The Dual Interactions of p53 with MDM2 and p300: Implications for the Design of MDM2 Inhibitors

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. p53_TAD1 Binding with Mdm2
2.2. p53_TAD1 Binding with p300_Taz2
2.3. Comparison of p53_TAD1 Binding with Mdm2 and p300_Taz2
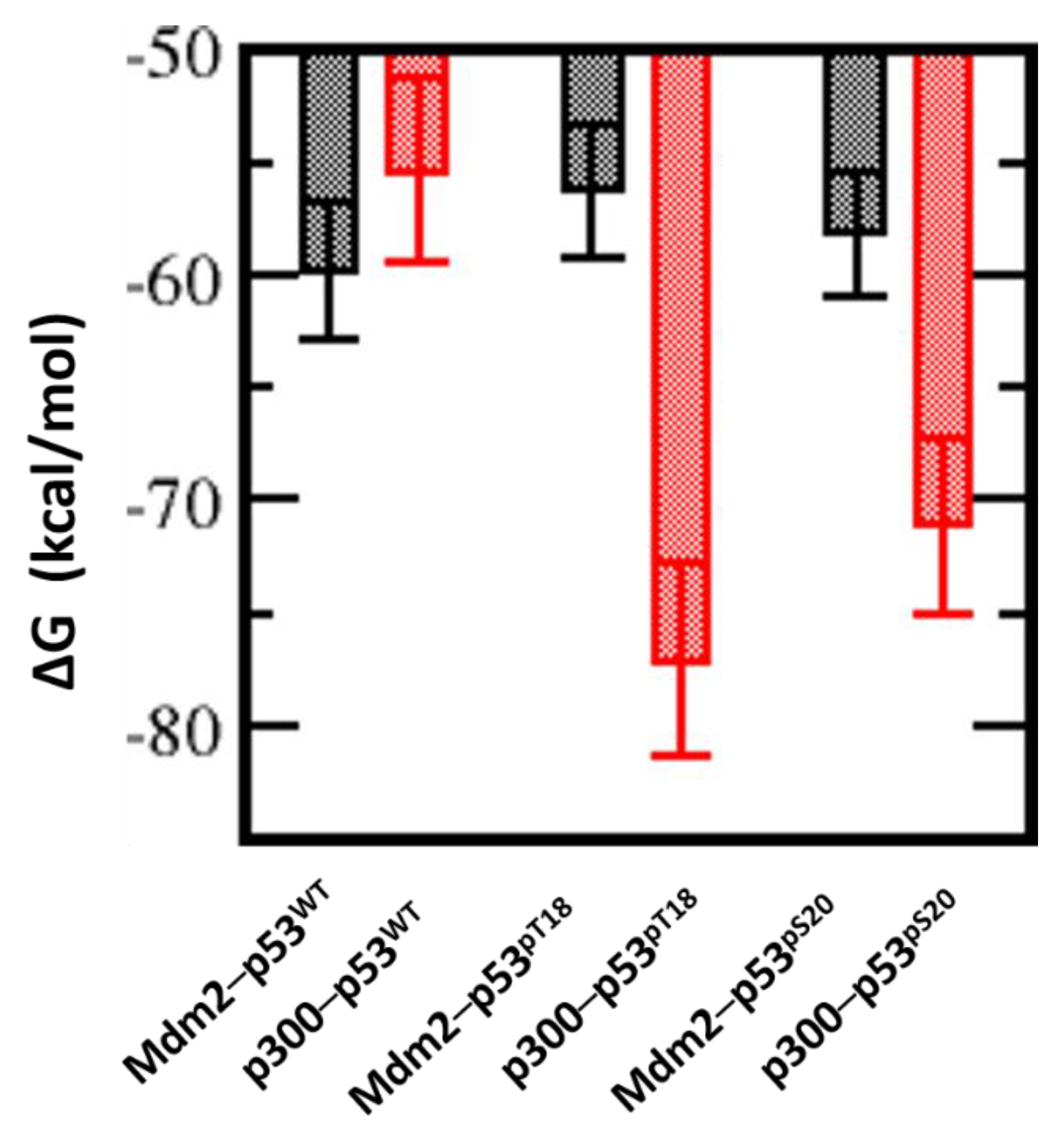
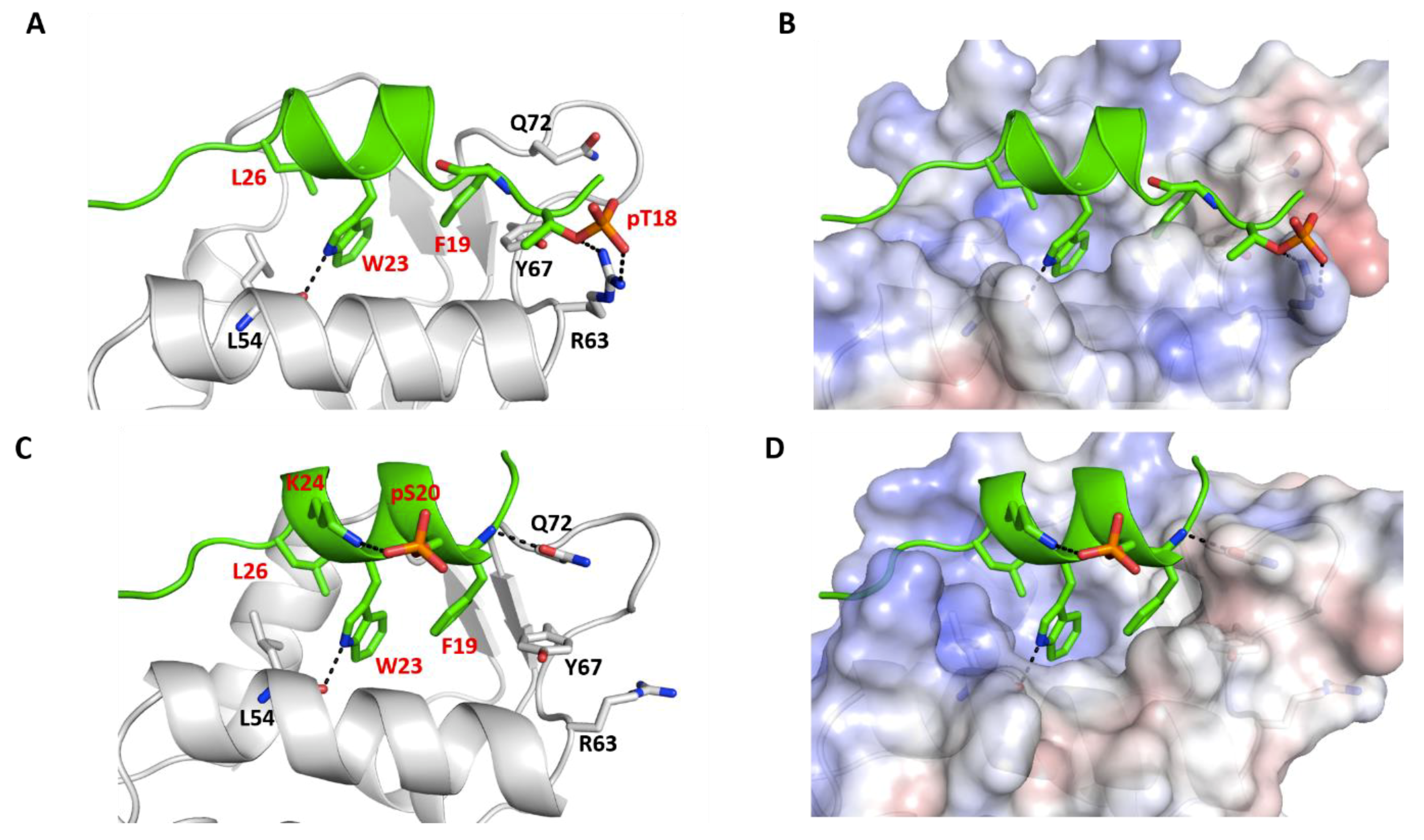
2.4. Effect of Phosphorylation on the Binding of Peptides with p300_Taz2 and Mdm2
2.5. Effect of Phosphorylation on the Structural Dynamics of p53 Peptide
2.6. Structure and Dynamics of ATSP_7041–Mdm2 and ATSP_7041–p300 Complexes
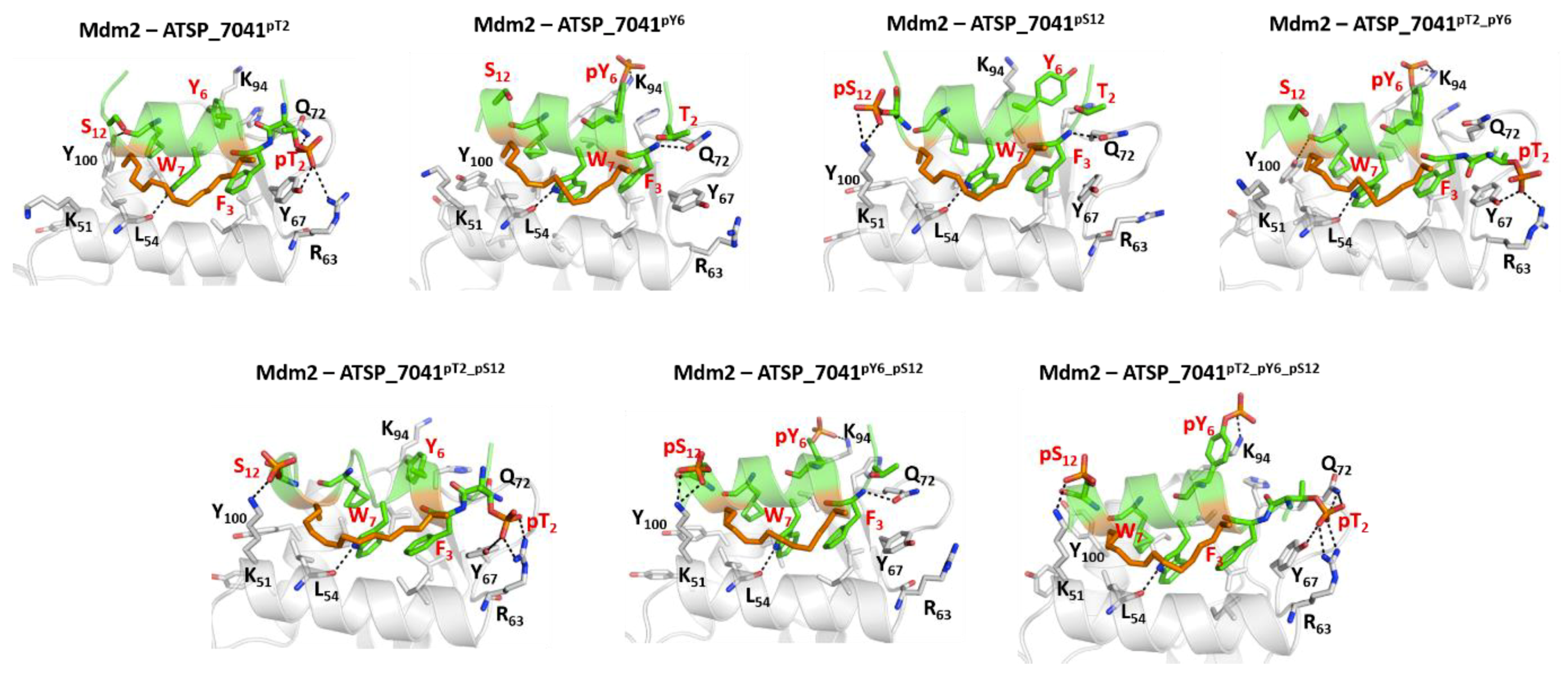
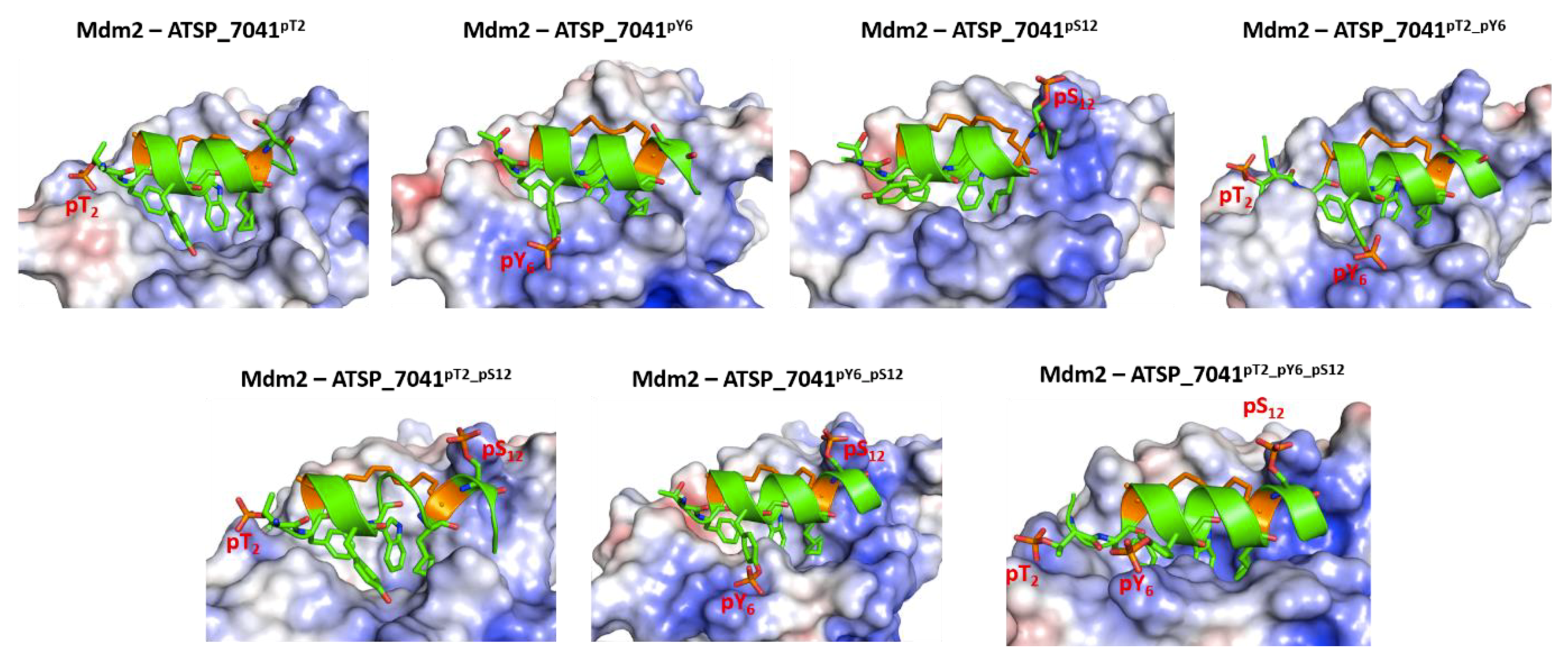
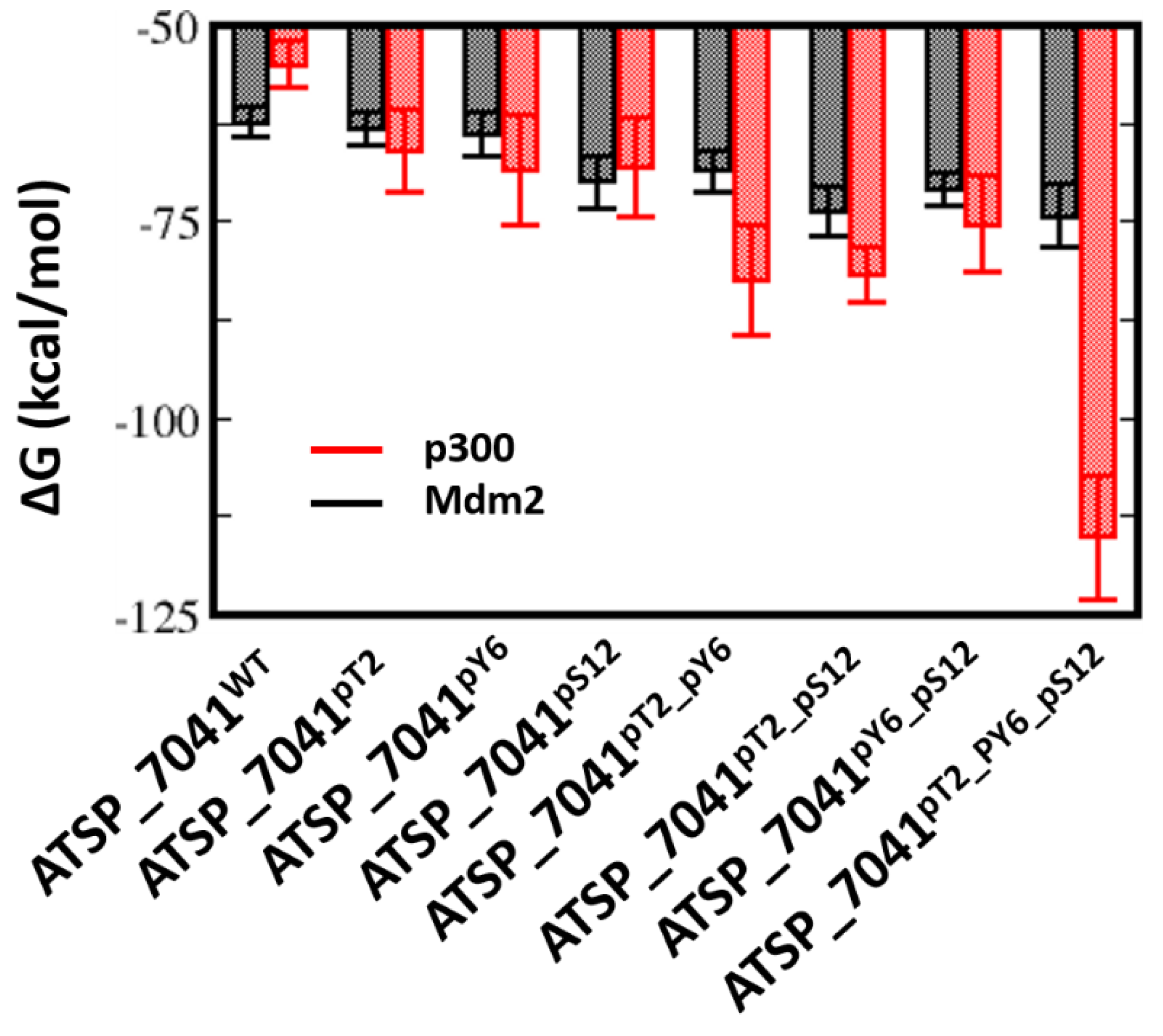
2.7. Effect of Phosphorylation on the Binding of ATSP_7041 with p300 and Mdm2
2.8. Effect of Phosphorylation on the Structural Dynamics of the ATSP_7014 Peptide
3. Discussion
4. Materials and Methods
4.1. Model Preparation
4.2. Molecular Dynamics (MD) Simulations
4.3. Binding Energy Calculations and Energy Decomposition Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Toledo, F.; Wahl, G.M. Regulating the p53 pathway: In vitro hypotheses, in vivo veritas. Nat. Rev. Cancer 2006, 6, 909–923. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.J.; Lain, S.; Verma, C.S.; Fersht, A.R.; Lane, D.P. Awakening guardian angels: Drugging the p53 pathway. Nat. Rev. Cancer 2009, 9, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.J.; Cheok, C.F.; Verma, C.S.; Lane, D.P. Reactivation of p53: From peptides to small molecules. Trends Pharmacol. Sci. 2011, 32, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.P. Cancer. p53, guardian of the genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef]
- Cheok, C.F.; Lane, D.P. Exploiting the p53 Pathway for Therapy. Cold Spring Harb. Perspect. Med. 2017, 7, a02631. [Google Scholar] [CrossRef]
- Freed-Pastor, W.A.; Prives, C. Mutant p53: One name, many proteins. Genes Dev. 2012, 26, 1268–1286. [Google Scholar] [CrossRef]
- Baud, M.G.J.; Bauer, M.R.; Verduci, L.; Dingler, F.A.; Patel, K.J.; Horil Roy, D.; Joerger, A.C.; Fersht, A.R. Aminobenzothiazole derivatives stabilize the thermolabile p53 cancer mutant Y220C and show anticancer activity in p53-Y220C cell lines. Euro. J. Med. Chem. 2018, 152, 101–114. [Google Scholar] [CrossRef]
- Wassman, C.D.; Baronio, R.; Demir, Ö.; Wallentine, B.D.; Chen, C.K.; Hall, L.V.; Salehi, F.; Lin, D.W.; Chung, B.P.; Hatfield, G.W.; et al. Computational identification of a transiently open L1/S3 pocket for reactivation of mutant p53. Nat. Commun. 2013, 4, 1407. [Google Scholar] [CrossRef]
- Bykov, V.J.N.; Eriksson, S.E.; Bianchi, J.; Wiman, K.G. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer 2018, 18, 89–102. [Google Scholar] [CrossRef]
- Khoo, K.H.; Verma, C.S.; Lane, D.P. Drugging the p53 pathway: Understanding the route to clinical efficacy. Nat. Rev. Drug Discov. 2014, 13, 217–236. [Google Scholar] [CrossRef]
- Meek, D.W.; Anderson, C.W. Posttranslational Modification of p53: Cooperative Integrators of Function. Cold Spring Harb. Perspect. Biol. 2009, 1, a000950. [Google Scholar] [CrossRef]
- Meek, D.W.; Hupp, T.R. The regulation of MDM2 by multisite phosphorylation--opportunities for molecular-based intervention to target tumours? Semin. Cancer Biol. 2010, 20, 19–28. [Google Scholar] [CrossRef]
- Barlev, N.A.; Liu, L.; Chehab, N.H.; Mansfield, K.; Harris, K.G.; Halazonetis, T.D.; Berger, S.L. Acetylation of p53 activates transcription through recruitment of coactivators/histone acetyltransferases. Mol. Cell 2001, 8, 1243–1254. [Google Scholar] [CrossRef]
- Liu, G.; Xia, T.; Chen, X. The activation domains, the proline-rich domain, and the C-terminal basic domain in p53 are necessary for acetylation of histones on the proximal p21 promoter and interaction with p300/CREB-binding protein. J. Biol. Chem. 2003, 278, 17557–17565. [Google Scholar] [CrossRef]
- Gu, W.; Roeder, R.G. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 1997, 90, 595–606. [Google Scholar] [CrossRef]
- Ito, A.; Lai, C.H.; Zhao, X.; Saito, S.; Hamilton, M.H.; Appella, E.; Yao, T.P. p300/CBP-mediated p53 acetylation is commonly induced by p53-activating agents and inhibited by MDM2. EMBO J. 2001, 20, 1331–1340. [Google Scholar] [CrossRef]
- Sakaguchi, K.; Saito, S.; Higashimoto, Y.; Roy, S.; Anderson, C.W.; Appella, E. Damage-mediated phosphorylation of human p53 threonine 18 through a cascade mediated by a casein 1-like kinase. Effect on Mdm2 binding. J. Biol. Chem. 2000, 275, 9278–9283. [Google Scholar] [CrossRef]
- Bedford, D.C.; Kasper, L.H.; Fukuyama, T.; Brindle, P.K. Target gene context influences the transcriptional requirement for the KAT3 family of CBP and p300 histone acetyltransferases. Epigenetics 2010, 5, 9–15. [Google Scholar] [CrossRef]
- Dyson, H.J.; Wright, P.E. Role of intrinsic protein disorder in the function and interactions of the transcriptional coactivators CREB-binding protein (CBP) and p300. J. Biol. Chem. 2016, 291, 6714–6722. [Google Scholar] [CrossRef]
- Lee, C.W.; Sørensen, T.S.; Shikama, N.; La Thangue, N.B. Functional interplay between p53 and E2F through co-activator p300. Oncogene 1998, 16, 2695–2710. [Google Scholar] [CrossRef]
- Perissi, V.; Dasen, J.S.; Kurokawa, R.; Wang, Z.; Korzus, E.; Rose, D.W.; Glass, C.K.; Rosenfeld, M.G. Factor-specific modulation of CREB-binding protein acetyltransferase activity. Proc. Natl. Acad. Sci. USA 1999, 96, 3652–3657. [Google Scholar] [CrossRef]
- Avantaggiati, M.L.; Ogryzko, V.; Gardner, K.; Giordano, A.; Levine, A.S.; Kelly, K. Recruitment of p300/CBP in p53-dependent signal pathways. Cell 1997, 89, 1175–1184. [Google Scholar] [CrossRef]
- Grossman, S.R.; Perez, M.; Kung, A.L.; Joseph, M.; Mansur, C.; Xiao, Z.X.; Kumar, S.; Howley, P.M.; Livingston, D.M. p300/MDM2 complexes participate in MDM2-mediated p53 degradation. Mol. Cell 1998, 2, 405–415. [Google Scholar] [CrossRef]
- Candau, R.; Scolnick, D.M.; Darpino, P.; Ying, C.Y.; Halazonetis, T.D.; Berger, S.L. Two tandem and independent sub-activation domains in the amino terminus of p53 require the adaptor complex for activity. Oncogene 1997, 15, 807–816. [Google Scholar] [CrossRef]
- Polley, S.; Guha, S.; Kar, S.; Roy, N.S.; Sakaguchi, K.; Chuman, Y.S.V.; Kundu, T.; Roy, S. Differential recognition of phosphorylated transactivation domains of p53 by different p300 domains. J. Mol. Biol. 2008, 376, 8–12. [Google Scholar] [CrossRef]
- Teufel, D.P.; Freund, S.M.; Bycroft, M.; Fersht, A.R. Four domains of p300 each bind tightly to a sequence spanning both transactivation subdomains of p53. Proc. Natl. Acad. Sci. USA 2007, 104, 7009–7014. [Google Scholar] [CrossRef]
- Jenkins, L.M.M.; Yamaguchi, H.; Hayashi, R.; Cherry, S.; Tropea, J.E.; Miller, M.; Wlodawer, A.; Appella, E.; Mazur, S.J. Two distinct motifs within the p53 transactivation domain bind to the Taz2 domain of p300 and are differentially affected by phosphorylation. Biochemistry 2009, 48, 1244–1255. [Google Scholar] [CrossRef]
- Espinosa, J.M.; Emerson, B.M. Transcriptional regulation by p53 through intrinsic DNA/chromatin binding and site-directed cofactor recruitment. Mol. Cell 2001, 8, 57–69. [Google Scholar] [CrossRef]
- Hsu, C.H.; Chang, M.D.; Tai, K.Y.; Yang, Y.T.; Wang, P.S.; Chen, C.J.; Wang, Y.H.; Lee, S.C.; Wu, C.W.; Juan, L.J. HCMV IE2-mediated inhibition of HAT activity downregulates p53 function. EMBO J. 2004, 23, 2269–2280. [Google Scholar] [CrossRef]
- Luo, J.; Nikolaev, A.Y.; Imai, S.; Chen, D.; Su, F.; Shiloh, A.; Guarente, L.; Gu, W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell 2001, 107, 137–148. [Google Scholar] [CrossRef] [Green Version]
- Vaziri, H.; Dessain, S.K.; Ng Eaton, E.; Imai, S.I.; Frye, R.A.; Pandita, T.K.; Guarente, L.; Weinberg, R.A. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 2001, 107, 149–159. [Google Scholar] [CrossRef] [Green Version]
- Scolnick, D.M.; Chehab, N.H.; Stavridi, E.S.; Lien, M.C.; Caruso, L.; Moran, E.; Berger, S.L.; Halazonetis, T.D. CREB-binding protein and p300/CBP-associated factor are transcriptional coactivators of the p53 tumor suppressor protein. Cancer Res. 1997, 57, 3693–3696. [Google Scholar]
- Ferreon, J.C.; Lee, C.W.; Arai, M.; Martinez-Yamout, M.A.; Dyson, H.J.; Wright, P.E. Cooperative regulation of p53 by modulation of ternary complex formation with CBP/p300 and HDM2. Proc. Natl. Acad. Sci. USA 2009, 106, 6591–6596. [Google Scholar] [CrossRef] [Green Version]
- Grossman, S.R. p300/CBP/p53 interaction and regulation of the p53 response. Eur. J. Biochem. 2001, 268, 2773–2778. [Google Scholar] [CrossRef]
- Teufel, D.P.; Bycroft, M.; Fersht, A.R. Regulation by phosphorylation of the relative affinities of the N-terminal transactivation domains of p53 for p300 domains and Mdm2. Oncogene 2009, 28, 2112–2118. [Google Scholar] [CrossRef] [Green Version]
- Krois, A.S.; Ferreon, J.C.; Martinez-Yamout, M.A.; Dyson, H.J.; Wright, P.E. Recognition of the disordered p53 transactivation domain by the transcriptional adapter zinc finger domains of CREB-binding protein. Proc. Natl. Acad. Sci. USA 2016, 113, E1853–E1862. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.W.; Ferreon, J.C.; Ferreon, A.C.; Arai, M.; Wright, P.E. Graded enhancement of p53 binding to CREB-binding protein (CBP) by multisite phosphorylation. Proc. Natl. Acad. Sci. USA 2010, 107, 19290–19295. [Google Scholar] [CrossRef] [Green Version]
- Feng, H.; Jenkins, L.M.; Durell, S.R.; Hayashi, R.; Mazur, S.J.; Cherry, S.; Tropea, J.E.; Miller, M.; Wlodawer, A.; Appella, E.; et al. Structural basis for p300 Taz2-p53 TAD1 binding and modulation by phosphorylation. Structure 2009, 17, 202–210. [Google Scholar] [CrossRef] [Green Version]
- Dunker, A.K.; Cortese, M.S.; Romero, P.; Iakoucheva, L.; Uversky, V.N. Flexible nets. The roles of intrinsic disorder in protein interaction networks. FEBS J. 2005, 272, 5129–5148. [Google Scholar] [CrossRef]
- Vise, P.; Baral, B.; Stancik, A.; Lowry, D.F.; Daughdrill, G.W. Identifying long-range structure in the intrinsically unstructured transactivation domain of p53. Proteins 2007, 67, 526–530. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.J.; Quah, S.T.; Jong, J.; Goh, A.M.; Chiam, P.C.; Khoo, K.H.; Choong, M.L.; Lee, M.A.; Yurlova, L.; Zolghadr, K.; et al. Stapled peptides with improved potency and specificity that activate p53. ACS Chem. Biol. 2013, 8, 506–512. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.S.; Graves, B.; Guerlavais, V.; Tovar, C.; Packman, K.; To, K.H.; Olson, K.A.; Kesavan, K.; Gangurde, P.; Mukherjee, A.; et al. Stapled a-helical peptide drug development: A potent dual inhibitor of MDM2 and MDM4 for p53-dependent cancer therapy. Proc. Natl. Acad. Sci. USA 2013, 110, E3445–E3455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tisato, V.; Voltan, R.; Gonelli, A.; Secchiero, P.; Zauli, G. MDM2/X inhibitors under clinical evaluation: Perspectives for the management of hematological malignancies and pediatric cancer. J. Hematol. Oncol. 2017, 10, 133. [Google Scholar] [CrossRef] [Green Version]
- Tan, Y.S.; Mhoumadi, Y.; Verma, C.S. Roles of computational modelling in understanding p53 structure, biology, and its therapeutic targeting. J. Mol. Cell Biol. 2019, 11, 306–316. [Google Scholar] [CrossRef] [Green Version]
- De Guzman, R.N.; Liu, H.Y.; Martinez-Yamout, M.; Dyson, H.J.; Wright, P.E. Solution structure of the TAZ2 (CH3) domain of the transcriptional adaptor protein CBP. J. Mol. Biol. 2000, 303, 243–253. [Google Scholar] [CrossRef]
- Lee, H.J.; Srinivasan, D.; Coomber, D.; Lane, D.P.; Verma, C.S. Modulation of the p53-MDM2 interaction by phosphorylation of Thr18: A computational study. Cell Cycle 2007, 6, 2604–2611. [Google Scholar] [CrossRef]
- Yadahalli, S.; Neira, J.L.; Johnson, C.M.; Tan, Y.S.; Rowling, P.J.E.; Chattopadhyay, A.; Verma, C.S.; Itzhaki, L.S. Kinetic and thermodynamic effects of phosphorylation on p53 binding to MDM2. Sci. Rep. 2019, 9, 693. [Google Scholar] [CrossRef]
- García-Echeverría, C.; Chène, P.; Blommers, M.J.J.; Furet, P. Discovery of potent antagonists of the interaction between human double minute 2 and tumor suppressor p53. J. Med. Chem. 2000, 43, 3205–3208. [Google Scholar] [CrossRef]
- Patridge, A.; Kaan, H.Y.K.; Juang, Y.C.; Sadruddin, A.; Lim, S.; Brown, C.J.; Ng, S.; Thean, D.; Ferrer, F.; Johannes, C.; et al. Incorporation of Putative Helix-Breaking Amino Acids in the Design of Novel Stapled Peptides: Exploring Biophysical and Cellular Permeability Properties. Molecules 2019, 24, 2292. [Google Scholar] [CrossRef] [Green Version]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; et al. Amber 18; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Tan, Y.S.; Reeks, J.; Brown, C.J.; Thean, D.; Ferrer Gago, F.J.; Yuen, T.Y.; Goh, E.T.; Lee, X.E.; Jennings, C.E.; Joseph, T.L.; et al. Benzene Probes in Molecular Dynamics Simulations Reveal Novel Binding Sites for Ligand Design. J. Phys. Chem. Lett. 2016, 7, 3452–3457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Homeyer, N.; Horn, A.H.C.; Lanig, H.; Sticht, H. AMBER force field parameters for phosphorylated amino acids in different protonation states: Phosphoserine, phosphothreonine, phosphotyrosine and phosphohistidine. J. Mol. Model. 2006, 12, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theor. Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N_log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Kannan, S.; Zacharias, M. Enhanced sampling of peptide and protein conformations using replica exchange simulations with a peptide backbone biasing-potential. Proteins 2007, 66, 697–706. [Google Scholar] [CrossRef]
- Ostermeir, K.; Zacharias, M. Hamiltonian replica-exchange simulations with adaptive biasing of peptide backbone and side chain dihedral angles. J. Comput. Chem. 2014, 35, 150–158. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- De Lano, W. The PyMOL Molecular Graphics System; De Lano Scientific: San Carlos, CA, USA, 2002. [Google Scholar]
- Homeyer, N.; Gohlke, H. Free Energy Calculations by the Molecular Mechanics Poisson-Boltzmann Surface Area Method. Mol. Inform. 2012, 31, 114–122. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the molecular mechanics/Poisson Boltzmann surface area and molecular mechanics/generalized Born surface area methods. II. The accuracy of ranking poses generated from docking. J. Comput. Chem. 2011, 32, 866–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kannan, S.; Partridge, A.W.; Lane, D.P.; Verma, C.S. The Dual Interactions of p53 with MDM2 and p300: Implications for the Design of MDM2 Inhibitors. Int. J. Mol. Sci. 2019, 20, 5996. https://doi.org/10.3390/ijms20235996
Kannan S, Partridge AW, Lane DP, Verma CS. The Dual Interactions of p53 with MDM2 and p300: Implications for the Design of MDM2 Inhibitors. International Journal of Molecular Sciences. 2019; 20(23):5996. https://doi.org/10.3390/ijms20235996
Chicago/Turabian StyleKannan, Srinivasaraghavan, Anthony W. Partridge, David P. Lane, and Chandra S. Verma. 2019. "The Dual Interactions of p53 with MDM2 and p300: Implications for the Design of MDM2 Inhibitors" International Journal of Molecular Sciences 20, no. 23: 5996. https://doi.org/10.3390/ijms20235996