Thermo-Responsive Polymer Brushes with Side Graft Chains: Relationship Between Molecular Architecture and Underwater Adherence

Abstract
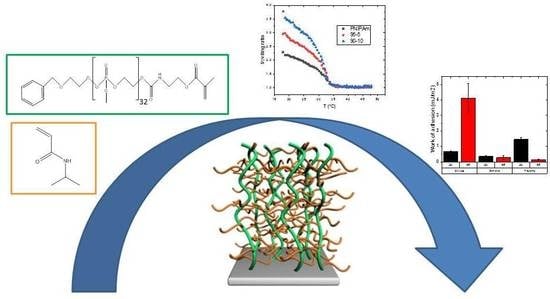
:
1. Introduction
2. Results and Discussion
2.1. Synthesis of PPE Macromonomer
2.2. Synthesis of Graft PNIPAm-g-PMEP Polymer Brushes
2.3. Determination of Turbidity/Cloud Point
2.4. Characterization of the Modified Surfaces
2.5. Underwater Adherence between Native SiO2 Colloidal Probe and Graft Polymer Brushes
2.6. Underwater Adhesion against Different Substrates
3. Materials and Methods
3.1. Materials
3.2. Synthesis of Poly(Methyl Ethylene Phosphate) Methacrylate (PMEP)
3.2.1. 2-Methoxy-2-oxo-1,3,2-dioxaphospholane (MEP)
3.2.2. Polymerization to PMEP
3.3. Synthesis of Grafted Brushes PNIPAm-g-PMEP
3.4. Null Ellipsometry
3.5. In Situ Spectroscopic Ellipsometry
3.6. Dynamic Light Scattering
3.7. Gel Permeation Chromatography
3.8. Turbidity Measurements via UV–Vis Spectroscopy
3.9. Wettability via Captive Bubble Method
3.10. Streaming Potential Measurements
3.11. AFM-CP Force Measurements
3.12. Chemical Modification of the Colloidal Probes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| PNIPAm | Poly(N-isopropylacrylamide) |
| LCST | Lower Critical Solution Temperature |
| PMEP | Poly(methyl ethylene phosphate) |
| Dopa | 3,4-dihydroxyphenylalanine |
| PPEs | Polyphosphoesters |
| PEEP | Poly(ethyl ethylene phosphate) |
| AFM-CP | Atomic force microscopy–colloidal probe technique |
| ROP | Ring-opening polymerization |
| DBU | 1,8-diazabicyclo(5.4.0)undec-7-ene |
| TU | 1-(3,5-bis(trifluoromethyl)phenyl)-3-cyclohexylthiourea |
| PMEP MA | Poly(methyl ethylene phosphate) methacrylate |
| APTES | 3-aminopropyl-triethoxysilane |
| BrIn | A-bromoisobutyryl bromide |
| ARGET- | Activators regenerated by electron transfer |
| ATRP | Atom transfer radical polymerization |
| PMDTA | N,N,N′,N″,N″-pentamethyldiethylenetriamine |
| NMR | Nuclear magnetic resonance |
| IEP | Isoelectric point |
References
- Silverman, H.G.; Roberto, F.F. Understanding marine mussel adhesion. Mar. Biotechnol. 2007, 9, 661–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waite, J.H. Mussel adhesion—Essential footwork. J. Exp. Biol. 2017, 220, 517–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waite, J.H.; Qin, X. Polyphosphoprotein from the adhesive pads of Mytilus edulis. Biochemistry 2001, 40, 2887–2893. [Google Scholar] [CrossRef] [PubMed]
- Stewart, R.J.; Weaver, J.C.; Morse, D.E.; Waite, J.H. The tube cement of Phragmatopoma californica: A solid foam. J. Exp. Biol. 2004, 207, 4727–4734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; Sun, C.; Stewart, R.J.; Waite, J.H. Cement proteins of the tube-building polychaete Phragmatopoma californica. J. Biol. Chem. 2005, 280, 42938–42944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, R.J.; Wang, C.S.; Song, I.T.; Jones, J.P. The role of coacervation and phase transitions in the sandcastle worm adhesive system. Adv. Colloid Interface Sci. 2017, 239, 88–96. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Dellatore, S.M.; Miller, W.M.; Messersmith, P.B. Mussel-inspired surface chemistry for multifunctional coatings. Science 2007, 318, 426–430. [Google Scholar] [CrossRef] [Green Version]
- Ahn, B.K.; Lee, D.W.; Israelachvili, J.N.; Waite, J.H. Surface-initiated self-healing of polymers in aqueous media. Nat. Mater. 2014, 13, 867. [Google Scholar] [CrossRef]
- Jenkins, C.L.; Siebert, H.M.; Wilker, J.J. Integrating mussel chemistry into a bio-based polymer to create degradable adhesives. Macromolecules 2017, 50, 561–568. [Google Scholar] [CrossRef]
- Zhao, Y.; Wu, Y.; Wang, L.; Zhang, M.; Chen, X.; Liu, M.; Fan, J.; Liu, J.; Zhou, F.; Wang, Z. Bio-inspired reversible underwater adhesive. Nat. Commun. 2017, 8, 2218. [Google Scholar] [CrossRef]
- Jones, J.P.; Sima, M.; O’Hara, R.G.; Stewart, R.J. Water-borne endovascular embolics inspired by the undersea adhesive of marine sandcastle worms. Adv. Healthc. Mater. 2016, 5, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Shao, H.; Bachus, K.N.; Stewart, R.J. A water-borne adhesive modeled after the sandcastle glue of P. californica. Macromol. Biosci. 2009, 9, 464–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Q.; Lee, D.W.; Ahn, B.K.; Seo, S.; Kaufman, Y.; Israelachvili, J.N.; Waite, J.H. Underwater contact adhesion and microarchitecture in polyelectrolyte complexes actuated by solvent exchange. Nat. Mater. 2016, 15, 407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waite, J.H. Nature’s underwater adhesive specialist. Int. J. Adhes. Adhes. 1987, 7, 9–14. [Google Scholar] [CrossRef]
- Stewart, R.J.; Ransom, T.C.; Hlady, V. Natural underwater adhesives. J. Polym. Sci. Part B Polym. Phys. 2011, 49, 757–771. [Google Scholar] [CrossRef] [Green Version]
- Milner, S.T. Polymer brushes. Science 1991, 251, 905–914. [Google Scholar] [CrossRef]
- Brittain, W.J.; Minko, S. A structural definition of polymer brushes. J. Polym. Sci. Part A Polym. Chem. 2007, 45, 3505–3512. [Google Scholar] [CrossRef]
- Raynor, J.E.; Capadona, J.R.; Collard, D.M.; Petrie, T.A.; García, A.J. Polymer brushes and self-assembled monolayers: Versatile platforms to control cell adhesion to biomaterials. Biointerphases 2009, 4, FA3–FA16. [Google Scholar] [CrossRef] [Green Version]
- Hucknall, A.; Rangarajan, S.; Chilkoti, A. In pursuit of zero: Polymer brushes that resist the adsorption of proteins. Adv. Mater. 2009, 21, 2441–2446. [Google Scholar] [CrossRef]
- Ladd, J.; Zhang, Z.; Chen, S.; Hower, J.C.; Jiang, S. Zwitterionic polymers exhibiting high resistance to nonspecific protein adsorption from human serum and plasma. Biomacromolecules 2008, 9, 1357–1361. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, M.; Chen, S.; Horbett, T.A.; Ratner, B.D.; Jiang, S. Blood compatibility of surfaces with superlow protein adsorption. Biomaterials 2008, 29, 4285–4291. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, O.J.; Calius, E.; Kennedy, J.V.; Travas-Sejdic, J. Polymer brushes for improvement of dry adhesion in biomimetic dry adhesives. Int. J. Nanotechnol. 2014, 11, 636–644. [Google Scholar] [CrossRef]
- Drechsler, A.; Elmahdy, M.M.; Uhlmann, P.; Stamm, M. PH and salt response of mixed brushes made of oppositely charged polyelectrolytes studied by in situ AFM force measurements and imaging. Langmuir 2018, 34, 4739–4749. [Google Scholar] [CrossRef] [PubMed]
- Drechsler, A.; Synytska, A.; Uhlmann, P.; Elmahdy, M.M.; Stamm, M.; Kremer, F. Interaction forces between microsized silica particles and weak polyelectrolyte brushes at varying pH and salt concentration. Langmuir 2009, 26, 6400–6410. [Google Scholar] [CrossRef] [PubMed]
- Synytska, A.; Svetushkina, E.; Puretskiy, N.; Stoychev, G.; Berger, S.; Ionov, L.; Bellmann, C.; Eichhorn, K.-J.; Stamm, M. Biocompatible polymeric materials with switchable adhesion properties. Soft Matter 2010, 6, 5907–5914. [Google Scholar] [CrossRef]
- Bauer, K.N.; Tee, H.T.; Velencoso, M.M.; Wurm, F.R. Main-chain poly(phosphoester)s: History, syntheses, degradation, bio-and flame-retardant applications. Prog. Polym. Sci. 2017, 73, 61–122. [Google Scholar] [CrossRef]
- Schöttler, S.; Becker, G.; Winzen, S.; Steinbach, T.; Mohr, K.; Landfester, K.; Mailänder, V.; Wurm, F.R. Protein adsorption is required for stealth effect of poly(ethylene glycol)- and poly(phosphoester)-coated nanocarriers. Nat. Nanotechnol. 2016, 11, 372. [Google Scholar] [CrossRef]
- Gao, H.; Tan, Y.; Guan, Q.; Cai, T.; Liang, G.; Wu, Q. Synthesis, characterization and micellization of amphiphilic polyethylene-b-polyphosphoester block copolymers. RSC Adv. 2015, 5, 49376–49384. [Google Scholar] [CrossRef]
- Synytska, A.; Biehlig, E.; Ionov, L. Adaptive PEG–PDMS brushes: Effect of architecture on adhesiveness in air and under water. Macromolecules 2014, 47, 8377–8385. [Google Scholar] [CrossRef]
- Svetushkina, E.; Puretskiy, N.; Ionov, L.; Stamm, M.; Synytska, A. A comparative study on switchable adhesion between thermoresponsive polymer brushes on flat and rough surfaces. Soft Matter 2011, 7, 5691–5696. [Google Scholar] [CrossRef]
- Penczek, S.; Libiszowski, J. Polymerization of 2-methoxy-2-oxo-1,3,2-dioxaphospholane. Kinetics and polymer microstructure. Die Makromol. Chem. 1988, 189, 1765–1785. [Google Scholar] [CrossRef]
- Wang, D.-A.; Williams, C.G.; Li, Q.; Sharma, B.; Elisseeff, J.H. Synthesis and characterization of a novel degradable phosphate-containing hydrogel. Biomaterials 2003, 24, 3969–3980. [Google Scholar] [CrossRef]
- Wang, Y.-C.; Tang, L.-Y.; Sun, T.-M.; Li, C.-H.; Xiong, M.-H.; Wang, J. Self-assembled micelles of biodegradable triblock copolymers based on poly(ethyl ethylene phosphate) and poly(ε-caprolactone) as drug carriers. Biomacromolecules 2007, 9, 388–395. [Google Scholar] [CrossRef] [PubMed]
- Matyjaszewski, K.; Dong, H.; Jakubowski, W.; Pietrasik, J.; Kusumo, A. Grafting from surfaces for “everyone”: ARGET ATRP in the presence of air. Langmuir 2007, 23, 4528–4531. [Google Scholar] [CrossRef] [PubMed]
- Kaneyoshi, H.; Inoue, Y.; Matyjaszewski, K. Synthesis of block and graft copolymers with linear polyethylene segments by combination of degenerative transfer coordination polymerization and atom transfer radical polymerization. Macromolecules 2005, 38, 5425–5435. [Google Scholar] [CrossRef]
- Marschelke, C.; Raguzin, I.; Matura, A.; Fery, A.; Synytska, A. Controlled and tunable design of polymer interface for immobilization of enzymes: Does curvature matter? Soft Matter 2017, 13, 1074–1084. [Google Scholar] [CrossRef] [PubMed]
- Jain, K.; Vedarajan, R.; Watanabe, M.; Ishikiriyama, M.; Matsumi, N. Tunable LCST behavior of poly(N-isopropylacrylamide/ionic liquid) copolymers. Polym. Chem. 2015, 6, 6819–6825. [Google Scholar] [CrossRef]
- Wu, T.; Zhang, Y.; Wang, X.; Liu, S. Fabrication of hybrid silica nanoparticles densely grafted with thermoresponsive poly(N-isopropylacrylamide) brushes of controlled thickness via surface-initiated atom transfer radical polymerization. Chem. Mater. 2007, 20, 101–109. [Google Scholar] [CrossRef]
- Zhang, Y.; Furyk, S.; Bergbreiter, D.E.; Cremer, P.S. Specific ion effects on the water solubility of macromolecules: PNIPAM and the Hofmeister series. J. Am. Chem. Soc. 2005, 127, 14505–14510. [Google Scholar] [CrossRef]
- Kessel, S.; Schmidt, S.; Müller, R.; Wischerhoff, E.; Laschewsky, A.; Lutz, J.-F.O.; Uhlig, K.; Lankenau, A.; Duschl, C.; Fery, A. Thermoresponsive peg-based polymer layers: Surface characterization with AFM force measurements. Langmuir 2009, 26, 3462–3467. [Google Scholar] [CrossRef]
- Biesalski, M.; Johannsmann, D.; Rühe, J.N. Neutral and charged polymer brushes at solid surfaces. Macromol. Symp. 1999, 145, 113–124. [Google Scholar] [CrossRef]
- Furchner, A.; Bittrich, E.; Uhlmann, P.; Eichhorn, K.-J.; Hinrichs, K. In-situ characterization of the temperature-sensitive swelling behavior of poly(N-isopropylacrylamide) brushes by infrared and visible ellipsometry. Thin Solid Films 2013, 541, 41–45. [Google Scholar] [CrossRef]
- Zhuang, P.; Dirani, A.; Glinel, K.; Jonas, A.M. Temperature dependence of the surface and volume hydrophilicity of hydrophilic polymer brushes. Langmuir 2016, 32, 3433–3444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plunkett, K.N.; Zhu, X.; Moore, J.S.; Leckband, D.E. PNIPAM chain collapse depends on the molecular weight and grafting density. Langmuir 2006, 22, 4259–4266. [Google Scholar] [CrossRef] [PubMed]
- Pelton, R. Poly(N-isopropylacrylamide)(PNIPAM) is never hydrophobic. J. Colloid Interface Sci. 2010, 348, 673–674. [Google Scholar] [CrossRef] [PubMed]
- Malham, I.B.; Bureau, L. Density effects on collapse, compression, and adhesion of thermoresponsive polymer brushes. Langmuir 2009, 26, 4762–4768. [Google Scholar] [CrossRef] [Green Version]
- Sun, T.; Wang, G.; Feng, L.; Liu, B.; Ma, Y.; Jiang, L.; Zhu, D. Reversible switching between superhydrophilicity and superhydrophobicity. Angew. Chem. Int. Ed. 2004, 43, 357–360. [Google Scholar] [CrossRef]
- Gilcreest, V.P.; Carroll, W.M.; Rochev, Y.A.; Blute, I.; Dawson, K.A.; Gorelov, A.V. Thermoresponsive poly(N-isopropylacrylamide) copolymers: Contact angles and surface energies of polymer films. Langmuir 2004, 20, 10138–10145. [Google Scholar] [CrossRef]
- Jacobasch, H.-J. Characterization of solid surfaces by electrokinetic measurements. Prog. Org. Coat. 1989, 17, 115–133. [Google Scholar] [CrossRef]
- Delgado, Á.V.; González-Caballero, F.; Hunter, R.; Koopal, L.; Lyklema, J. Measurement and interpretation of electrokinetic phenomena. J. Colloid Interface Sci. 2007, 309, 194–224. [Google Scholar] [CrossRef]
- Binnig, G.; Quate, C.F.; Gerber, C. Atomic force microscope. Phys. Rev. Lett. 1986, 56, 930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ducker, W.A.; Senden, T.J.; Pashley, R.M. Direct measurement of colloidal forces using an atomic force microscope. Nature 1991, 353, 239. [Google Scholar] [CrossRef]
- Butt, H.-J. Measuring electrostatic, van der Waals, and hydration forces in electrolyte solutions with an atomic force microscope. Biophys. J. 1991, 60, 1438–1444. [Google Scholar] [CrossRef] [Green Version]
- Erath, J.; Schmidt, S.; Fery, A. Characterization of adhesion phenomena and contact of surfaces by soft colloidal probe AFM. Soft Matter 2010, 6, 1432–1437. [Google Scholar] [CrossRef]
- Kappl, M.; Butt, H.J. The colloidal probe technique and its application to adhesion force measurements. Part. Part. Syst. Charact. 2002, 19, 129–143. [Google Scholar] [CrossRef]
- Hertz, H. Über die Berührung fester elastischer Körper. J. für die Reine und Angewandte Mathematik 1881, 92, 156–171. [Google Scholar]
- Hertz, H. On the contact of elastic solids. In Miscellaneous Papers; Jones, D.E., Schaott, G.A., Eds.; Macmillan: London, UK; New York, NY, USA, 1896. [Google Scholar]
- Johnson, K.L.; Kendall, K.; Roberts, A. Surface energy and the contact of elastic solids. Proc. R. Soc. A Math. Phys. Sci. 1971, 324, 301–313. [Google Scholar] [CrossRef] [Green Version]
- Derjaguin, B.V.; Muller, V.M.; Toporov, Y.P. Effect of contact deformations on the adhesion of particles. J. Colloid Interface Sci. 1975, 53, 314–326. [Google Scholar] [CrossRef]
- Tabor, D. Surface forces and surface interactions. In Plenary and Invited Lectures; Kerker, M., Zettlemoyer, A.C., Rowell, R.L., Eds.; Academic Press: Cambridge, MA, USA, 1977; pp. 3–14. [Google Scholar]
- Senden, T.; Di Meglio, J.-M.; Auroy, P. Anomalous adhesion in adsorbed polymer layers. Eur. Phys. J. B Condens. Matter Complex Syst. 1998, 3, 211–216. [Google Scholar] [CrossRef]
- Clement, B.; Grignard, B.; Koole, L.; Jérôme, C.; Lecomte, P. Metal-free strategies for the synthesis of functional and well-defined polyphosphoesters. Macromolecules 2012, 45, 4476–4486. [Google Scholar] [CrossRef]
- Hutter, J.L.; Bechhoefer, J. Calibration of atomic-force microscope tips. Rev. Sci. Instrum. 1993, 64, 1868–1873. [Google Scholar] [CrossRef] [Green Version]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Layer | Thickness (nm) |
|---|---|
| SiO2 | 1.4 ± 0.2 |
| 3-aminopropyl- triethoxysilane | 0.7 ± 0.1 |
| α-bromoisobutyryl bromide | 0.5 ± 0.1 |
| Ratio Pnipam:Phosphate Units | σ (Chains/nm2) | D (nm) | Mn (kDa) | Dry Thickness (nm) |
|---|---|---|---|---|
| 100:0 | 0.22 | 2.1 | 83 | 30.4 ± 0.2 |
| 95:5 | 0.26 | 1.8 | 55 | 35.1 ± 0.7 |
| 90:10 | 0.23 | 2.0 | 69 | 32.9 ± 0.9 |
| Ratio PNIPAm:Phosphate Units | Dry Thickness (nm) | Underwater Thickness at 20 °C (nm) |
|---|---|---|
| 100:0 | 30.4 ± 0.2 | 69.5 ± 1.8 |
| 95:5 | 35.1 ± 0.7 | 104.8 ± 3.5 |
| 90:10 | 32.9 ± 0.9 | 120.7 ± 4.1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sidoli, U.; Tee, H.T.; Raguzin, I.; Mühldorfer, J.; Wurm, F.R.; Synytska, A. Thermo-Responsive Polymer Brushes with Side Graft Chains: Relationship Between Molecular Architecture and Underwater Adherence. Int. J. Mol. Sci. 2019, 20, 6295. https://doi.org/10.3390/ijms20246295
Sidoli U, Tee HT, Raguzin I, Mühldorfer J, Wurm FR, Synytska A. Thermo-Responsive Polymer Brushes with Side Graft Chains: Relationship Between Molecular Architecture and Underwater Adherence. International Journal of Molecular Sciences. 2019; 20(24):6295. https://doi.org/10.3390/ijms20246295
Chicago/Turabian StyleSidoli, Ugo, Hisaschi T. Tee, Ivan Raguzin, Jakob Mühldorfer, Frederik R. Wurm, and Alla Synytska. 2019. "Thermo-Responsive Polymer Brushes with Side Graft Chains: Relationship Between Molecular Architecture and Underwater Adherence" International Journal of Molecular Sciences 20, no. 24: 6295. https://doi.org/10.3390/ijms20246295