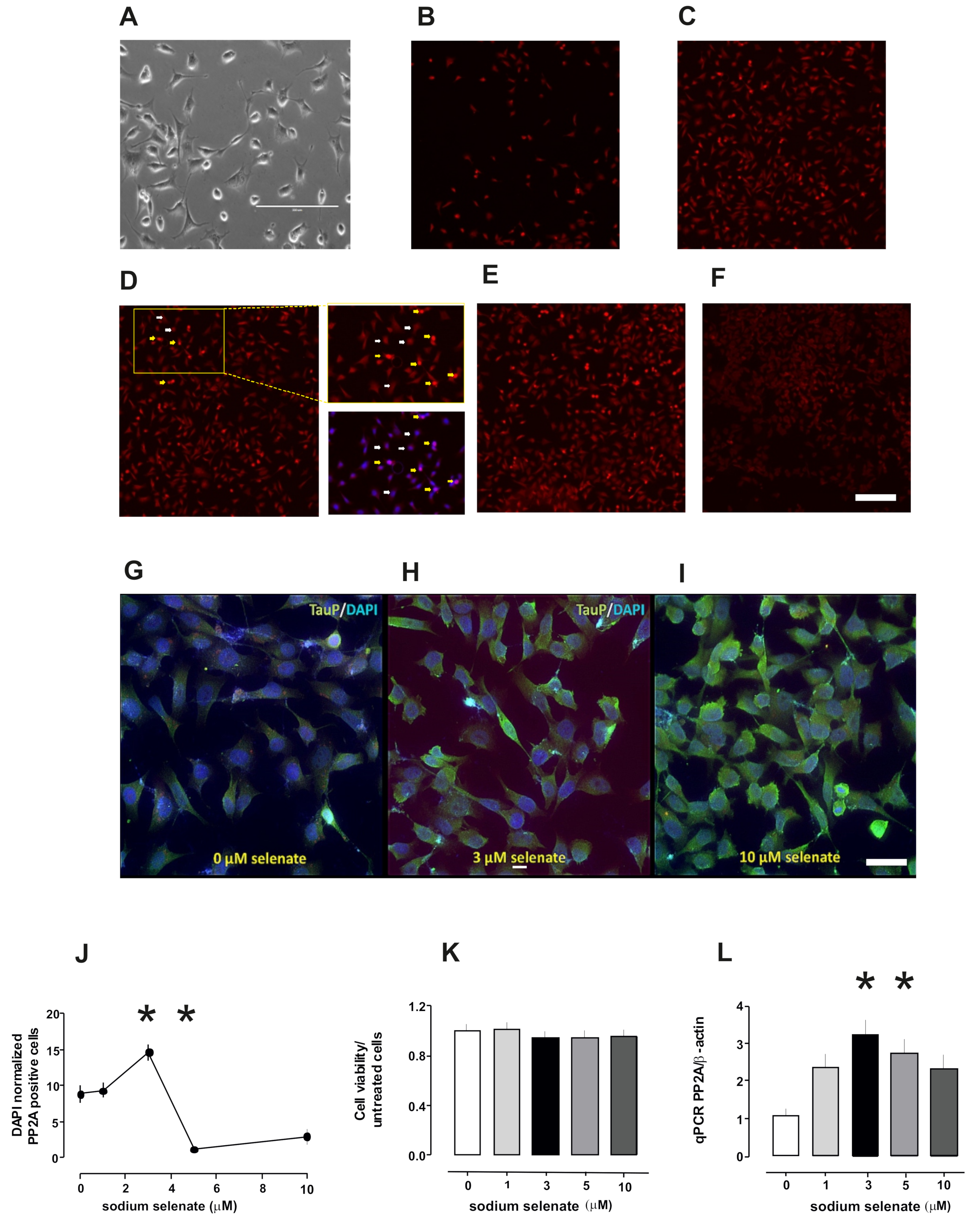
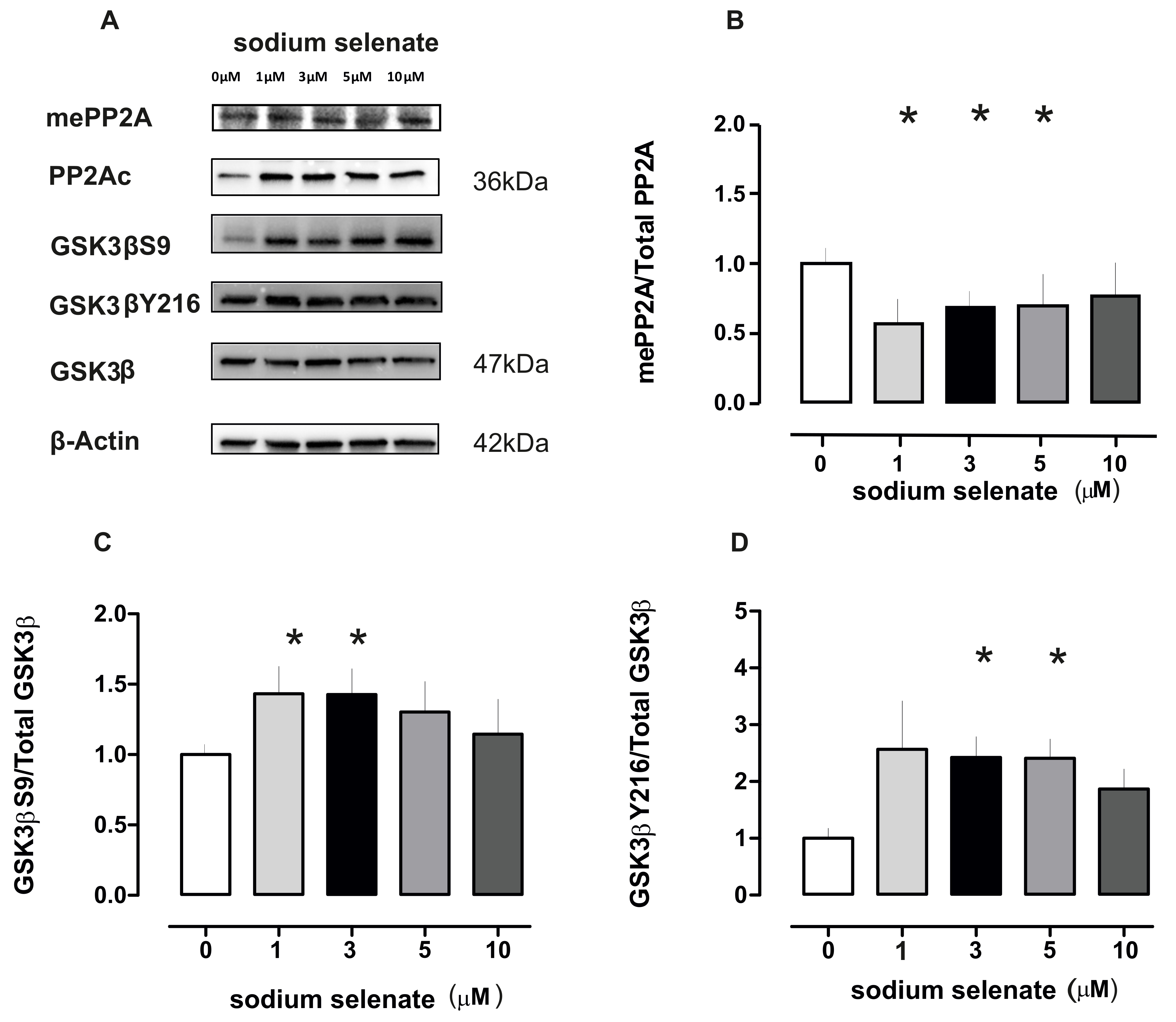
2.1. PP2A Levels Alter in Cells After Sodium Selenate Treatment
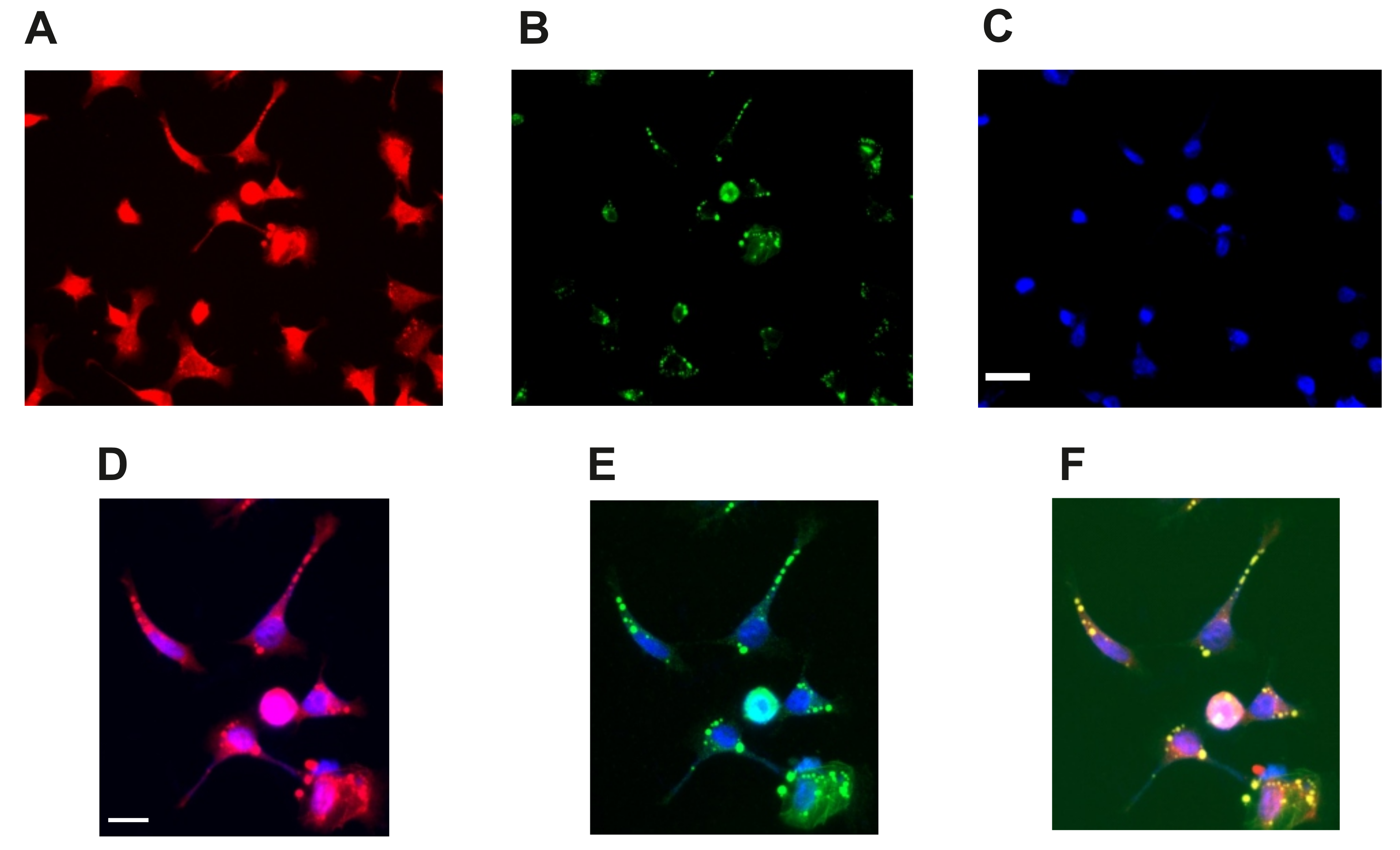
2.2. Active PP2A Decreases, Whilst Total GSK3β and Tau, Including Phosphorylation States Increase with Sodium Selenate Treatment
2.3. Electrophysiology Studies
3. Discussion
4. Material Methods
4.1. Cell Culture
4.2. Immunofluorescence Microscopy
4.3. Flow Cytometery
4.4. qRT-PCR
4.5. Immunoblotting
4.6. Antibodies
4.7. Electrophysiology
4.8. Statistics
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bellinger, F.P.; Raman, A.V.; Reeves, M.A.; Berry, M.J. Regulation and function of selenoproteins in human disease. Biochem. J. 2009, 422, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Sontag, E.; Hladik, C.; Montgomery, L.; Luangpirom, A.; Mudrak, I.; Ogris, E.; White, C.L., 3rd. Downregulation of protein phosphatase 2A carboxyl methylation and methyltransferase may contribute to Alzheimer disease pathogenesis. J. Neuropathol. Exp. Neurol. 2004, 63, 1080–1091. [Google Scholar] [CrossRef] [PubMed]
- Sontag, J.M.; Sontag, E. Protein phosphatase 2A dysfunction in Alzheimer’s disease. Front. Mol. Neurosci. 2014, 7, 16. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, B.R.; Roberts, B.R.; Bush, A.I.; Hare, D.J. Selenium, selenoproteins and neurodegenerative diseases. Metal. Integr. Biomet. Sci. 2015, 7, 1213–1228. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, N.M.; Martin, D.; Hutter-Paier, B.; Windisch, M.; Nguyen, T.; Nheu, L.; Sundstrom, L.E.; Costello, A.J.; Hovens, C.M. Sodium selenate specifically activates PP2A phosphatase, dephosphorylates Tau and reverses memory deficits in an Alzheimer’s disease model. J. Clin. Neurosci. 2010, 17, 1025–1033. [Google Scholar] [CrossRef]
- Jones, N.C.; Nguyen, T.; Corcoran, N.M.; Velakoulis, D.; Chen, T.; Grundy, R.; O’Brien, T.J.; Hovens, C.M. Targeting hyperphosphorylated tau with sodium selenate suppresses seizures in rodent models. Neurobiol. Dis. 2012, 45, 897–901. [Google Scholar] [CrossRef]
- Liu, S.J.; Zheng, P.; Wright, D.K.; Dezsi, G.; Braine, E.; Nguyen, T.; Corcoran, N.M.; Johnston, L.A.; Hovens, C.M.; Mayo, J.N.; et al. Sodium selenate retards epileptogenesis in acquired epilepsy models reversing changes in protein phosphatase 2a and hyperphosphorylated tau. Brain 2016, 139, 1919–1938. [Google Scholar] [CrossRef] [PubMed]
- Shultz, S.R.; Wright, D.K.; Zheng, P.; Stuchbery, R.; Liu, S.J.; Sashindranath, M.; Medcalf, R.L.; Johnston, L.A.; Hovens, C.M.; Jones, N.C.; et al. Sodium selenate reduces hyperphosphorylated tau and improves outcomes after traumatic brain injury. Brain 2015, 138, 1297–1313. [Google Scholar] [CrossRef]
- Tan, X.L.; Wright, D.K.; Liu, S.; Hovens, C.; O’Brien, T.J.; Shultz, S.R. Sodium selenate, a protein phosphatase 2a activator, mitigates hyperphosphorylated tau and improves repeated mild traumatic brain injury outcomes. Neuropharmacology 2016, 108, 382–393. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, J.; Zhang, K.; Jin, N.; Zhao, Y.; Liu, Q.; Ni, J.; Shen, L. Selenium positively affects the proteome of 3xTg AD mice cortex by altering the expression of various key proteins: Unveiling the mechanistic role of selenium in ad prevention. J. Neurosci. Res. 2018, 96, 1798–1815. [Google Scholar] [CrossRef]
- Song, G.; Zhang, Z.; Wen, L.; Chen, C.; Shi, Q.; Zhang, Y.; Ni, J.; Liu, Q. Selenomethionine ameliorates cognitive decline, reduces Tau hyperphosphorylation, and reverses synaptic deficit in the triple transgenic mouse model of Alzheimer’s disease. J. Alzheimers Dis. 2014, 41, 85–99. [Google Scholar] [CrossRef] [PubMed]
- Van Eersel, J.; Ke, Y.D.; Liu, X.; Delerue, F.; Kril, J.J.; Gotz, J.; Ittner, L.M. Sodium selenate mitigates Tau pathology, neurodegeneration, and functional deficits in Alzheimer’s disease models. Proc. Natl. Acad. Sci. USA 2010, 107, 13888–13893. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, T.; Blum, D.; Burnouf, S.; Demeyer, D.; Buee-Scherrer, V.; D’Hooge, R.; Buee, L.; Balschun, D. Rescue of impaired late-phase long-term depression in a tau transgenic mouse model. Neurobiol. Aging 2015, 36, 730–739. [Google Scholar] [CrossRef] [PubMed]
- Gotz, J.; Schild, A. Transgenic and knockout models of PP2A. Methods Enzymol. 2003, 366, 390–403. [Google Scholar] [PubMed]
- Liu, F.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X. Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur. J. Neurosci. 2005, 22, 1942–1950. [Google Scholar] [CrossRef] [PubMed]
- Janssens, V.; Goris, J. Protein phosphatase 2A: A highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem. J. 2001, 353, 417–439. [Google Scholar] [CrossRef] [PubMed]
- Kins, S.; Kurosinski, P.; Nitsch, R.M.; Gotz, J. Activation of the erk and JNK signaling pathways caused by neuron-specific inhibition of PP2A in transgenic mice. Am. J. Pathol. 2003, 163, 833–843. [Google Scholar] [CrossRef]
- Sontag, E. Protein phosphatase 2A: The trojan horse of cellular signaling. Cell Signal. 2001, 13, 7–16. [Google Scholar] [CrossRef]
- Ruvolo, P.P. The broken “off” switch in cancer signaling: PP2A as a regulator of tumorigenesis, drug resistance, and immune surveillance. BBA Clin. 2016, 6, 87–99. [Google Scholar] [CrossRef]
- Hoffman, A.; Taleski, G.; Sontag, E. The protein serine/threonine phosphatases PP2A, PP1 and calcineurin: A triple threat in the regulation of the neuronal cytoskeleton. Mol. Cell Neurosci. 2017, 84, 119–131. [Google Scholar] [CrossRef]
- Brion, J.P. Neurofibrillary tangles and Alzheimer’s disease. Eur. Neurol. 1998, 40, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Lee, K.W.; Park, E.S.; Oh, S.; Yan, R.; Zhang, J.; Beach, T.G.; Adler, C.H.; Voronkov, M.; Braithwaite, S.P.; et al. Dysregulation of protein phosphatase 2A in parkinson disease and dementia with lewy bodies. Ann. Clin. Transl. Neurol. 2016, 3, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Blum, D.; Herrera, F.; Francelle, L.; Mendes, T.; Basquin, M.; Obriot, H.; Demeyer, D.; Sergeant, N.; Gerhardt, E.; Brouillet, E.; et al. Mutant huntingtin alters Tau phosphorylation and subcellular distribution. Hum. Mol. Genet. 2015, 24, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Qian, W.; Shi, J.; Yin, X.; Iqbal, K.; Grundke-Iqbal, I.; Gong, C.X.; Liu, F. PP2A regulates tau phosphorylation directly and also indirectly via activating GSK-3β. J. Alzheimers Dis. 2010, 19, 1221–1229. [Google Scholar] [CrossRef] [PubMed]
- Houge, G.; Haesen, D.; Vissers, L.E.; Mehta, S.; Parker, M.J.; Wright, M.; Vogt, J.; McKee, S.; Tolmie, J.L.; Cordeiro, N.; et al. B56Δ-related protein phosphatase 2A dysfunction identified in patients with intellectual disability. J. Clin. Investig. 2015, 125, 3051–3062. [Google Scholar] [CrossRef] [PubMed]
- Gotz, J.; Kues, W. The role of protein phosphatase 2A catalytic subunit calpha in embryogenesis: Evidence from sequence analysis and localization studies. Biol. Chem. 1999, 380, 1117–1120. [Google Scholar] [CrossRef] [PubMed]
- Kins, S.; Crameri, A.; Evans, D.R.; Hemmings, B.A.; Nitsch, R.M.; Gotz, J. Reduced protein phosphatase 2A activity induces hyperphosphorylation and altered compartmentalization of tau in transgenic mice. J. Biol. Chem. 2001, 276, 38193–38200. [Google Scholar] [PubMed]
- Nicholls, R.E.; Sontag, J.M.; Zhang, H.; Staniszewski, A.; Yan, S.; Kim, C.Y.; Yim, M.; Woodruff, C.M.; Arning, E.; Wasek, B.; et al. PP2A methylation controls sensitivity and resistance to β-amyloid-induced cognitive and electrophysiological impairments. Proc. Natl. Acad. Sci. USA 2016, 113, 3347–3352. [Google Scholar] [CrossRef] [PubMed]
- Sontag, E.; Nunbhakdi-Craig, V.; Lee, G.; Bloom, G.S.; Mumby, M.C. Regulation of the phosphorylation state and microtubule-binding activity of Tau by protein phosphatase 2A. Neuron 1996, 17, 1201–1207. [Google Scholar] [CrossRef]
- Louis, J.V.; Martens, E.; Borghgraef, P.; Lambrecht, C.; Sents, W.; Longin, S.; Zwaenepoel, K.; Pijnenborg, R.; Landrieu, I.; Lippens, G.; et al. Mice lacking phosphatase PP2A subunit PR61/B’Δ (PPP2R5D) develop spatially restricted tauopathy by deregulation of CDK5 and GSK3β. Proc. Natl. Acad. Sci. USA 2011, 108, 6957–6962. [Google Scholar] [CrossRef]
- Taleski, G.; Sontag, E. Protein phosphatase 2A and Tau: An orchestrated “pas de deux”. FEBS Lett. 2018, 592, 1079–1095. [Google Scholar] [CrossRef] [PubMed]
- De Baere, I.; Derua, R.; Janssens, V.; van Hoof, C.; Waelkens, E.; Merlevede, W.; Goris, J. Purification of porcine brain protein phosphatase 2A leucine carboxyl methyltransferase and cloning of the human homologue. Biochemistry 1999, 38, 16539–16547. [Google Scholar] [CrossRef] [PubMed]
- Longin, S.; Jordens, J.; Martens, E.; Stevens, I.; Janssens, V.; Rondelez, E.; De Baere, I.; Derua, R.; Waelkens, E.; Goris, J.; et al. An inactive protein phosphatase 2A population is associated with methylesterase and can be re-activated by the phosphotyrosyl phosphatase activator. Biochem. J. 2004, 380, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Gotz, J. How it all started: Tau and protein phosphatase 2A. J. Alzheimers Dis. 2013, 37, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Sangodkar, J.; Farrington, C.C.; McClinch, K.; Galsky, M.D.; Kastrinsky, D.B.; Narla, G. All roads lead to PP2A: Exploiting the therapeutic potential of this phosphatase. FEBS J. 2016, 283, 1004–1024. [Google Scholar] [CrossRef] [PubMed]
- Sontag, E.; Nunbhakdi-Craig, V.; Lee, G.; Brandt, R.; Kamibayashi, C.; Kuret, J.; White, C.L., 3rd.; Mumby, M.C.; Bloom, G.S. Molecular interactions among protein phosphatase 2A, Tau, and microtubules. Implications for the regulation of Tau phosphorylation and the development of tauopathies. J. Biol. Chem. 1999, 274, 25490–25498. [Google Scholar] [CrossRef] [PubMed]
- Sontag, J.M.; Nunbhakdi-Craig, V.; Mitterhuber, M.; Ogris, E.; Sontag, E. Regulation of protein phosphatase 2A methylation by lcmt1 and PME-1 plays a critical role in differentiation of neuroblastoma cells. J. Neurochem. 2010, 115, 1455–1465. [Google Scholar] [CrossRef] [PubMed]
- Sontag, J.M.; Wasek, B.; Taleski, G.; Smith, J.; Arning, E.; Sontag, E.; Bottiglieri, T. Altered protein phosphatase 2A methylation and tau phosphorylation in the young and aged brain of methylenetetrahydrofolate reductase (MTHFR) deficient mice. Front. Aging Neurosci. 2014, 6, 214. [Google Scholar] [CrossRef] [PubMed]
- Chou, D.M.; Petersen, P.; Walter, J.C.; Walter, G. Protein phosphatase 2A regulates binding of CDC45 to the prereplication complex. J. Biol. Chem. 2002, 277, 40520–40527. [Google Scholar] [CrossRef] [PubMed]
- Aarts, M.; Liu, Y.; Liu, L.; Besshoh, S.; Arundine, M.; Gurd, J.W.; Wang, Y.T.; Salter, M.W.; Tymianski, M. Treatment of ischemic brain damage by perturbing nmda receptor-PSD-95 protein interactions. Science 2002, 298, 846–850. [Google Scholar] [CrossRef]
- Ruediger, R.; Pham, H.T.; Walter, G. Disruption of protein phosphatase 2A subunit interaction in human cancers with mutations in the A α subunit gene. Oncogene 2001, 20, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, N.M.; Hovens, C.M.; Michael, M.; Rosenthal, M.A.; Costello, A.J. Open-label, phase I dose-escalation study of sodium selenate, a novel activator of PP2A, in patients with castration-resistant prostate cancer. Br. J. Cancer 2010, 103, 462–468. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, N.M.; Najdovska, M.; Costello, A.J. Inorganic selenium retards progression of experimental hormone refractory prostate cancer. J. Urol. 2004, 171, 907–910. [Google Scholar] [CrossRef] [PubMed]
- Brozmanova, J.; Manikova, D.; Vlckova, V.; Chovanec, M. Selenium: A double-edged sword for defense and offence in cancer. Arch. Toxicol. 2010, 84, 919–938. [Google Scholar] [CrossRef] [PubMed]
- Westermarck, J.; Hahn, W.C. Multiple pathways regulated by the tumor suppressor PP2A in transformation. Trends Mol. Med. 2008, 14, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Grech, G.; Baldacchino, S.; Saliba, C.; Grixti, M.P.; Gauci, R.; Petroni, V.; Fenech, A.G.; Scerri, C. Deregulation of the protein phosphatase 2A, PP2A in cancer: Complexity and therapeutic options. Tumour Biol. 2016, 37, 11691–11700. [Google Scholar] [CrossRef] [PubMed]
- Vinceti, M.; Mandrioli, J.; Borella, P.; Michalke, B.; Tsatsakis, A.; Finkelstein, Y. Selenium neurotoxicity in humans: Bridging laboratory and epidemiologic studies. Toxicol. Lett. 2014, 230, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Vinceti, M.; Filippini, T.; Del Giovane, C.; Dennert, G.; Zwahlen, M.; Brinkman, M.; Zeegers, M.P.; Horneber, M.; D’Amico, R.; Crespi, C.M. Selenium for preventing cancer. Cochrane Database Syst. Rev. 2018, 29, CD005195. [Google Scholar] [CrossRef] [PubMed]
- Bartolini, D.; Sancineto, L.; Fabro de Bem, A.; Tew, K.D.; Santi, C.; Radi, R.; Toquato, P.; Galli, F. Selenocompounds in cancer therapy: An overview. Adv. Cancer Res. 2017, 136, 259–302. [Google Scholar] [PubMed]
- Encinas, M.; Iglesias, M.; Liu, Y.; Wang, H.; Muhaisen, A.; Cena, V.; Gallego, C.; Comella, J.X. Sequential treatment of SH-SY5Y cells with retinoic acid and brain-derived neurotrophic factor gives rise to fully differentiated, neurotrophic factor-dependent, human neuron-like cells. J. Neurochem. 2000, 75, 991–1003. [Google Scholar] [CrossRef] [PubMed]
- Van der Jeugd, A.; Parra-Damas, A.; Baeta-Corral, R.; Soto-Faguas, C.M.; Ahmed, T.; LaFerla, F.M.; Gimenez-Llort, L.; D’Hooge, R.; Saura, C.A. Reversal of memory and neuropsychiatric symptoms and reduced tau pathology by selenium in 3xTg AD mice. Sci. Rep. 2018, 8, 6431. [Google Scholar] [CrossRef] [PubMed]
- Augustinack, J.C.; Schneider, A.; Mandelkow, E.M.; Hyman, B.T. Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer’s disease. Acta Neuropathol. 2002, 103, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, P.R.; Berry, M.J. Selenoprotein synthesis: A unique translational mechanism used by a diverse family of proteins. Thyroid 2005, 15, 769–775. [Google Scholar] [CrossRef] [PubMed]
- Zheng, R.; Zhang, Z.H.; Chen, C.; Chen, Y.; Jia, S.Z.; Liu, Q.; Ni, J.Z.; Song, G.L. Selenomethionine promoted hippocampal neurogenesis via the PI3K-AKT-GSK3β-WNT pathway in a mouse model of Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2017, 485, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Nairn, A.C.; Shenolikar, S. The role of protein phosphatases in synaptic transmission, plasticity and neuronal development. Curr. Opin. Neurobiol. 1992, 2, 296–301. [Google Scholar] [CrossRef]
- Winder, D.G.; Sweatt, J.D. Roles of serine/threonine phosphatases in hippocampal synaptic plasticity. Nat. Rev. Neurosci. 2001, 2, 461–474. [Google Scholar] [CrossRef]
- Strack, S.; Zaucha, J.A.; Ebner, F.F.; Colbran, R.J.; Wadzinski, B.E. Brain protein phosphatase 2A: Developmental regulation and distinct cellular and subcellular localization by B subunits. J. Comp. Neurol. 1998, 392, 515–527. [Google Scholar] [CrossRef]
- Agarwal-Mawal, A.; Qureshi, H.Y.; Cafferty, P.W.; Yuan, Z.; Han, D.; Lin, R.; Paudel, H.K. 14-3-3 connects glycogen synthase kinase-3β to Tau within a brain microtubule-associated tau phosphorylation complex. J. Biol. Chem. 2003, 278, 12722–12728. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Agarwal-Mawal, A.; Paudel, H.K. 14-3-3 binds to and mediates phosphorylation of microtubule-associated tau protein by ser9-phosphorylated glycogen synthase kinase 3β in the brain. J. Biol. Chem. 2004, 279, 26105–26114. [Google Scholar] [CrossRef]
- Viquez, N.M.; Fuger, P.; Valakh, V.; Daniels, R.W.; Rasse, T.M.; di Antonio, A. PP2A and GSK-3β act antagonistically to regulate active zone development. J. Neurosci. 2009, 29, 11484–11494. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, R.; Gu, J.; Yin, X.; Jin, N.; Xie, S.; Wang, Y.; Chang, H.; Qian, W.; Shi, J.; et al. Cross talk between PI3K-AKT-GSK-3β and PP2A pathways determines tau hyperphosphorylation. Neurobiol. Aging 2015, 36, 188–200. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Gu, Z.; Liu, W.; Yan, Z. Glycogen synthase kinase 3 regulates N-methyl-d-aspartate receptor channel trafficking and function in cortical neurons. Mol. Pharmacol. 2007, 72, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Wildburger, N.C.; Laezza, F. Control of neuronal ion channel function by glycogen synthase kinase-3: New prospective for an old kinase. Front. Mol. Neurosci. 2012, 5, 80. [Google Scholar] [CrossRef] [PubMed]
- Sontag, E.; Luangpirom, A.; Hladik, C.; Mudrak, I.; Ogris, E.; Speciale, S.; White, C.L., 3rd. Altered expression levels of the protein phosphatase 2A abalphac enzyme are associated with alzheimer disease pathology. J. Neuropathol. Exp. Neurol. 2004, 63, 287–301. [Google Scholar] [CrossRef]
- Zhang, B.Y.; Saijilafu; Liu, C.M.; Wang, R.Y.; Zhu, Q.; Jiao, Z.; Zhou, F.Q. Akt-independent GSK3 inactivation downstream of PI3K signaling regulates mammalian axon regeneration. Biochem. Biophys. Res. Commun. 2014, 443, 743–748. [Google Scholar] [CrossRef]
- Zhang, Z.H.; Chen, C.; Wu, Q.Y.; Zheng, R.; Liu, Q.; Ni, J.Z.; Hoffmann, P.R.; Song, G.L. Selenomethionine reduces the deposition of beta-amyloid plaques by modulating β-secretase and enhancing selenoenzymatic activity in a mouse model of Alzheimer’s disease. Metal. Integr. Biomet. Sci. 2016, 8, 782–789. [Google Scholar] [CrossRef]
- Zhang, Z.H.; Wu, Q.Y.; Zheng, R.; Chen, C.; Chen, Y.; Liu, Q.; Hoffmann, P.R.; Ni, J.Z.; Song, G.L. Selenomethionine mitigates cognitive decline by targeting both Tau hyperphosphorylation and autophagic clearance in an Alzheimer’s disease mouse model. J. Neurosci. 2017, 37, 2449–2462. [Google Scholar] [CrossRef] [PubMed]
- Krishnankutty, A.; Kimura, T.; Saito, T.; Aoyagi, K.; Asada, A.; Takahashi, S.I.; Ando, K.; Ohara-Imaizumi, M.; Ishiguro, K.; Hisanaga, S.I. In vivo regulation of glycogen synthase kinase 3β activity in neurons and brains. Sci. Rep. 2017, 7, 8602. [Google Scholar] [CrossRef] [PubMed]
- Mancinelli, R.; Carpino, G.; Petrungaro, S.; Mammola, C.L.; Tomaipitinca, L.; Filippini, A.; Facchiano, A.; Ziparo, E.; Giampietri, C. Multifaceted roles of GSK-3 in cancer and autophagy-related diseases. Oxid. Med. Cell. Long. 2017, 2017, 4629495. [Google Scholar] [CrossRef] [PubMed]
- Encinas, M.; Iglesias, M.; Llecha, N.; Comella, J.X. Extracellular-regulated kinases and phosphatidylinositol 3-kinase are involved in brain-derived neurotrophic factor-mediated survival and neuritogenesis of the neuroblastoma cell line SH-SY5Y. J. Neurochem. 1999, 73, 1409–1421. [Google Scholar] [CrossRef] [PubMed]
- Ward, M.P.; Spiers, J.P. Protein phosphatase 2A regulation of markers of extracellular matrix remodelling in hepatocellular carcinoma cells: Functional consequences for tumour invasion. Br. J. Pharmacol. 2017, 174, 1116–1130. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Habbab, W.; Aoudé, I.; Palangi, F.; Abdulla, S.; Ahmed, T. The Anti-Tumor Agent Sodium Selenate Decreases Methylated PP2A, Increases GSK3βY216 Phosphorylation, Including Tau Disease Epitopes and Reduces Neuronal Excitability in SHSY-5Y Neurons. Int. J. Mol. Sci. 2019, 20, 844. https://doi.org/10.3390/ijms20040844
Habbab W, Aoudé I, Palangi F, Abdulla S, Ahmed T. The Anti-Tumor Agent Sodium Selenate Decreases Methylated PP2A, Increases GSK3βY216 Phosphorylation, Including Tau Disease Epitopes and Reduces Neuronal Excitability in SHSY-5Y Neurons. International Journal of Molecular Sciences. 2019; 20(4):844. https://doi.org/10.3390/ijms20040844
Chicago/Turabian StyleHabbab, Wesal, Imad Aoudé, Freshteh Palangi, Sara Abdulla, and Tariq Ahmed. 2019. "The Anti-Tumor Agent Sodium Selenate Decreases Methylated PP2A, Increases GSK3βY216 Phosphorylation, Including Tau Disease Epitopes and Reduces Neuronal Excitability in SHSY-5Y Neurons" International Journal of Molecular Sciences 20, no. 4: 844. https://doi.org/10.3390/ijms20040844