High-Throughput Virtual Screening, Molecular Dynamics Simulation, and Enzyme Kinetics Identified ZINC84525623 as a Potential Inhibitor of NDM-1

 ,
, 
Abstract
:
1. Introduction
2. Results and Discussion
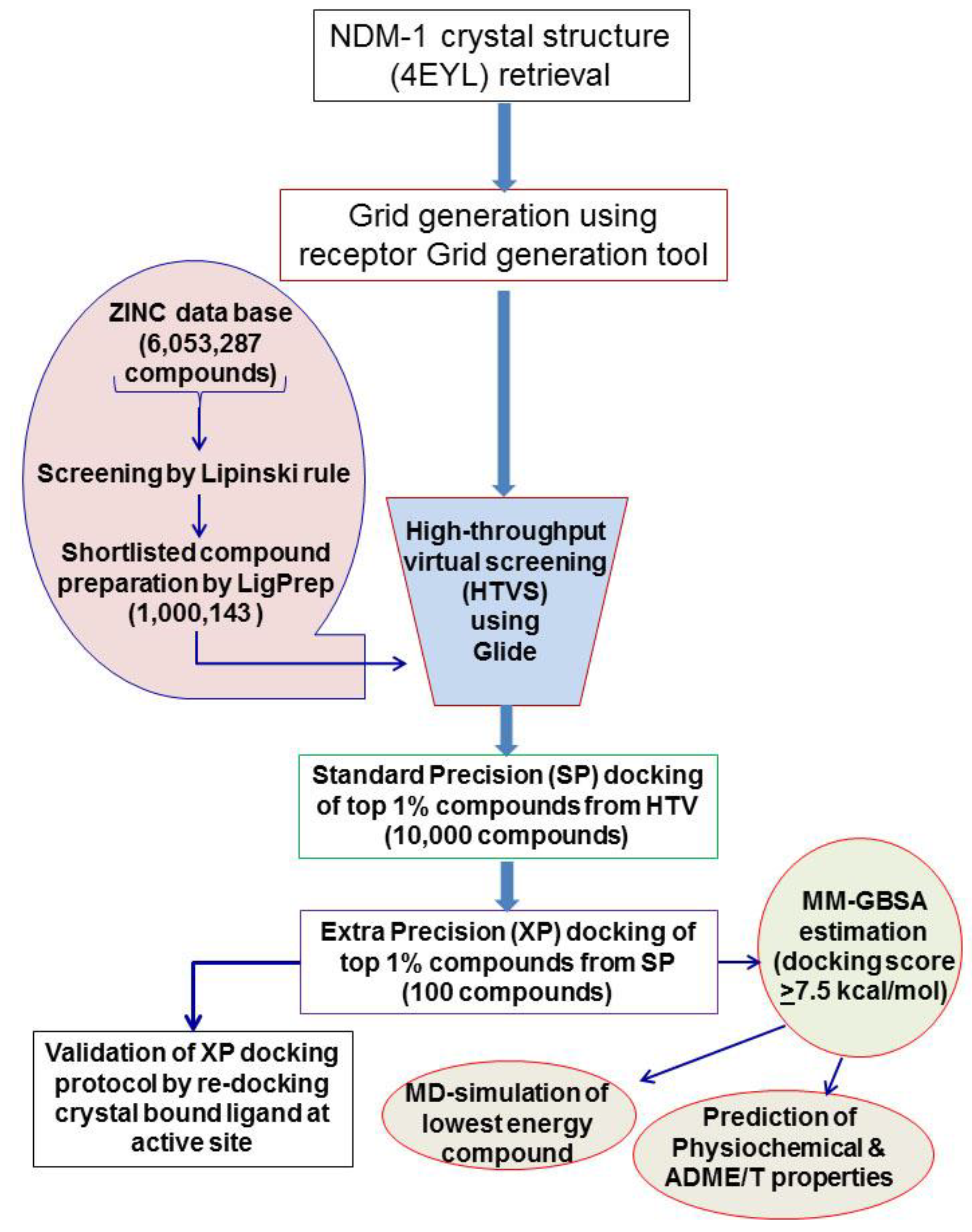
2.1. Virtual Screening and Molecular Docking of ZINC Lead-Like Compounds
2.2. Physiochemical and ADME/T Properties
2.3. Analyses of the NDM-1 and Screened Inhibitors Interaction
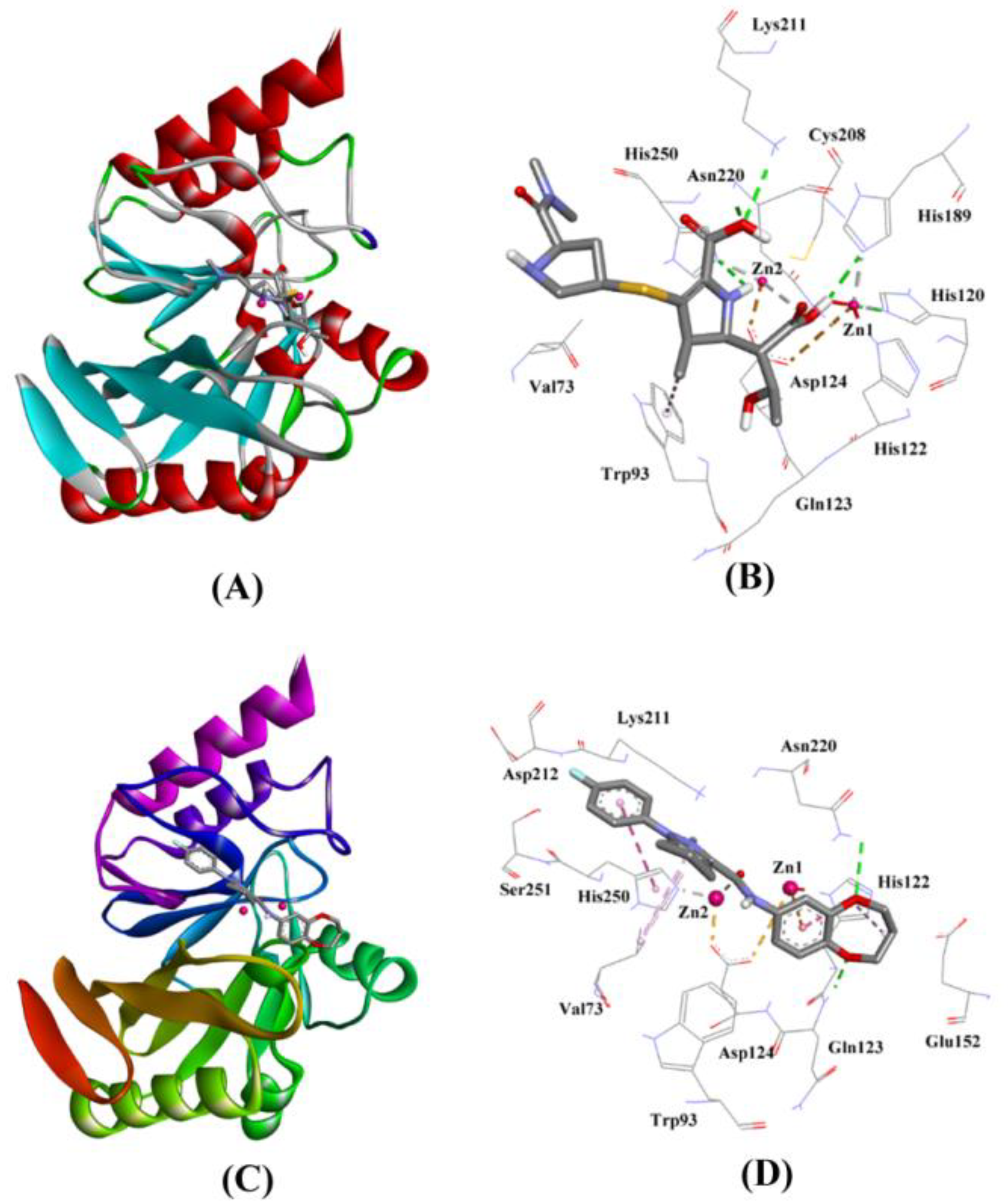
2.3.1. NDM-1 and Meropenem Interaction
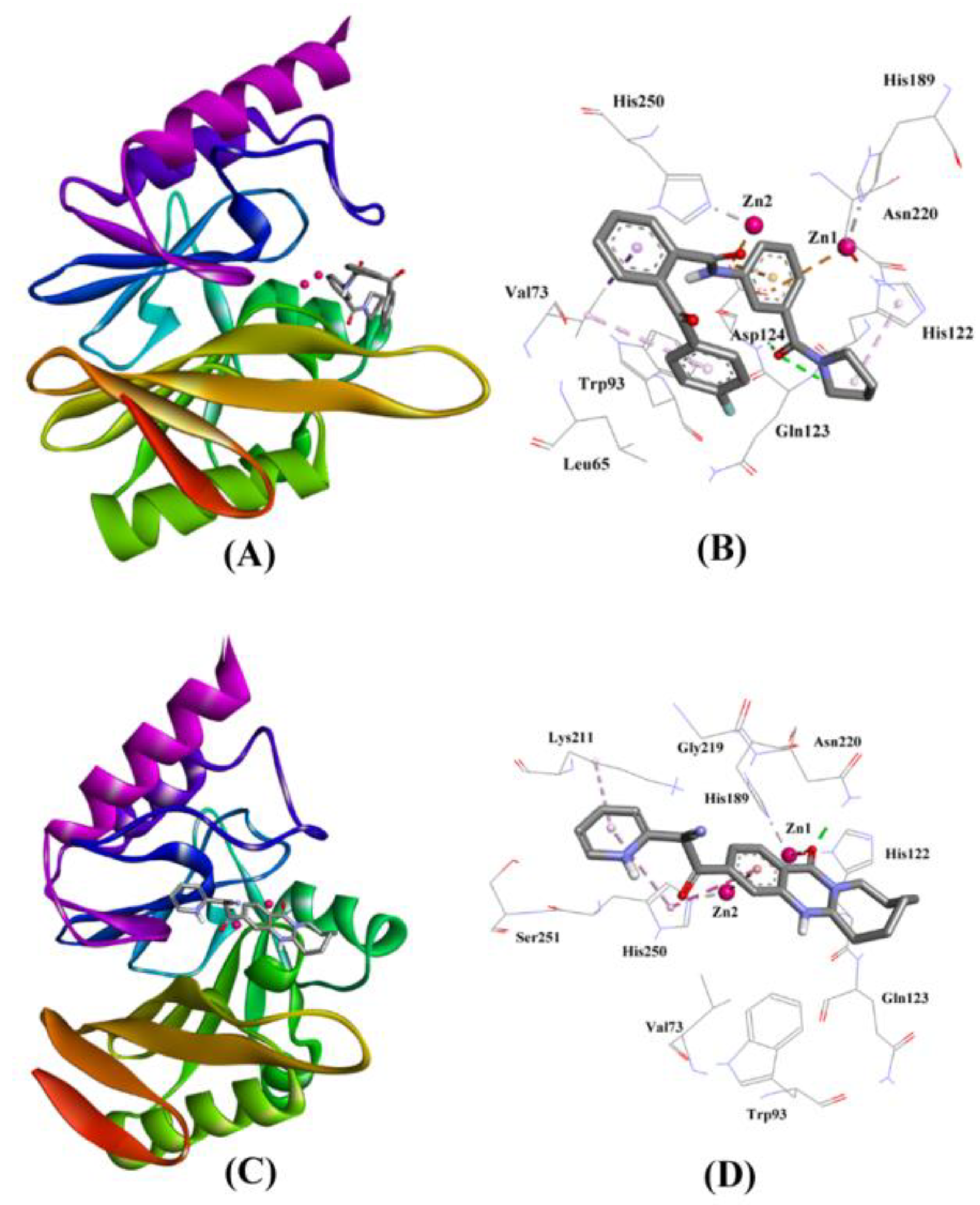
2.3.2. NDM-1 and ZINC10936382 Interaction
2.3.3. NDM-1 and ZINC30479078 Interaction
2.3.4. NDM-1 and ZINC41493045 Interaction
2.3.5. NDM-1 and ZINC7424911 Interaction
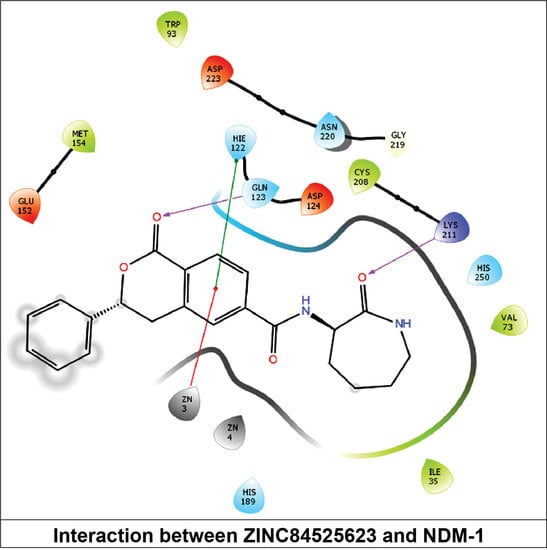
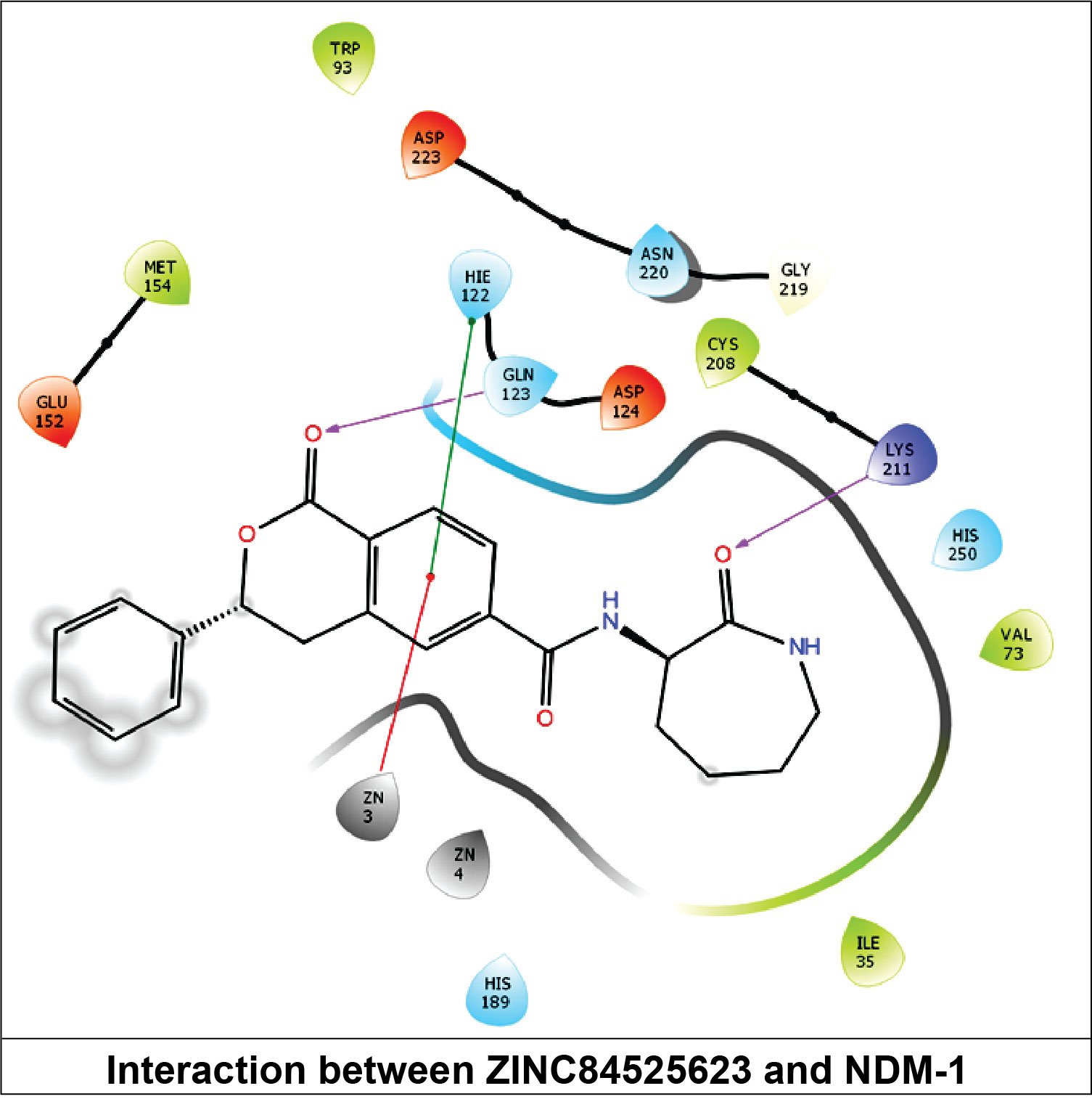
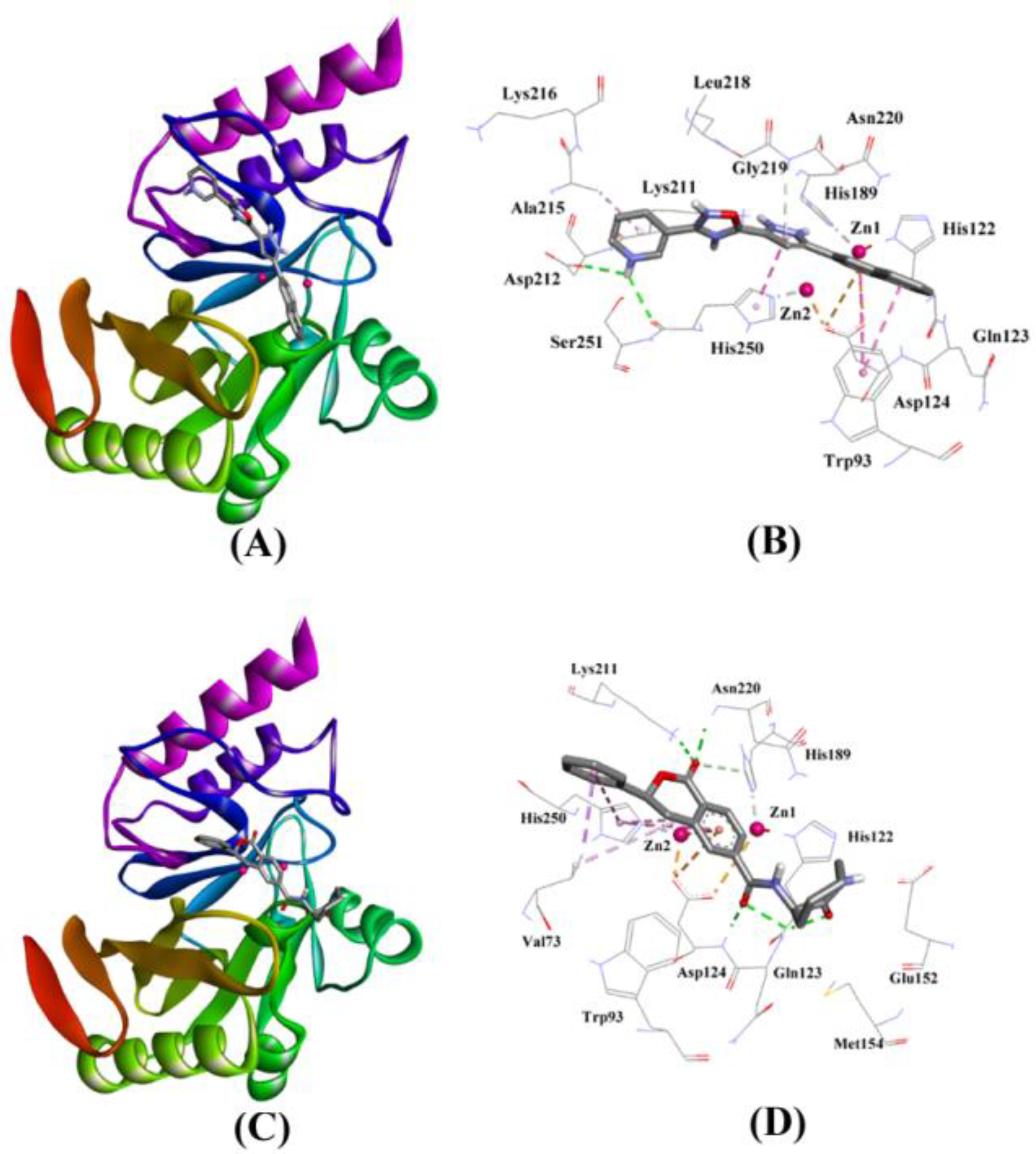
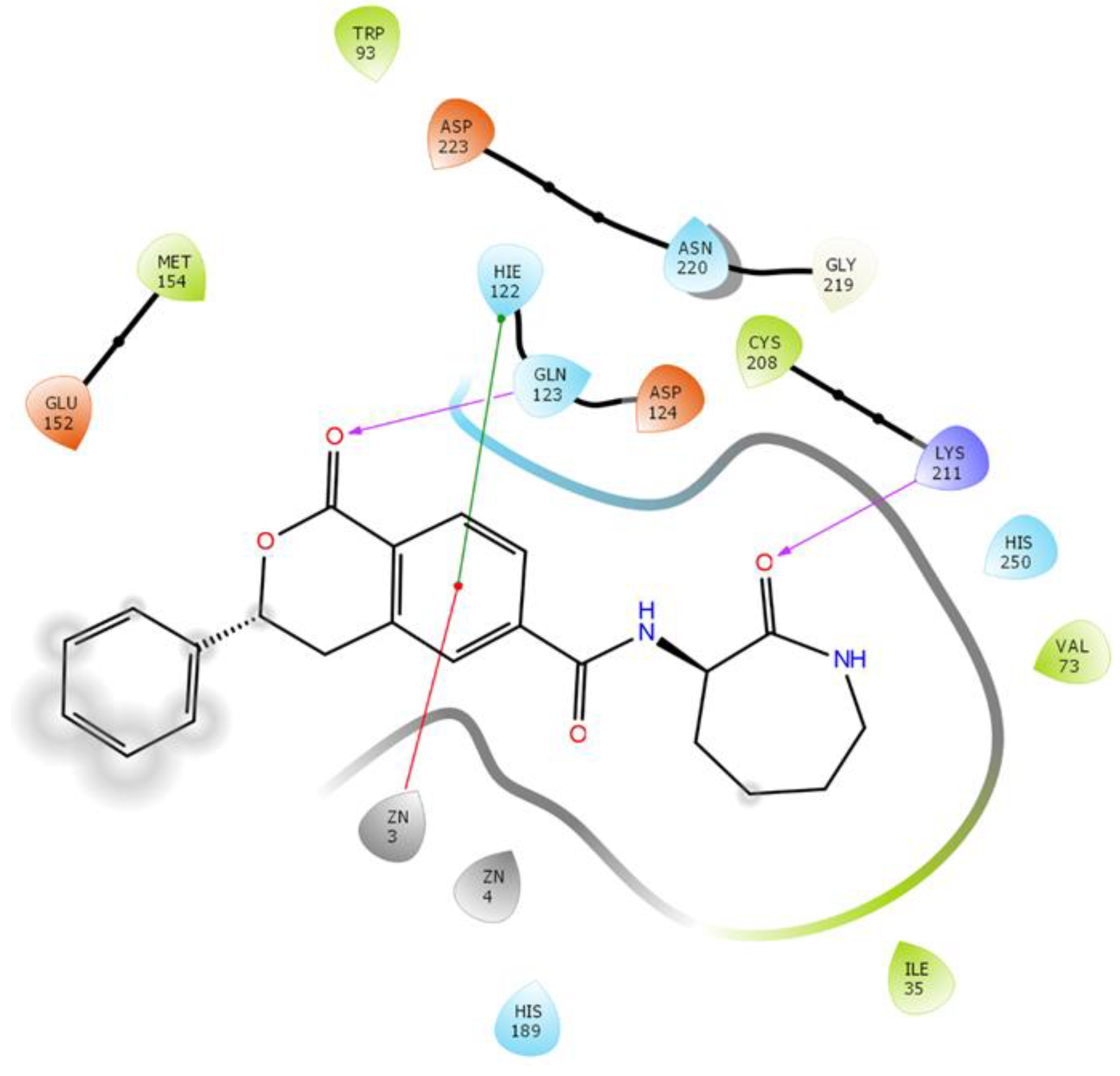
2.3.6. NDM-1 and ZINC84525623 Interaction
2.4. Molecular Mechanics—General Born Surface Area (MM-GBSA) Estimation
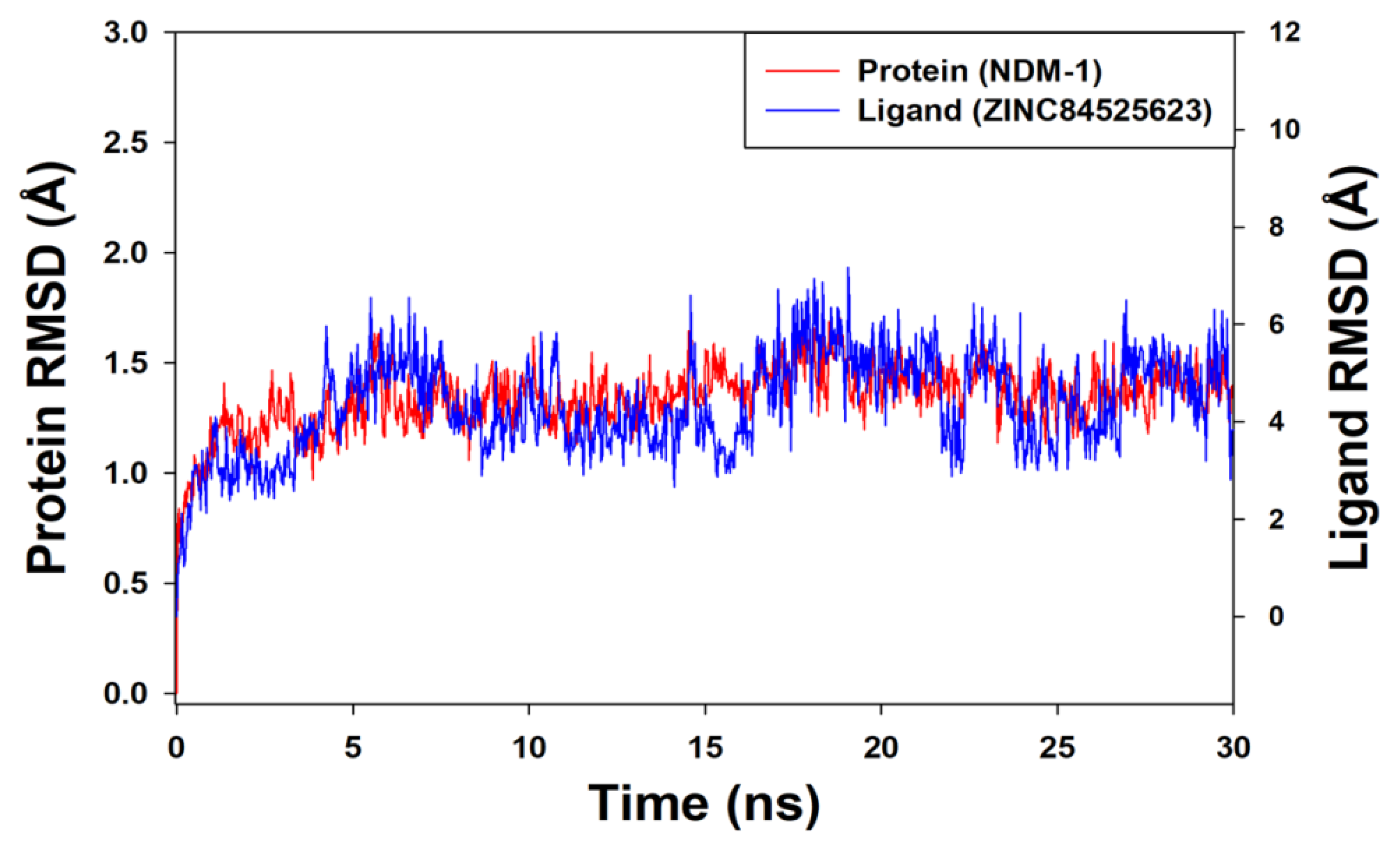
Molecular Dynamics (MD) Simulation
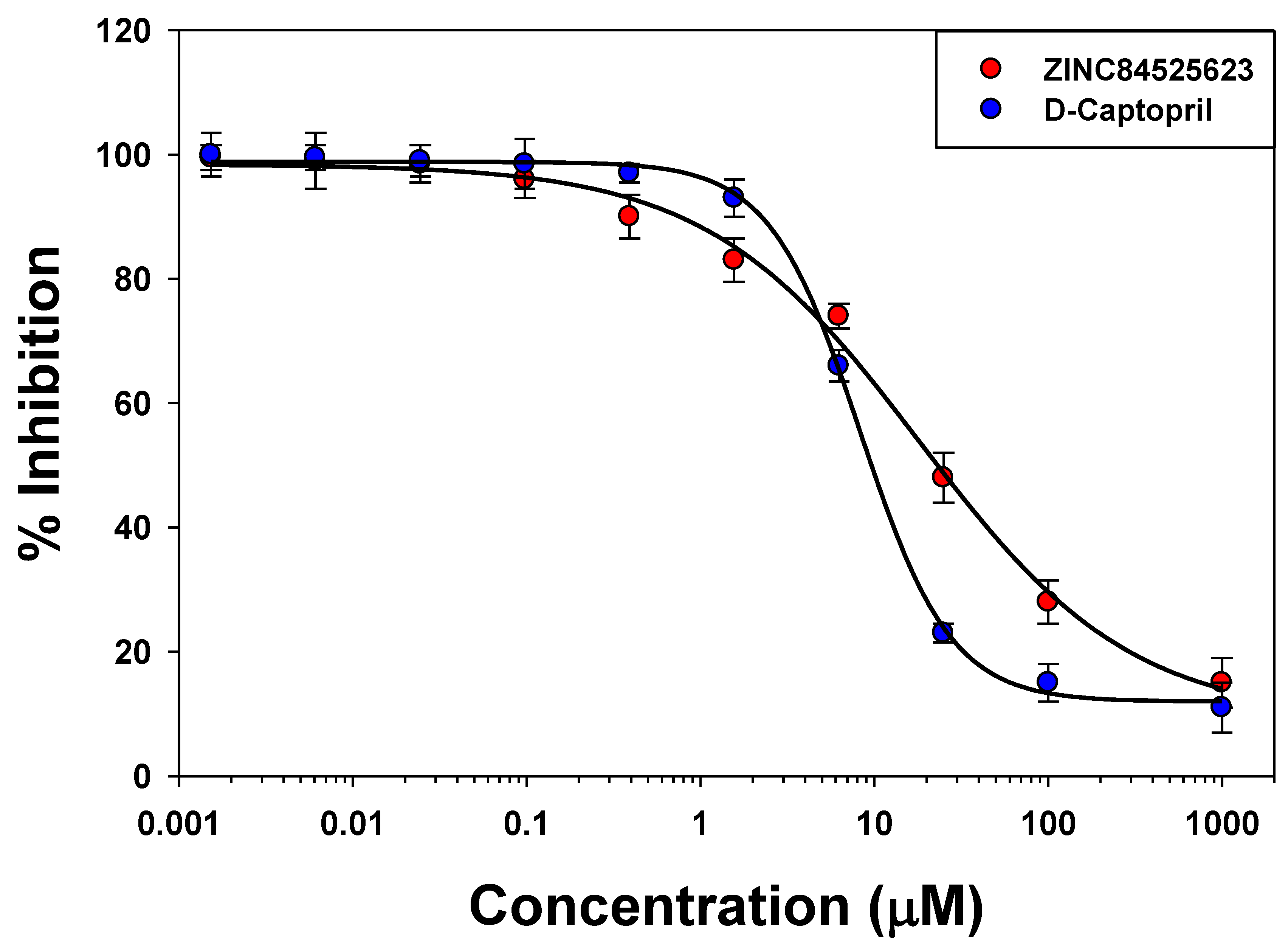
2.5. IC50 Value Determination
2.6. Steady State Enzyme Kinetics
3. Materials and Methods
3.1. Materials
3.2. Experimental Procedures
3.3. Retrieval and Preparation of Ligands
3.4. Preparation of Protein, Active Site Prediction, and Grid Generation
3.5. Virtual Screening and Molecular Docking
3.6. Validation of Docking Protocol
3.7. Determination of Physiochemical and ADME/T Properties
3.8. Molecular Mechanics—Generalized Born Surface Area (MM-GBSA) Calculations
3.9. Molecular Dynamics (MD) Simulation
3.10. Cloning, Expression and Purification of NDM-1
3.11. Determination of IC50
3.12. Enzyme Kinetics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Patel, G.; Bonomo, R.A. “Stormy waters ahead”: Global emergence of carbapenemases. Front. Microbiol. 2013, 4, 48. [Google Scholar] [CrossRef]
- Bush, K. Bench-to-bedside review: The role of b-lactamases in antibiotic resistant Gram-negative infections. Crit. Care 2010, 14, 224. [Google Scholar] [CrossRef] [PubMed]
- Bush, K. Alarming b-lactamase-mediated resistance in multi-drug resistant Enterobacteriaceae. Curr. Opin. Microbiol. 2010, 13, 558–564. [Google Scholar] [CrossRef] [PubMed]
- Faheem, M.; Rehman, M.T.; Danishuddin, M.; Khan, A.U. Biochemical characterization of CTX-M-15 from Enterobacter cloacae and designing a novel non-β-lactam based β-lactamase inhibitor. PLoS ONE 2013, 8, e56926. [Google Scholar] [CrossRef]
- Ambler, R.P. The structure of β-lactamases. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1980, 289, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Bush, K.; Jacoby, G.A.; Medeiros, A.A. A functional classification scheme for beta-lactamases and its correlation with molecular structure. Antimicrob. Agents Chemother. 1995, 39, 1211–1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bush, K.; Jacoby, G. A Updated functional classification of beta-lactamases. Antimicrob. Agents Chemother. 2010, 54, 969–976. [Google Scholar] [CrossRef]
- Kumarasamy, K.K.; Toleman, M.A.; Walsh, T.R.; Bagaria, J.; Butt, F.; Balakrishnan, R.; Chaudhary, U.; Doumith, M.; Giske, C.G.; Irfan, S.; et al. Emergence of a new antibiotic resistance mechanism in India, Pakistan, and the UK: A molecular, biological, and epidemiological study. Lancet Infect. Dis. 2010, 10, 597–602. [Google Scholar] [CrossRef]
- Khan, A.U.; Maryam, L.; Zarrilli, R. Structure, Genetics and Worldwide Spread of New Delhi Metallo-β-lactamase (NDM): A threat to public health. BMC Microbiol. 2017, 17, 101. [Google Scholar] [CrossRef]
- Drawza, S.M.; Papp-Wallace, K.M.; Bonomo, R.A. New β-Lactamase Inhibitors: A Therapeutic Renaissance in an MDR World. Antimicrob. Agents Chemother. 2014, 58, 1835–1846. [Google Scholar] [CrossRef]
- AlAjmi, M.F.; Rehman, M.T.; Hussain, A.; Rather, G.M. Screening and identification of inhibitors from natural source against Polo-like kinase (PLK): An In silico study against cancer. Int. J. Biol. Macromol. 2018, 116, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Jani, K.S.; Dalafave, D.S. Computational design of targeted inhibitors of polo-like kinase 1 (PLK-1). Bioinform. Biol. Insights 2012, 6, 23–31. [Google Scholar]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Verma, P.; Tiwari, M.; Tiwari, V. In silico high-throughput virtual screening and molecular dynamics simulation study to identify inhibitor for AdeABC efflux pump of Acinetobacter baumannii. J. Biomol. Struct. Dyn. 2018, 36, 1182–1194. [Google Scholar] [CrossRef] [PubMed]
- King, D.T.; Worrall, L.J.; Gruninger, R.; Strynadka, N.C. New Delhi Metallo-Beta-Lactamase: Structural Insights into Beta-Lactam Recognition and Inhibition. J. Am. Chem. Soc. 2012, 134, 11362–11365. [Google Scholar] [CrossRef] [PubMed]
- King, D.; Strynadka, N. Crystal structure of New Delhi metallo-β-lactamase reveals molecular basis for antibiotic resistance. Protein Sci. 2011, 20, 1484–1491. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Cunningham, M.A.; Mire, J.; Tesar, C.; Sacchettini, J.; Joachimiak, A. NDM-1, the ultimate promiscuous enzyme: Substrate recognition and catalytic mechanism. FASEB J. 2013, 27, 1917–1927. [Google Scholar] [CrossRef]
- Concha, N.O.; Rasmussen, B.A.; Bush, K.; Herzberg, O. Crystal structure of the wide-spectrum binuclear zinc beta-lactamase from Bacteroides fragilis. Structure 1996, 4, 823–836. [Google Scholar] [CrossRef]
- Khan, A.U.; Rehman, M.T. Role of non-active-site residue Trp-93 in the function and stability of New Delhi metallo-β-lactamase 1. Antimicrob. Agents Chemother. 2016, 60, 356–360. [Google Scholar] [CrossRef]
- Crisp, J.; Conners, R.; Garrity, J.D.; Carenbauer, A.L.; Crowder, M.W.; Spencer, J. Structural basis for the role of Asp-120 in metallo-beta-lactamases. Biochemistry 2007, 18, 10664–10674. [Google Scholar] [CrossRef]
- Page, M.I.; Badarau, A. The mechanisms of catalysis by metallo b-lactamases. Bioinorg. Chem. Appl. 2008, 2008, 576297. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Fast, W.; Valentine, A.M.; Benkovic, S.J. Metallo-beta-lactamase: Structure and mechanism. Curr. Opin. Microbiol. 1999, 3, 614–622. [Google Scholar]
- Das, C.K.; Nair, N.N. Hydrolysis of cephalexin and meropenem by New Delhi metallo-β-lactamase: The substrate protonation mechanism is drug dependent. Phys. Chem. Chem. Phys. 2017, 19, 13111–13121. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Wang, J.; Niu, G.; Shui, W.; Sun, Y.; Zhou, H.; Zhang, Y.; Yang, C.; Lou, Z.; Rao, Z. A structural view of the antibiotic degradation enzyme NDM-1 from a superbug. Protein Cell 2011, 2, 384–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- AlShabib, N.A.; Khan, J.M.; Malik, A.; AlSenaidy, M.A.; Rehman, M.T.; AlAjmi, M.F.; AlSenaidy, A.M.; Husain, F.M.; Khan, R.H. Molecular insight into binding behavior of polyphenol (rutin) with beta lactoglobulin: Spectroscopic and computational studies. J. Mol. Liq. 2018, 269, 511–520. [Google Scholar] [CrossRef]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A force field providing broad coverage of drug-like small molecules and proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Rehman, M.T.; Ahmed, S.; Khan, A.U. Interaction of meropenem with ‘N’ and ‘B’ isoforms of Human Serum Albumin: A spectroscopic and molecular docking study. J. Biomol. Struct. Dyn. 2016, 34, 1849–1864. [Google Scholar] [CrossRef]
- Li, J.; Abel, R.; Zhu, K.; Cao, Y.; Zhao, S.; Friesner, R.A. The VSGB 2.0 Model: A next generation energy model for high resolution protein structure modeling. Proteins 2011, 79, 2794–2812. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Branka, A.C. Nose-Hoover chain method for non-equilibrium molecular dynamics simulation. Phys. Rev. E Stat. Phys. Plasmas Fluids Relat. Interdiscip. Top. 2000, 61, 4769–4773. [Google Scholar]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Stachyra, T.; Pechereau, M.C.; Bruneau, J.M.; Claudon, M.; Frere, J.M.; Miossec, C.; Coleman, K.; Black, M.T. Mechanistic studies of the inactivation of TEM-1 and P99 by NXL104, a novel non-b-lactam b-lactamase inhibitor. Antimicrob. Agents Chemother. 2010, 54, 5132–5138. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Yu, Y.; Chen, H.; Cao, X.; Lao, X.; Fang, Y.; Shi, Y.; Chen, J.; Zheng, H. Inhibitor discovery of full-length New Delhi metallo-β-lactamase-1 (NDM-1). PLoS ONE 2013, 8, e62955. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No. | ZINC ID | Docking Score * | Glide g-Score * | Glide e-Model * | XP g-Score * |
|---|---|---|---|---|---|
| 1. | ZINC10936382 | −8.322 | −8.322 | −68.183 | −8.322 |
| 2. | ZINC30479078 | −9.046 | −9.046 | −66.578 | −9.046 |
| 3. | ZINC41493045 | −7.714 | −7.714 | −64.597 | −7.714 |
| 4. | ZINC7424911 | −8.254 | −8.265 | −63.254 | −8.254 |
| 5. | ZINC84525623 | −8.790 | −8.790 | −64.740 | −8.790 |
| 6. | Control (Meropenem) | −6.413 | −6.413 | −56.140 | −6.413 |
| Structure/Name of Compounds | Mol. Weight (g/mol) | HBD b | HBA c | Tpsa d (Å2) | Net Charge | RB e |
|---|---|---|---|---|---|---|
 ZINC10936382 | 393.418 | 1 | 5 | 65.4 | 0 | 3 |
 ZINC30479078 | 416.452 | 1 | 4 | 66.5 | 0 | 5 |
 ZINC41493045 | 372.428 | 0 | 6 | 89.2 | 0 | 3 |
 ZINC7424911 | 339.358 | 1 | 5 | 80.5 | 0 | 3 |
 ZINC84525623 | 378.428 | 2 | 4 | 84.5 | 0 | 3 |
| ZINC Id | QPpolrz (13 to 70) | QPlogS (−6 to 0.5) | QPlogPC16 (4 to 18) | QPlogPoct (8 to 43) | QPlogPw (5 to 48) | QPlogPo/w (−2 to 6) | QPlogKp (−8 to −1) | QPlog Khsa (−1.5 to 1.2) |
|---|---|---|---|---|---|---|---|---|
| ZINC10936382 | 43.299 | −6.736 | 11.702 | 18.741 | 9.631 | 4.843 | −1.346 | 0.813 |
| ZINC30479078 | 48.327 | −5.251 | 12.043 | 20.834 | 10.492 | 2.723 | −2.811 | 0.636 |
| ZINC41493045 | 40.603 | −4.524 | 11.974 | 18.573 | 11.572 | 2.070 | −2.946 | −0.429 |
| ZINC7424911 | 41.415 | −5.717 | 12.778 | 19.790 | 12.387 | 3.337 | −2.263 | 0.450 |
| ZINC84525623 | 42.420 | −3.863 | 12.527 | 21.290 | 15.816 | 2.284 | −3.107 | −0.003 |
| Compounds/ZINC IDs | Molecular Interactions | Nature of Interactions | Distance (Å) | Docking Binding Energy (∆G), kcal/mol | Docking Binding Affinity (Kd), M−1 | MM-GBSA Binding Energy, kcal/mol |
|---|---|---|---|---|---|---|
| Control (Hydrolyzed Meropenem) | Lys211:HZ1–Lig:O Asn220:HN–Lig:O Asn220:HD21–Lig:O Lig:H–His250:NE2 Lig:H–His120:NE2 Lig:H–His189:NE2 ZN2–Lig:O Trp93–Lig:C | Hydrogen Bond Hydrogen Bond Hydrogen Bond Hydrogen Bond Hydrogen Bond Hydrogen Bond Other (Metal–Acceptor) Hydrophobic (Pi–Alkyl) | 2.72 1.99 1.85 2.59 2.76 2.32 2.92 5.25 | −6.413 | 5.05 × 104 | −52.971 |
| ZINC10936382 | Gln123:HN–Lig:O Asn220:HD22–Lig:O ZN2–Lig:O ZN1–Lig His122–Lig His250–Lig Val73–Lig His122–Lig Lig–Val73 | Hydrogen Bond Hydrogen Bond Other (Metal–Acceptor) Electrostatic Hydrophobic (Pi–Pi Stacked) Hydrophobic (Pi–Pi Stacked) Hydrophobic (Alkyl) Hydrophobic (Pi–Alkyl) Hydrophobic (Pi–Alkyl) | 2.08 2.93 2.86 4.97 4.94 4.80 3.98 4.26 5.18 | −8.322 | 1.27 × 106 | −61.432 |
| ZINC30479078 | Gln123:HN–Lig:O Asp124:HN–Lig:O Asp124:OD2–Lig Val73:CG1–Lig His122–Lig Lig–Val73 | Hydrogen Bond Hydrogen Bond Electrostatic Hydrophobic (Pi–Sigma) Hydrophobic (Pi–Alkyl) Hydrophobic (Pi–Alkyl) | 2.67 2.18 3.70 3.61 4.04 5.00 | −9.046 | 4.31 × 106 | −70.643 |
| ZINC41493045 | His122:HD1–Lig:O Asn220:HD21–Lig:O ZN1–Lig:O ZN2–Lig His250–Lig Lys211–Lig His250–Lig | Hydrogen Bond Hydrogen Bond Other (Metal–Acceptor) Electrostatic Hydrophobic (Pi–Pi Stacked) Hydrophobic (Alkyl) Hydrophobic (Pi–Alkyl) | 2.66 1.82 3.08 3.76 4.42 5.26 5.15 | −7.714 | 4.54 × 105 | −62.523 |
| ZINC07424911 | Lig:HN–Asp212:OD2 Lig:HN–His250:O Asn220:HN–Lig Asp124:OD2–Lig His250–Lig Trp93–Lig Trp93–Lig Ala215–Lig | Hydrogen Bond Hydrogen Bond Hydrogen Bond Electrostatic Hydrophobic (Pi–Pi Stacked) Hydrophobic (Pi–Pi T–shaped) Hydrophobic (Pi–Pi T–shaped) Hydrophobic (Alkyl) | 2.76 2.72 2.93 4.24 3.97 5.47 4.84 4.34 | −8.254 | 1.13 ×106 | −60.619 |
| ZINC84525623 | Gln123:HN–Lig:O Gln123:HN–Lig:O Asp124:HN–Lig:O Lys211:HZ1–Lig:O Asn220:HN–Lig:O His189:CE1–Lig:O ZN2–Lig Asp124:OD2–Lig Val73:CG1–Lig His250–Lig His250–Lig Lig–Val73 His250–Lig | Hydrogen Bond Hydrogen Bond Hydrogen Bond Hydrogen Bond Hydrogen Bond Carbon–Hydrogen Bond Electrostatic Electrostatic Hydrophobic (Pi–Sigma) Hydrophobic (Pi–Pi Stacked) Hydrophobic (Pi–Pi T-shaped) Hydrophobic (Alkyl) Hydrophobic (Pi–Alkyl) | 2.66 2.16 2.08 2.71 2.10 3.57 3.66 4.33 3.58 5.47 5.26 5.46 4.72 | −8.790 | 2.80 × 106 | −96.388 |
| Substrates | NDM-1 | NDM-1 + ZINC84525623 * | ||||
|---|---|---|---|---|---|---|
| Km (µM) | kcat (s−1) | kcat/Km (µM−1 s−1) | Km (µM) | kcat (s−1) | kcat/Km (µM−1 s−1) | |
| Ampicillin Cefotaxime Imipenem Meropenem Nitrocefin | 88.9 ± 3.1 57.7 ± 3.3 68.4 ± 4.6 58.4 ± 4.1 29.0 ± 2.0 | 438.2 ± 10.5 330.8 ± 16.4 665.8 ± 14.4 285.8 ± 17.3 279.7 ± 18.2 | 4.93 ± 0.21 5.73 ± 0.43 9.73 ± 0.69 4.89 ± 0.45 9.64 ± 0.91 | 111.3 ± 6.2 87.9 ± 5.7 93.6 ± 5.1 80.3 ± 6.4 54.9 ± 4.7 | 97.8 ± 6.1 104.5 ± 6.3 123.1 ± 7.8 125.7 ± 6.0 123.5 ± 6.4 | 0.88 ± 0.07 1.19 ± 0.10 1.32 ± 0.11 1.56 ± 0.14 2.25 ± 0.22 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rehman, M.T.; AlAjmi, M.F.; Hussain, A.; Rather, G.M.; Khan, M.A. High-Throughput Virtual Screening, Molecular Dynamics Simulation, and Enzyme Kinetics Identified ZINC84525623 as a Potential Inhibitor of NDM-1. Int. J. Mol. Sci. 2019, 20, 819. https://doi.org/10.3390/ijms20040819
Rehman MT, AlAjmi MF, Hussain A, Rather GM, Khan MA. High-Throughput Virtual Screening, Molecular Dynamics Simulation, and Enzyme Kinetics Identified ZINC84525623 as a Potential Inhibitor of NDM-1. International Journal of Molecular Sciences. 2019; 20(4):819. https://doi.org/10.3390/ijms20040819
Chicago/Turabian StyleRehman, Md Tabish, Mohamed F AlAjmi, Afzal Hussain, Gulam Mohmad Rather, and Meraj A Khan. 2019. "High-Throughput Virtual Screening, Molecular Dynamics Simulation, and Enzyme Kinetics Identified ZINC84525623 as a Potential Inhibitor of NDM-1" International Journal of Molecular Sciences 20, no. 4: 819. https://doi.org/10.3390/ijms20040819