Species Identification of Oaks (Quercus L., Fagaceae) from Gene to Genome


Abstract
:1. Introduction
2. Results
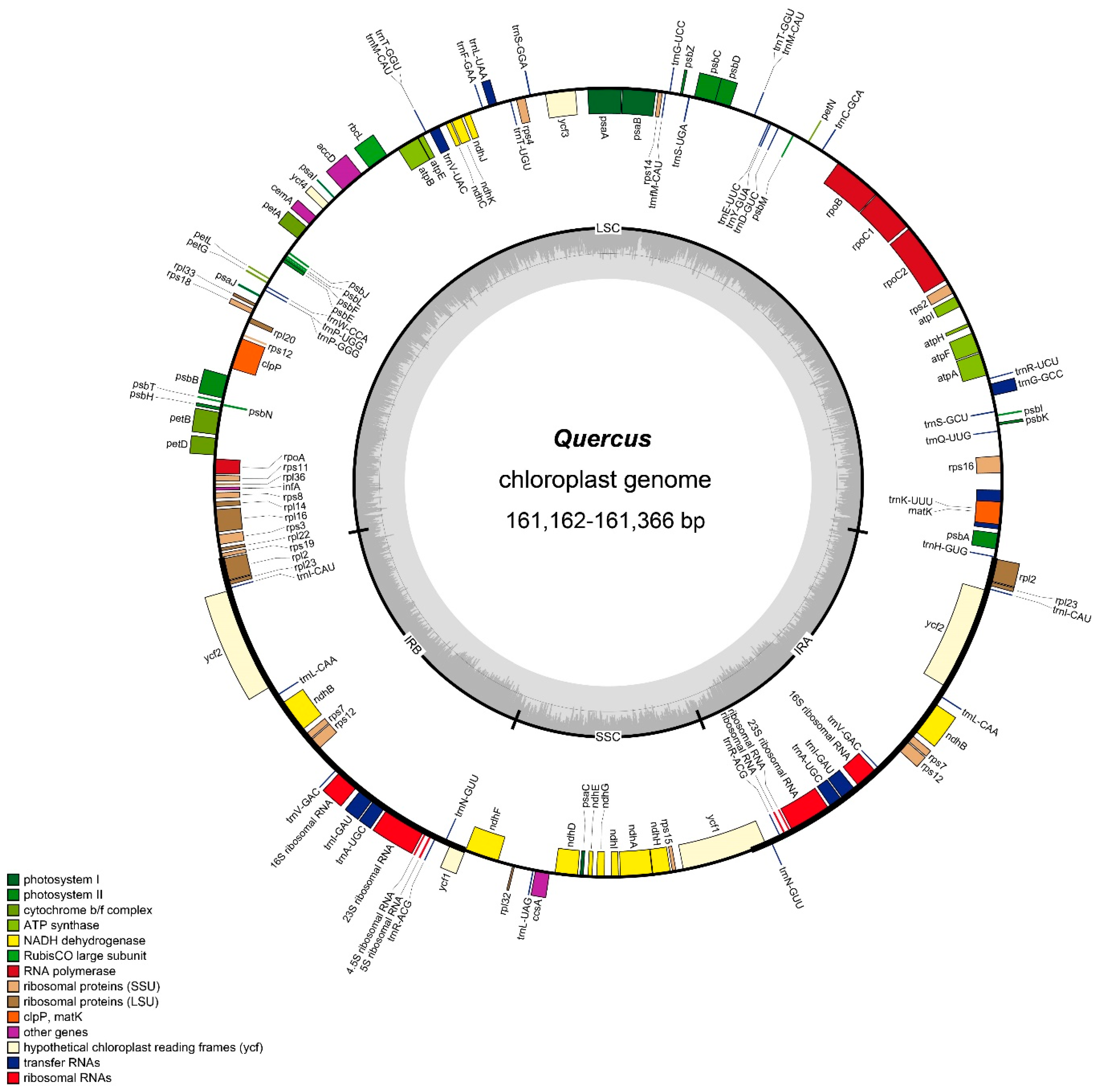
2.1. General Features of the Quercus Chloroplast Genome
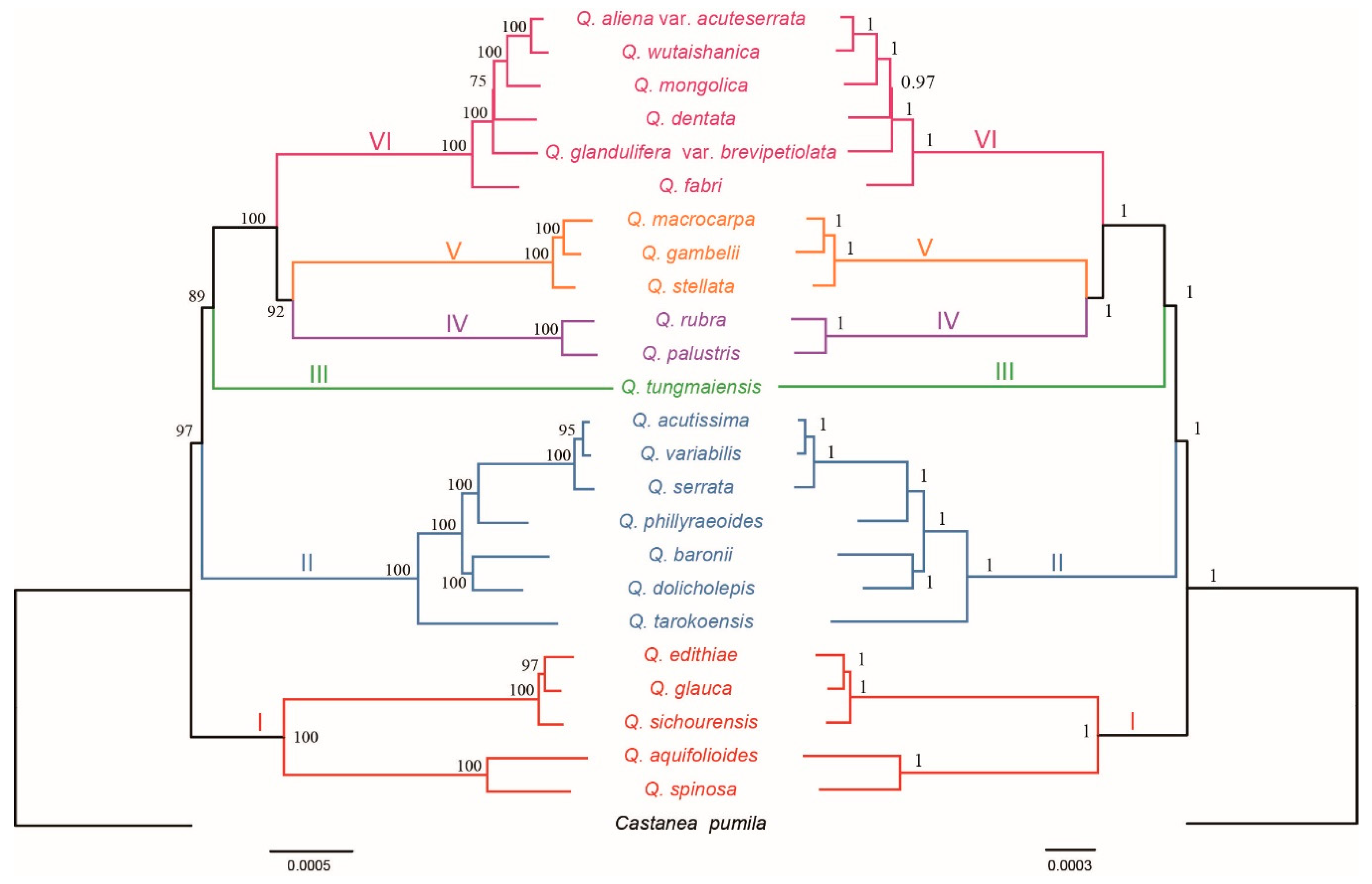
2.2. Phylogenetic Analyses
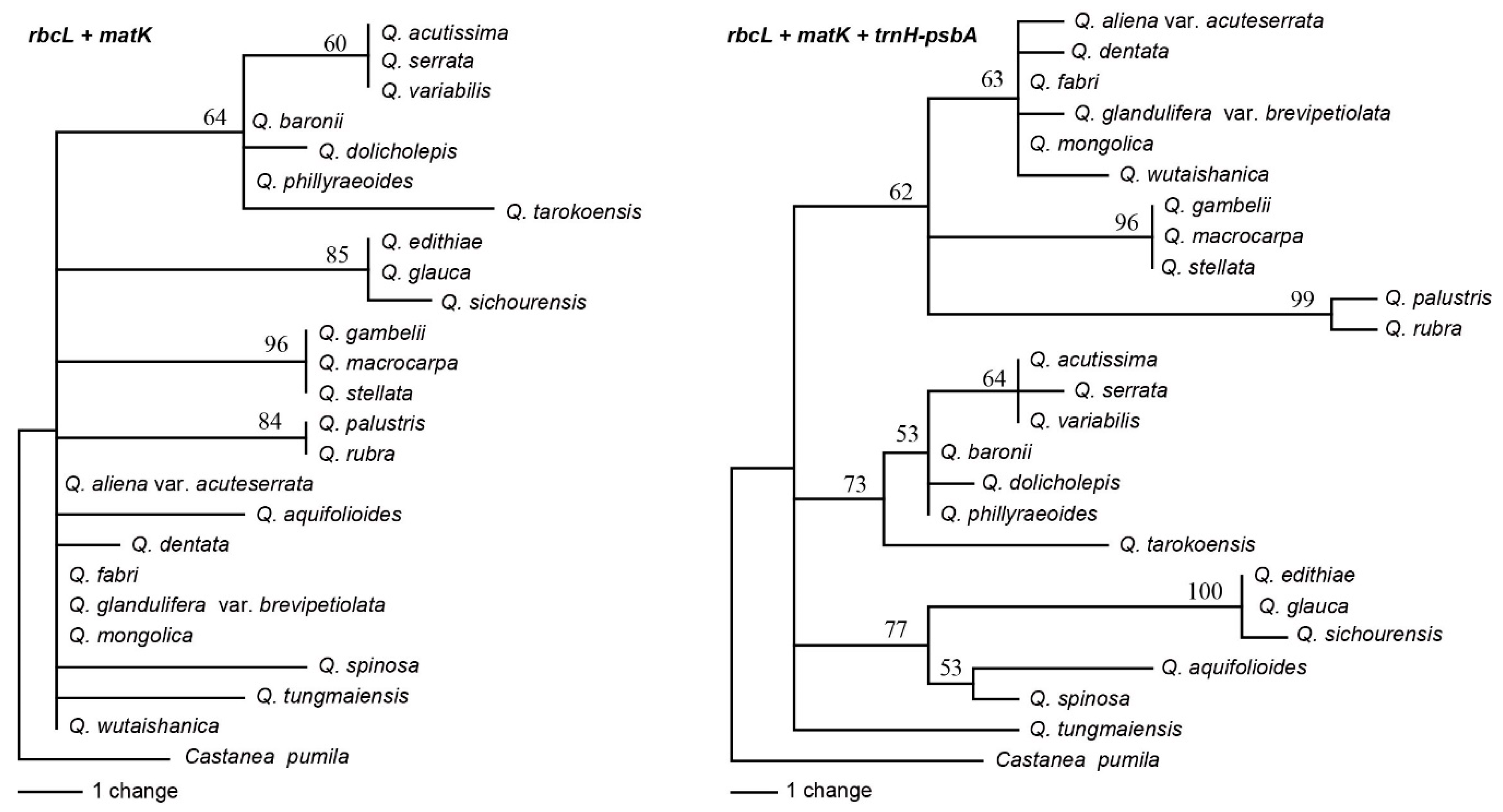
2.3. Analyses of the Standard DNA Barcodes
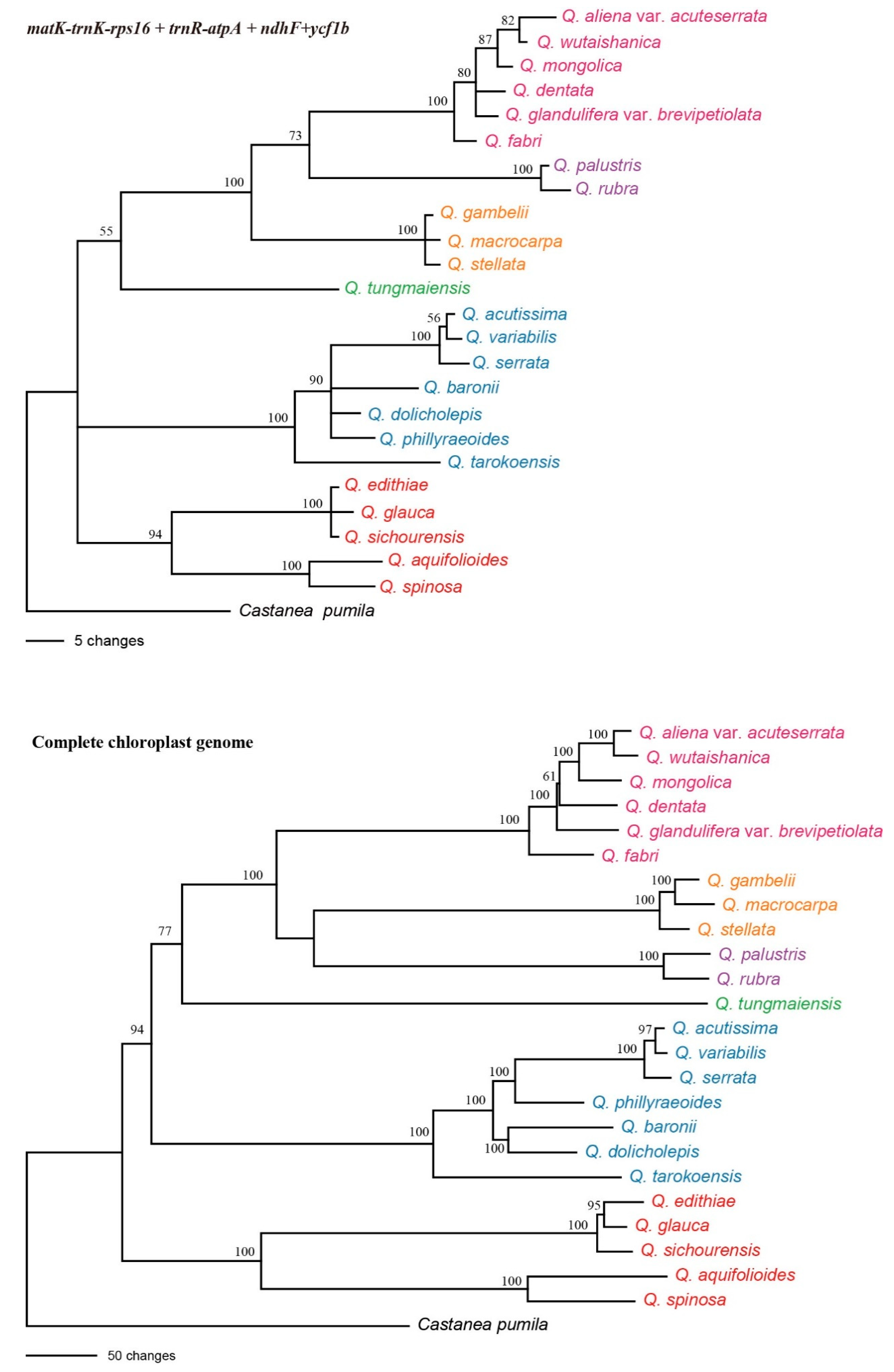
2.4. Analyses of Specific Barcodes
2.5. Super-Barcode
3. Discussion
4. Materials and Methods
4.1. Taxon Sampling
4.2. DNA Extraction and Sequencing
4.3. Genome Assembly and Genome Annotation
4.4. Phylogenetic Analyses
4.5. Sequence Divergence and Hotspot Identification
4.6. DNA Barcoding Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| LSC | Large single copy |
| SSC | Small single copy |
| IR | Inverted repeat |
References
- Hebert, P.D.N.; Cywinska, A.; Ball, S.L.; DeWaard, J.R. Biological identifications through DNA barcodes. Proc. R. Soc. Lond. B 2003, 270, 313–321. [Google Scholar] [CrossRef]
- Hollingsworth, P.M.; Graham, S.W.; Little, D.P. Choosing and using a plant DNA barcode. PLoS ONE 2011, 6, e19254. [Google Scholar] [CrossRef]
- Groups, C.P.B.; Li, D.Z.; Gao, L.M.; Li, H.T.; Wang, H.; Ge, X.J.; Liu, J.Q.; Chen, Z.D.; Zhou, S.L.; Chen, S.L.; et al. Comparative analysis of a large dataset indicates that internal transcribed spacer (ITS) should be incorporated into the core barcode for seed plants. Proc. Nat. Acad. Sci. USA 2011. [Google Scholar] [CrossRef]
- Simeone, M.C.; Piredda, R.; Attimonelli, M.; Bellarosa, R.; Schirone, B. Prospects of barcoding the Italian wild dendroflora: Oaks reveal severe limitations to tracking species identity. Mol. Ecol. Resour. 2011, 11, 72–83. [Google Scholar] [CrossRef]
- von Crautlein, M.; Korpelainen, H.; Pietilainen, M.; Rikkinen, J. DNA barcoding: A tool for improved taxon identification and detection of species diversity. Biodivers. Conserv. 2011, 20, 373–389. [Google Scholar] [CrossRef]
- Coissac, E.; Hollingsworth, P.M.; Lavergne, S.; Taberlet, P. From barcodes to genomes: Extending the concept of DNA barcoding. Mol. Ecol. 2016. [Google Scholar] [CrossRef]
- Li, X.; Yang, Y.; Henry, R.J.; Rossetto, M.; Wang, Y.; Chen, S. Plant DNA barcoding: From gene to genome. Biol. Rev. 2015, 90, 157–166. [Google Scholar] [CrossRef]
- Ruhsam, M.; Rai, H.S.; Mathews, S.; Ross, T.G.; Graham, S.W.; Raubeson, L.A.; Mei, W.; Thomas, P.I.; Gardner, M.F.; Ennos, R.A.; et al. Does complete plastid genome sequencing improve species discrimination and phylogenetic resolution in Araucaria? Mol. Ecol. Resour. 2015. [Google Scholar] [CrossRef]
- Fabiana, F.; Rizzo, Z.A.; Weismann, G.J.; Souza, O.R.; Lohmann, L.G.; Marie-Anne, V.S. Complete chloroplast genome sequences contribute to plant species delimitation: A case study of the Anemopaegma species complex. Am. J. Bot. 2017, 104, 1493–1509. [Google Scholar] [CrossRef]
- Wu, C.S.; Chaw, S.M.; Huang, Y.Y. Chloroplast phylogenomics indicates that Ginkgo biloba is sister to cycads. Genome Biol. Evol. 2013, 5, 243–254. [Google Scholar] [CrossRef]
- Cox, C.J.; Li, B.; Foster, P.G.; Embley, T.M.; Civan, P. Conflicting phylogenies for early land plants are caused by composition biases among synonymous substitutions. Syst. Biol. 2014, 63, 272–279. [Google Scholar] [CrossRef]
- Carbonell-Caballero, J.; Alonso, R.; Ibañez, V.; Terol, J.; Talon, M.; Dopazo, J. A phylogenetic analysis of 34 chloroplast genomes elucidates the relationships between wild and domestic species within the genus Citrus. Mol. Biol. Evol. 2015, 32, 2015–2035. [Google Scholar] [CrossRef]
- Yang, J.; Vazquez, L.; Chen, X.; Li, H.; Zhang, H.; Liu, Z.; Zhao, G. Development of Chloroplast and Nuclear DNA Markers for Chinese Oaks (Quercus Subgenus Quercus) and Assessment of Their Utility as DNA Barcodes. Front. Plant Sci. 2017, 8, 816. [Google Scholar] [CrossRef]
- Dong, W.; Xu, C.; Li, C.; Sun, J.; Zuo, Y.; Shi, S.; Cheng, T.; Guo, J.; Zhou, S. ycf1, the most promising plastid DNA barcode of land plants. Sci. Rep. 2015, 5, 8348. [Google Scholar] [CrossRef]
- Mayol, M.; Rossello, J.A. Why nuclear ribosomal DNA spacers (ITS) tell different stories in Quercus. Mol. Phylogenet. Evol. 2001, 19, 167–176. [Google Scholar] [CrossRef]
- Bellarosa, R.; Simeone, M.C.; Papini, A.; Schirone, B. Utility of ITS sequence data for phylogenetic reconstruction of Italian Quercus spp. Mol. Phylogenet. Evol. 2005, 34, 355–370. [Google Scholar] [CrossRef]
- Simeone, M.C.; Piredda, R.; Papini, A.; Vessella, F.; Schirone, B. Application of plastid and nuclear markers to DNA barcoding of Euro-Mediterranean oaks (Quercus, Fagaceae): Problems, prospects and phylogenetic implications. Bot. J. Linn. Soc. 2013, 172, 478–499. [Google Scholar] [CrossRef]
- Fineschi, S.; Taurchini, D.; Grossoni, P.; Petit, R.J.; Vendramin, G.G. Chloroplast DNA variation of white oaks in Italy. For. Ecol. Manage. 2002, 156, 103–114. [Google Scholar] [CrossRef]
- Lumaret, R.; Jabbour-Zahab, R. Ancient and current gene flow between two distantly related Mediterranean oak species, Quercus suber and Q. ilex. Ann. Bot. 2009, 104, 725–736. [Google Scholar] [CrossRef]
- McVay, J.D.; Hipp, A.L.; Manos, P.S. A genetic legacy of introgression confounds phylogeny and biogeography in oaks. Proc. Biol. Sci. 2017, 284. [Google Scholar] [CrossRef]
- Eaton, D.A.R.; Hipp, A.L.; González-Rodríguez, A.; Cavender-Bares, J. Historical introgression among the American live oaks and the comparative nature of tests for introgression. Evolution 2015, 69, 2587–2601. [Google Scholar] [CrossRef]
- de Groot, G.A.; During, H.J.; Maas, J.W.; Schneider, H.; Vogel, J.C.; Erkens, R.H. Use of rbcL and trnL-F as a two-locus DNA barcode for identification of NW-European ferns: An ecological perspective. PLoS ONE 2011, 6, e16371. [Google Scholar] [CrossRef]
- Pang, X.; Liu, C.; Shi, L.; Liu, R.; Liang, D.; Li, H.; Cherny, S.S.; Chen, S. Utility of the trnH-psbA intergenic spacer region and its combinations as plant DNA barcodes: A meta-analysis. PLoS ONE 2012, 7, e48833. [Google Scholar] [CrossRef]
- Saarela, J.M.; Sokoloff, P.C.; Gillespie, L.J.; Consaul, L.L.; Bull, R.D. DNA barcoding the Canadian Arctic flora: Core plastid barcodes (rbcL + matK) for 490 vascular plant species. PLoS ONE 2013, 8, e77982. [Google Scholar] [CrossRef]
- Krawczyk, K.; Szczecińska, M.; Sawicki, J. Evaluation of 11 single-locus and seven multilocus DNA barcodes in Lamium L. (Lamiaceae). Mol. Ecol. Resour. 2014, 14, 272–285. [Google Scholar] [CrossRef]
- Group, C.P.W. A DNA barcode for land plants. Proc. Nat. Acad. Sci. USA 2009, 106, 12794–12797. [Google Scholar] [CrossRef]
- Dong, W.; Liu, J.; Yu, J.; Wang, L.; Zhou, S. Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PLoS ONE 2012, 7, e35071. [Google Scholar] [CrossRef]
- Dong, W.; Liu, H.; Xu, C.; Zuo, Y.; Chen, Z.; Zhou, S. A chloroplast genomic strategy for designing taxon specific DNA mini-barcodes: A case study on ginsengs. BMC Genet. 2014, 15, 138. [Google Scholar] [CrossRef]
- Xu, C.; Dong, W.; Li, W.; Lu, Y.; Xie, X.; Jin, X.; Shi, J.; He, K.; Suo, Z. Comparative Analysis of Six Lagerstroemia Complete Chloroplast Genomes. Front. Plant Sci. 2017, 8, 15. [Google Scholar] [CrossRef]
- Song, Y.; Wang, S.; Ding, Y.; Xu, J.; Li, M.F.; Zhu, S.; Chen, N. Chloroplast Genomic Resource of Paris for Species Discrimination. Sci. Rep. 2017, 7, 3427. [Google Scholar] [CrossRef]
- Li, W.; Liu, Y.; Yang, Y.; Xie, X.; Lu, Y.; Yang, Z.; Jin, X.; Dong, W.; Suo, Z. Interspecific chloroplast genome sequence diversity and genomic resources in Diospyros. BMC Plant Biol. 2018, 18, 210. [Google Scholar] [CrossRef]
- Zhao, Z.; Wang, X.; Yu, Y.; Yuan, S.; Jiang, D.; Zhang, Y.; Zhang, T.; Zhong, W.; Yuan, Q.; Huang, L. Complete chloroplast genome sequences of Dioscorea: Characterization, genomic resources, and phylogenetic analyses. PeerJ 2018, 6, e6032. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.J.; Jansen, R.K. NdhF sequence evolution and the major clades in the sunflower Family. Proc. Nat. Acad. Sci. USA 1995, 92, 10379–10383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.H. Phylogeny of Catalpa (Bignoniaceae) inferred from sequences of chloroplast ndhF and nuclear ribosomal DNA. J. Syst. Evol. 2008, 46, 341–348. [Google Scholar] [CrossRef]
- Park, S.J.; Korompai, E.J.; Francisco-Ortega, J.; Santos-Guerra, A.; Jansen, R.K. Phylogenetic relationships of Tolpis (Asteraceae: Lactuceae) based on ndhF sequence data. Plant Syst. Evol. 2001, 226, 23–33. [Google Scholar] [CrossRef]
- Ji, Y.; Liu, C.; Yang, Z.; Yang, L.; He, Z.; Wang, H.; Yang, J.; Yi, T. Testing and using complete plastomes and ribosomal DNA sequences as the next generation DNA barcodes in Panax (Araliaceae). Mol. Ecol. Resour. 2019, 19, 1333–1345. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Choi, H.; Shin, J.; Jo, A.; Lee, K.-E.; Cho, S.-S.; Hwang, Y.-P.; Choi, C. Molecular Discrimination of Cynanchum wilfordii and Cynanchum auriculatum by InDel Markers of Chloroplast DNA. Molecules 2018, 23, 1337. [Google Scholar] [CrossRef] [Green Version]
- Wang, A.; Wu, H.; Zhu, X.; Lin, J. Species Identification of Conyza bonariensis Assisted by Chloroplast Genome Sequencing. Front. Genet. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Kane, N.; Sveinsson, S.; Dempewolf, H.; Yang, J.Y.; Zhang, D.; Engels, J.M.; Cronk, Q. Ultra-barcoding in cacao (Theobroma spp.; Malvaceae) using whole chloroplast genomes and nuclear ribosomal DNA. Am. J. Bot. 2012. [Google Scholar] [CrossRef] [Green Version]
- Hollingsworth, P.M.; Li, D.Z.; van der Bank, M.; Twyford, A.D. Telling plant species apart with DNA: From barcodes to genomes. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2016, 371. [Google Scholar] [CrossRef]
- Fu, C.N.; Wu, C.S.; Ye, L.J.; Mo, Z.Q.; Liu, J.; Chang, Y.W.; Li, D.Z.; Chaw, S.M.; Gao, L.M. Prevalence of isomeric plastomes and effectiveness of plastome super-barcodes in yews (Taxus) worldwide. Sci. Rep. 2019, 9, 2773. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, S.; Jing, Y.; Wang, L.; Zhou, S. A modified CTAB protocol for plant DNA extraction. Chin. Bull. Bot. 2013, 48, 72–78. [Google Scholar]
- Patel, R.K.; Jain, M. NGS QC Toolkit: A toolkit for quality control of next generation sequencing data. PLoS ONE 2012, 7, e30619. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Dong, W.; Xu, C.; Cheng, T.; Lin, K.; Zhou, S. Sequencing angiosperm plastid genomes made easy: A complete set of universal primers and a case study on the phylogeny of Saxifragales. Genome Biol. Evol. 2013, 5, 989–997. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.I.; Cronk, Q.C.B. Plann: A command-line application for annotating plastome sequences. Appl. Plant Sci. 2015, 3, 1500026. [Google Scholar] [CrossRef] [Green Version]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Hohna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, S.D.; Collins, R.A.; Boyer, S.; Lefort, M.C.; Malumbres-Olarte, J.; Vink, C.J.; Cruickshank, R.H. Spider: An R package for the analysis of species identity and evolution, with particular reference to DNA barcoding. Mol. Ecol. Resour. 2012, 12, 562–565. [Google Scholar] [CrossRef] [PubMed]
- Swofford, D. PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods); Version 4.0 Beta; Sinauer Associates: Sunderland, MA, USA, 2002. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | LSC | IR | SSC | Total Size (bp) | Number of Genes | Protein Coding Genes | tRNA | rRNA | Accession Number in Genbank |
|---|---|---|---|---|---|---|---|---|---|
| Q. macrocarpa | 90594 | 25848 | 18946 | 161236 | 113 | 79 | 30 | 4 | MK105459 |
| Q. gambelii | 90570 | 25848 | 18947 | 161213 | 113 | 79 | 30 | 4 | MK105457 |
| Q. stellata | 90562 | 25848 | 18956 | 161214 | 113 | 79 | 30 | 4 | MK105467 |
| Q. palustris | 90624 | 25852 | 18956 | 161284 | 113 | 79 | 30 | 4 | MK105461 |
| Q. aliena var. acuteserrata | 90532 | 25837 | 18988 | 161194 | 113 | 79 | 30 | 4 | MK105452 |
| Q. phillyraeoides | 90363 | 25866 | 19037 | 161132 | 113 | 79 | 30 | 4 | MK105462 |
| Q. glandulifera var. brevipetiolata | 90534 | 25826 | 19038 | 161224 | 113 | 79 | 30 | 4 | MK105458 |
| Q. wutaishanica | 90520 | 25825 | 19041 | 161211 | 113 | 79 | 30 | 4 | MK105466 |
| Q. mongolica | 90504 | 25820 | 19047 | 161191 | 113 | 79 | 30 | 4 | MK105460 |
| Q. dentata | 90593 | 25826 | 19055 | 161300 | 113 | 79 | 30 | 4 | MK105453 |
| Q. fabri | 90557 | 25832 | 19064 | 161285 | 113 | 79 | 30 | 4 | MK105456 |
| Q. serrata | 90447 | 25817 | 19065 | 161146 | 113 | 79 | 30 | 4 | MK105464 |
| Q. variabilis | 90464 | 25817 | 19070 | 161168 | 113 | 79 | 30 | 4 | MK105451 |
| Q. rubra | 90553 | 25870 | 19073 | 161366 | 113 | 79 | 30 | 4 | MK105463 |
| Markers | Length | Variable Sites | Information Sites | Discrimination Success (%) Based on Distance Method | ||
|---|---|---|---|---|---|---|
| Numbers | % | Numbers | % | |||
| rbcL | 698 | 8 | 1.15% | 5 | 0.72% | 12.50% |
| matK | 744 | 21 | 2.82% | 11 | 1.48% | 25.00% |
| trnH-psbA | 574 | 27 | 4.70% | 16 | 2.79% | 37.50% |
| rbcL + matK | 1442 | 29 | 2.01% | 16 | 1.11% | 29.17% |
| rbcL + matK + trnH-psbA | 2016 | 56 | 2.78% | 32 | 1.59% | 50.00% |
| matK-trnK-rps16 | 2311 | 93 | 4.02% | 59 | 2.55% | 79.17% |
| trnR-atpA | 1309 | 57 | 4.35% | 35 | 2.67% | 66.67% |
| ndhF | 1536 | 74 | 4.82% | 45 | 2.93% | 83.33% |
| ycf1b | 1765 | 94 | 5.33% | 59 | 3.34% | 70.83% |
| ndhF+ycf1b | 3301 | 168 | 5.09% | 104 | 3.15% | 91.67% |
| matK-trnK-rps16 + trnR-atpA + ndhF + ycf1b | 6921 | 318 | 4.59% | 198 | 2.86% | 100.00% |
| Number of Sites | Variable Sites | Information Sites | Nucleotide Diversity | |||
|---|---|---|---|---|---|---|
| Numbers | % | Numbers | % | |||
| LSC | 92,888 | 2009 | 2.16% | 1257 | 1.35% | 0.0043 |
| SSC | 19,535 | 593 | 3.04% | 368 | 1.88% | 0.00624 |
| IR | 25,879 | 91 | 0.35% | 54 | 0.21% | 0.00073 |
| Complete chloroplast genome | 164,156 | 2778 | 1.69% | 1727 | 1.05% | 0.00335 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pang, X.; Liu, H.; Wu, S.; Yuan, Y.; Li, H.; Dong, J.; Liu, Z.; An, C.; Su, Z.; Li, B. Species Identification of Oaks (Quercus L., Fagaceae) from Gene to Genome. Int. J. Mol. Sci. 2019, 20, 5940. https://doi.org/10.3390/ijms20235940
Pang X, Liu H, Wu S, Yuan Y, Li H, Dong J, Liu Z, An C, Su Z, Li B. Species Identification of Oaks (Quercus L., Fagaceae) from Gene to Genome. International Journal of Molecular Sciences. 2019; 20(23):5940. https://doi.org/10.3390/ijms20235940
Chicago/Turabian StylePang, Xinbo, Hongshan Liu, Suran Wu, Yangchen Yuan, Haijun Li, Junsheng Dong, Zhaohua Liu, Chuanzhi An, Zhihai Su, and Bin Li. 2019. "Species Identification of Oaks (Quercus L., Fagaceae) from Gene to Genome" International Journal of Molecular Sciences 20, no. 23: 5940. https://doi.org/10.3390/ijms20235940