Thioridazine Enhances P62-Mediated Autophagy and Apoptosis Through Wnt/β-Catenin Signaling Pathway in Glioma Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
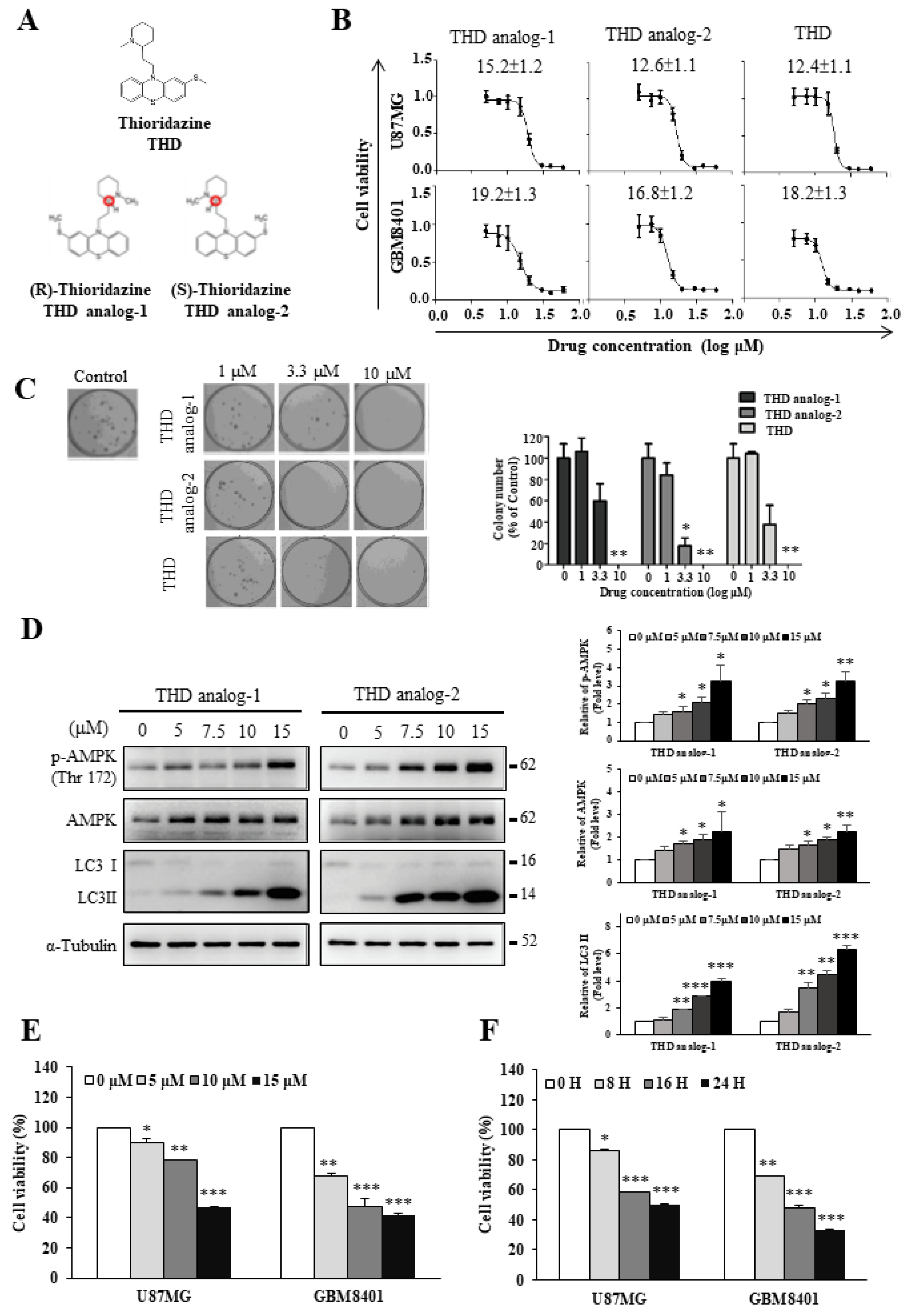
2.1. Both THD and THD Analogs Had Potent Effects on GBM Cell Viability
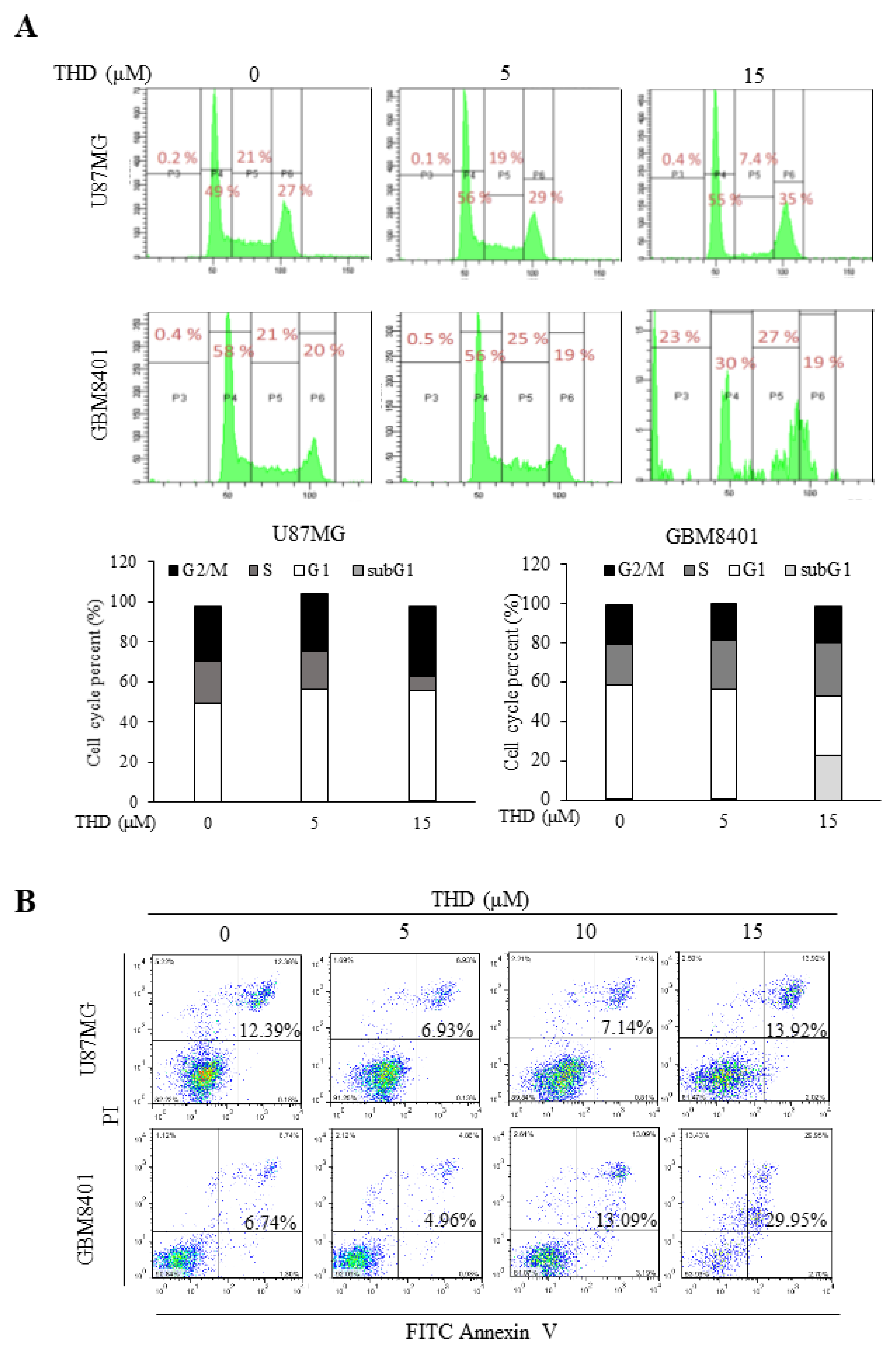
2.2. THD Induced Cell Cycle Arrest and Apoptosis in GBM Cells
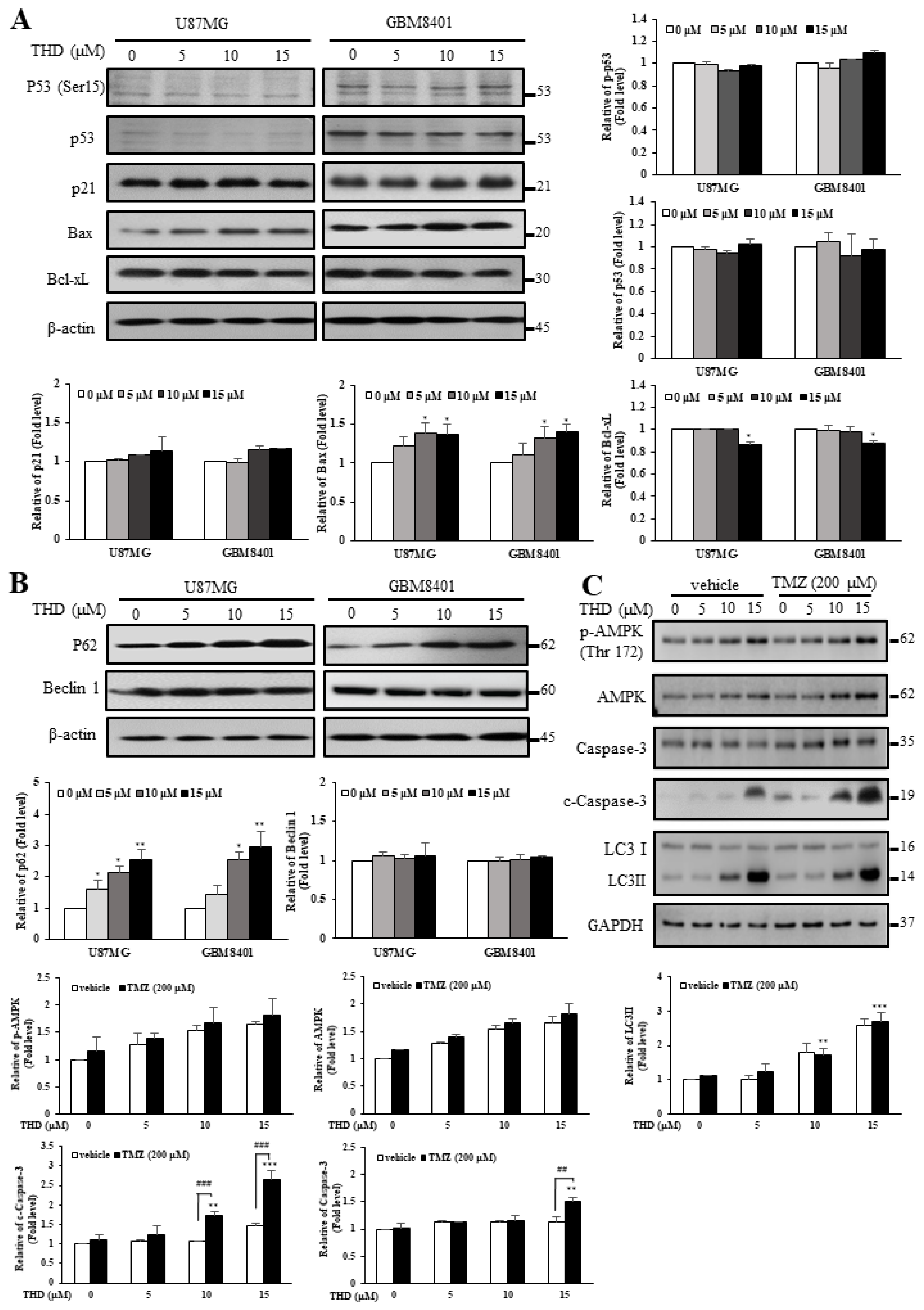
2.3. Effects of THD-Induced Apoptosis and Autophagy Were Mediated by P62 But Not P53 or Beclin-1
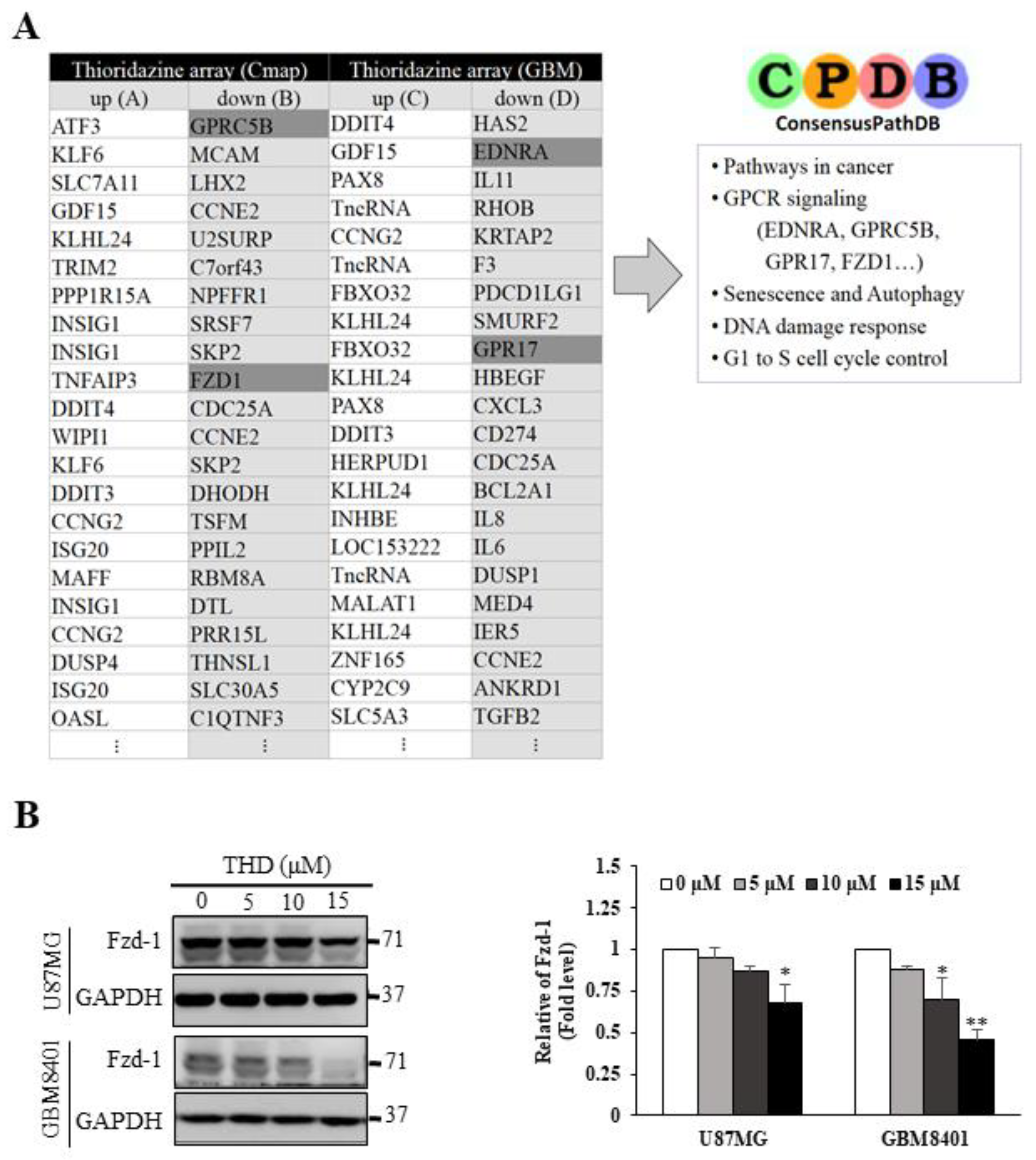
2.4. Pathway Analysis of Differentially Expressed Gene Signatures through Microarray Profiling
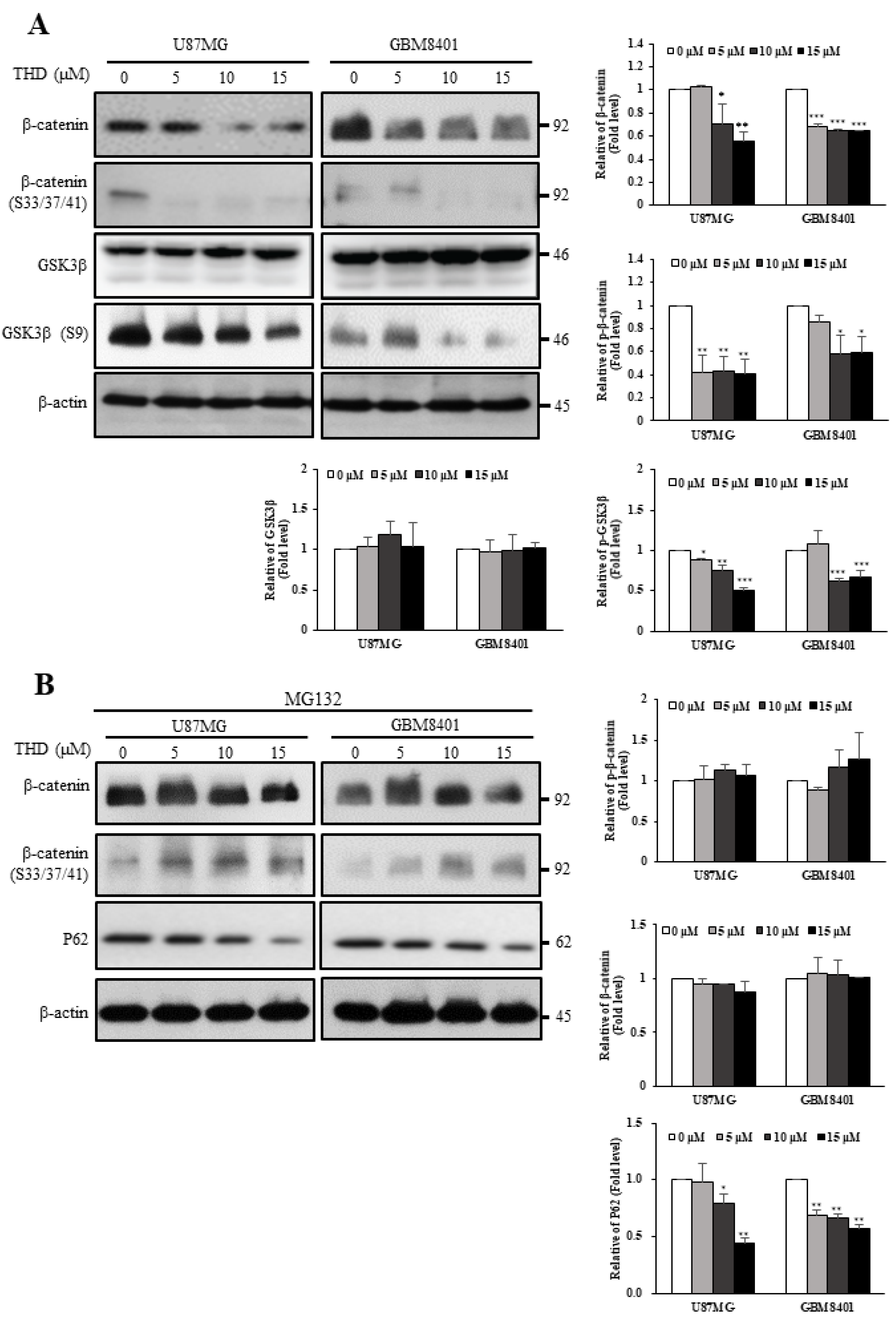
2.5. THD Mediated β-Catenin Degradation through the Wnt/β-Catenin Signaling Pathway
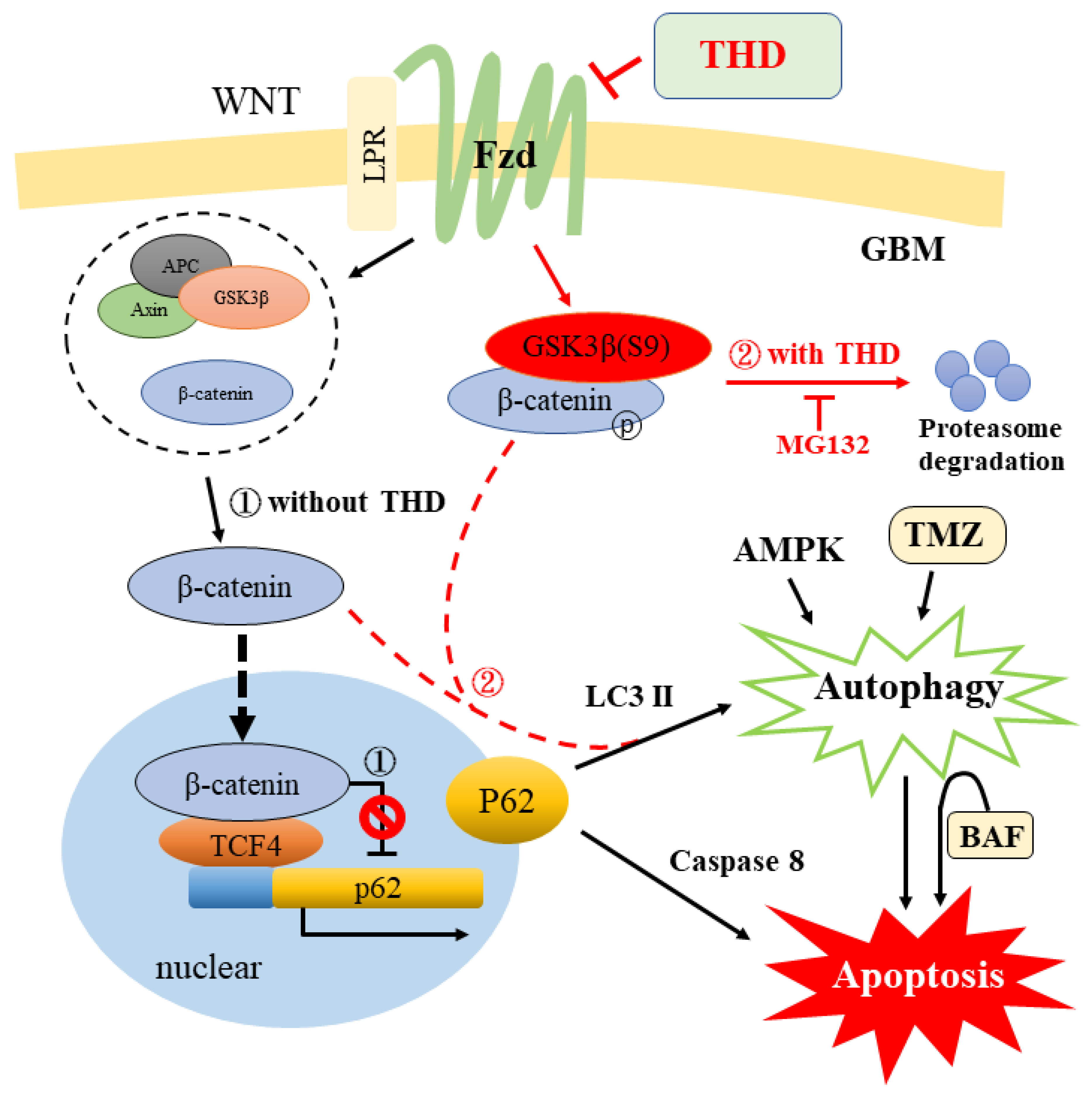
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Chemicals
4.2. Cell Viability Assay
4.3. Apoptosis Analysis
4.4. Cell Cycle Analysis
4.5. Western Blot Analysis
4.6. Clonogenic Assay
4.7. Microarray Analysis
4.8. Consensus PathDB Data Analysis
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Park, J.K.; Hodges, T.; Arko, L.; Shen, M.; Dello Iacono, D.; McNabb, A.; Olsen Bailey, N.; Kreisl, T.N.; Iwamoto, F.M.; Sul, J.; et al. Scale to predict survival after surgery for recurrent glioblastoma multiforme. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 3838–3843. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet. Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Chu, C.W.; Yang, M.C.; Chou, C.H.; Huang, W.S.; Hsiao, B.X.; Wang, Y.T.; Chiou, S.J.; Loh, J.K.; Hong, Y.R. GSK3betamediated Ser156 phosphorylation modulates a BH3like domain in BCL2L12 during TMZinduced apoptosis and autophagy in glioma cells. Int. J. Mol. Med. 2018, 42, 905–918. [Google Scholar] [PubMed]
- Yang, M.C.; Loh, J.K.; Li, Y.Y.; Huang, W.S.; Chou, C.H.; Cheng, J.T.; Wang, Y.T.; Lieu, A.S.; Howng, S.L.; Hong, Y.R.; et al. Bcl2L12 with a BH3-like domain in regulating apoptosis and TMZ-induced autophagy: A prospective combination of ABT-737 and TMZ for treating glioma. Int. J. Oncol. 2015, 46, 1304–1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chio, C.C.; Chen, K.Y.; Chang, C.K.; Chuang, J.Y.; Liu, C.C.; Liu, S.H.; Chen, R.M. Improved effects of honokiol on temozolomide-induced autophagy and apoptosis of drug-sensitive and -tolerant glioma cells. BMC Cancer 2018, 18, 379. [Google Scholar] [CrossRef] [PubMed]
- Knecht, E.; Aguado, C.; Carcel, J.; Esteban, I.; Esteve, J.M.; Ghislat, G.; Moruno, J.F.; Vidal, J.M.; Saez, R. Intracellular protein degradation in mammalian cells: Recent developments. Cell. Mol. Life Sci. CMLS 2009, 66, 2427–2443. [Google Scholar] [CrossRef] [PubMed]
- Doherty, J.; Baehrecke, E.H. Life, death and autophagy. Nat. Cell Biol. 2018, 20, 1110–1117. [Google Scholar] [CrossRef]
- Salminen, A.; Kaarniranta, K.; Haapasalo, A.; Hiltunen, M.; Soininen, H.; Alafuzoff, I. Emerging role of p62/sequestosome-1 in the pathogenesis of Alzheimer’s disease. Prog. Neurobiol. 2012, 96, 87–95. [Google Scholar] [CrossRef]
- Zhang, Y.B.; Gong, J.L.; Xing, T.Y.; Zheng, S.P.; Ding, W. Autophagy protein p62/SQSTM1 is involved in HAMLET-induced cell death by modulating apotosis in U87MG cells. Cell Death Dis. 2013, 4, e550. [Google Scholar] [CrossRef]
- Nager, M.; Sallan, M.C.; Visa, A.; Pushparaj, C.; Santacana, M.; Macia, A.; Yeramian, A.; Canti, C.; Herreros, J. Inhibition of WNT-CTNNB1 signaling upregulates SQSTM1 and sensitizes glioblastoma cells to autophagy blockers. Autophagy 2018, 14, 619–636. [Google Scholar] [CrossRef]
- Zhang, H.; Qi, Y.; Geng, D.; Shi, Y.; Wang, X.; Yu, R.; Zhou, X. Expression profile and clinical significance of Wnt signaling in human gliomas. Oncol. Lett. 2018, 15, 610–617. [Google Scholar] [CrossRef] [PubMed]
- Petherick, K.J.; Williams, A.C.; Lane, J.D.; Ordonez-Moran, P.; Huelsken, J.; Collard, T.J.; Smartt, H.J.; Batson, J.; Malik, K.; Paraskeva, C.; et al. Autolysosomal beta-catenin degradation regulates Wnt-autophagy-p62 crosstalk. EMBO J. 2013, 32, 1903–1916. [Google Scholar] [CrossRef] [PubMed]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H.; Overvatn, A.; Bjorkoy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Ouyang, H.; Zhu, T.; Lindvall, C.; Wang, Y.; Zhang, X.; Yang, Q.; Bennett, C.; Harada, Y.; Stankunas, K.; et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell 2006, 126, 955–968. [Google Scholar] [CrossRef] [PubMed]
- Varga, B.; Csonka, A.; Csonka, A.; Molnar, J.; Amaral, L.; Spengler, G. Possible Biological and Clinical Applications of Phenothiazines. Anticancer Res. 2017, 37, 5983–5993. [Google Scholar] [PubMed]
- Johannessen, T.C.; Hasan-Olive, M.M.; Zhu, H.; Denisova, O.; Grudic, A.; Latif, M.A.; Saed, H.; Varughese, J.K.; Rosland, G.V.; Yang, N.; et al. Thioridazine inhibits autophagy and sensitizes glioblastoma cells to temozolomide. Int. J. Cancer 2018. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Gong, P.; Liu, P.; Zhou, N.; Zhou, Y.; Wang, Y. Thioridazine elicits potent antitumor effects in colorectal cancer stem cells. Oncol. Rep. 2017, 37, 1168–1174. [Google Scholar] [CrossRef]
- Yue, H.; Huang, D.; Qin, L.; Zheng, Z.; Hua, L.; Wang, G.; Huang, J.; Huang, H. Targeting Lung Cancer Stem Cells with Antipsychological Drug Thioridazine. BioMed Res. Int. 2016, 2016, 6709828. [Google Scholar] [CrossRef]
- Kang, S.; Hong, J.; Lee, J.M.; Moon, H.E.; Jeon, B.; Choi, J.; Yoon, N.A.; Paek, S.H.; Roh, E.J.; Lee, C.J.; et al. Trifluoperazine, a Well-Known Antipsychotic, Inhibits Glioblastoma Invasion by Binding to Calmodulin and Disinhibiting Calcium Release Channel IP3R. Mol. Cancer Ther. 2017, 16, 217–227. [Google Scholar] [CrossRef]
- Oliva, C.R.; Zhang, W.; Langford, C.; Suto, M.J.; Griguer, C.E. Repositioning chlorpromazine for treating chemoresistant glioma through the inhibition of cytochrome c oxidase bearing the COX4-1 regulatory subunit. Oncotarget 2017, 8, 37568–37583. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.W.; Liang, Y.H.; Kuo, Y.L.; Chuu, C.P.; Lin, C.Y.; Lee, M.H.; Wu, A.T.; Yeh, C.T.; Chen, E.I.; Whang-Peng, J.; et al. Identification of thioridazine, an antipsychotic drug, as an antiglioblastoma and anticancer stem cell agent using public gene expression data. Cell Death Dis. 2015, 6, e1753. [Google Scholar] [PubMed]
- Kim, E.M.; Jung, C.H.; Kim, J.; Hwang, S.G.; Park, J.K.; Um, H.D. The p53/p21 Complex Regulates Cancer Cell Invasion and Apoptosis by Targeting Bcl-2 Family Proteins. Cancer Res. 2017, 77, 3092–3100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pattingre, S.; Espert, L.; Biard-Piechaczyk, M.; Codogno, P. Regulation of macroautophagy by mTOR and Beclin 1 complexes. Biochimie 2008, 90, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Hao, H.; Zhang, D.; Shi, J.; Wang, Y.; Chen, L.; Guo, Y.; Ma, J.; Jiang, X.; Jiang, H. Sorafenib induces autophagic cell death and apoptosis in hepatic stellate cell through the JNK and Akt signaling pathways. Anti-Cancer Drugs 2016, 27, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Hou, W.; Goldstein, L.A.; Lu, C.; Stolz, D.B.; Yin, X.M.; Rabinowich, H. Involvement of protective autophagy in TRAIL resistance of apoptosis-defective tumor cells. J. Biol. Chem. 2008, 283, 19665–19677. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.B.; Wang, Z.; Shu, F.; Jin, Y.H.; Liu, H.Y.; Wang, Q.J.; Yang, Y. Activation of AMP-activated protein kinase by temozolomide contributes to apoptosis in glioblastoma cells via p53 activation and mTORC1 inhibition. J. Biol. Chem. 2010, 285, 40461–40471. [Google Scholar] [CrossRef] [PubMed]
- O’Callaghan, K.; Kuliopulos, A.; Covic, L. Turning receptors on and off with intracellular pepducins: New insights into G-protein-coupled receptor drug development. J. Biol. Chem. 2012, 287, 12787–12796. [Google Scholar] [CrossRef]
- Bridges, T.M.; Lindsley, C.W. G-protein-coupled receptors: From classical modes of modulation to allosteric mechanisms. ACS Chem. Biol. 2008, 3, 530–541. [Google Scholar] [CrossRef]
- Aberle, H.; Bauer, A.; Stappert, J.; Kispert, A.; Kemler, R. beta-catenin is a target for the ubiquitin-proteasome pathway. EMBO J. 1997, 16, 3797–3804. [Google Scholar] [CrossRef]
- Flahaut, M.; Meier, R.; Coulon, A.; Nardou, K.A.; Niggli, F.K.; Martinet, D.; Beckmann, J.S.; Joseph, J.M.; Muhlethaler-Mottet, A.; Gross, N. The Wnt receptor FZD1 mediates chemoresistance in neuroblastoma through activation of the Wnt/beta-catenin pathway. Oncogene 2009, 28, 2245–2256. [Google Scholar] [CrossRef]
- Cohen, P.; Frame, S. The renaissance of GSK3. Nat. Rev. Mol. Cell Biol. 2001, 2, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Loughery, J.; Cox, M.; Smith, L.M.; Meek, D.W. Critical role for p53-serine 15 phosphorylation in stimulating transactivation at p53-responsive promoters. Nucleic Acids Res. 2014, 42, 7666–7680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjorkoy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Overvatn, A.; Stenmark, H.; Johansen, T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005, 171, 603–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Z.; Li, Y.; Pitti, R.; Lawrence, D.; Pham, V.C.; Lill, J.R.; Ashkenazi, A. Cullin3-based polyubiquitination and p62-dependent aggregation of caspase-8 mediate extrinsic apoptosis signaling. Cell 2009, 137, 721–735. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Han, J.; Lu, C.; Goldstein, L.A.; Rabinowich, H. Autophagic degradation of active caspase-8: A crosstalk mechanism between autophagy and apoptosis. Autophagy 2010, 6, 891–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, J.A.; Fan, Y.; Gandhirajan, R.K.; Madesh, M.; Zong, W.X. Hyperactivation of the mammalian degenerin MDEG promotes caspase-8 activation and apoptosis. J. Biol. Chem. 2013, 288, 2952–2963. [Google Scholar] [CrossRef] [PubMed]
- Zuccarini, M.; Giuliani, P.; Ziberi, S.; Carluccio, M.; Iorio, P.D.; Caciagli, F.; Ciccarelli, R. The Role of Wnt Signal in Glioblastoma Development and Progression: A Possible New Pharmacological Target for the Therapy of This Tumor. Genes 2018, 9, 105. [Google Scholar] [CrossRef]
- Fu, Q.; Chen, K.; Zhu, Q.; Wang, W.; Huang, F.; Miao, L.; Wu, X. beta-catenin promotes intracellular bacterial killing via suppression of Pseudomonas aeruginosa-triggered macrophage autophagy. J. Int. Med. Res. 2017, 45, 556–569. [Google Scholar] [CrossRef]
- Liu, C.; Li, Y.; Semenov, M.; Han, C.; Baeg, G.H.; Tan, Y.; Zhang, Z.; Lin, X.; He, X. Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell 2002, 108, 837–847. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chu, C.-W.; Ko, H.-J.; Chou, C.-H.; Cheng, T.-S.; Cheng, H.-W.; Liang, Y.-H.; Lai, Y.-L.; Lin, C.-Y.; Wang, C.; Loh, J.-K.; et al. Thioridazine Enhances P62-Mediated Autophagy and Apoptosis Through Wnt/β-Catenin Signaling Pathway in Glioma Cells. Int. J. Mol. Sci. 2019, 20, 473. https://doi.org/10.3390/ijms20030473
Chu C-W, Ko H-J, Chou C-H, Cheng T-S, Cheng H-W, Liang Y-H, Lai Y-L, Lin C-Y, Wang C, Loh J-K, et al. Thioridazine Enhances P62-Mediated Autophagy and Apoptosis Through Wnt/β-Catenin Signaling Pathway in Glioma Cells. International Journal of Molecular Sciences. 2019; 20(3):473. https://doi.org/10.3390/ijms20030473
Chicago/Turabian StyleChu, Cheng-Wei, Huey-Jiun Ko, Chia-Hua Chou, Tai-Shan Cheng, Hui-Wen Cheng, Yu-Hsin Liang, Yun-Ling Lai, Chen-Yen Lin, Chihuei Wang, Joon-Khim Loh, and et al. 2019. "Thioridazine Enhances P62-Mediated Autophagy and Apoptosis Through Wnt/β-Catenin Signaling Pathway in Glioma Cells" International Journal of Molecular Sciences 20, no. 3: 473. https://doi.org/10.3390/ijms20030473