GnRH Antagonists Produce Differential Modulation of the Signaling Pathways Mediated by GnRH Receptors


 , ,
, ,  and
and
Abstract
:1. Introduction
2. Results
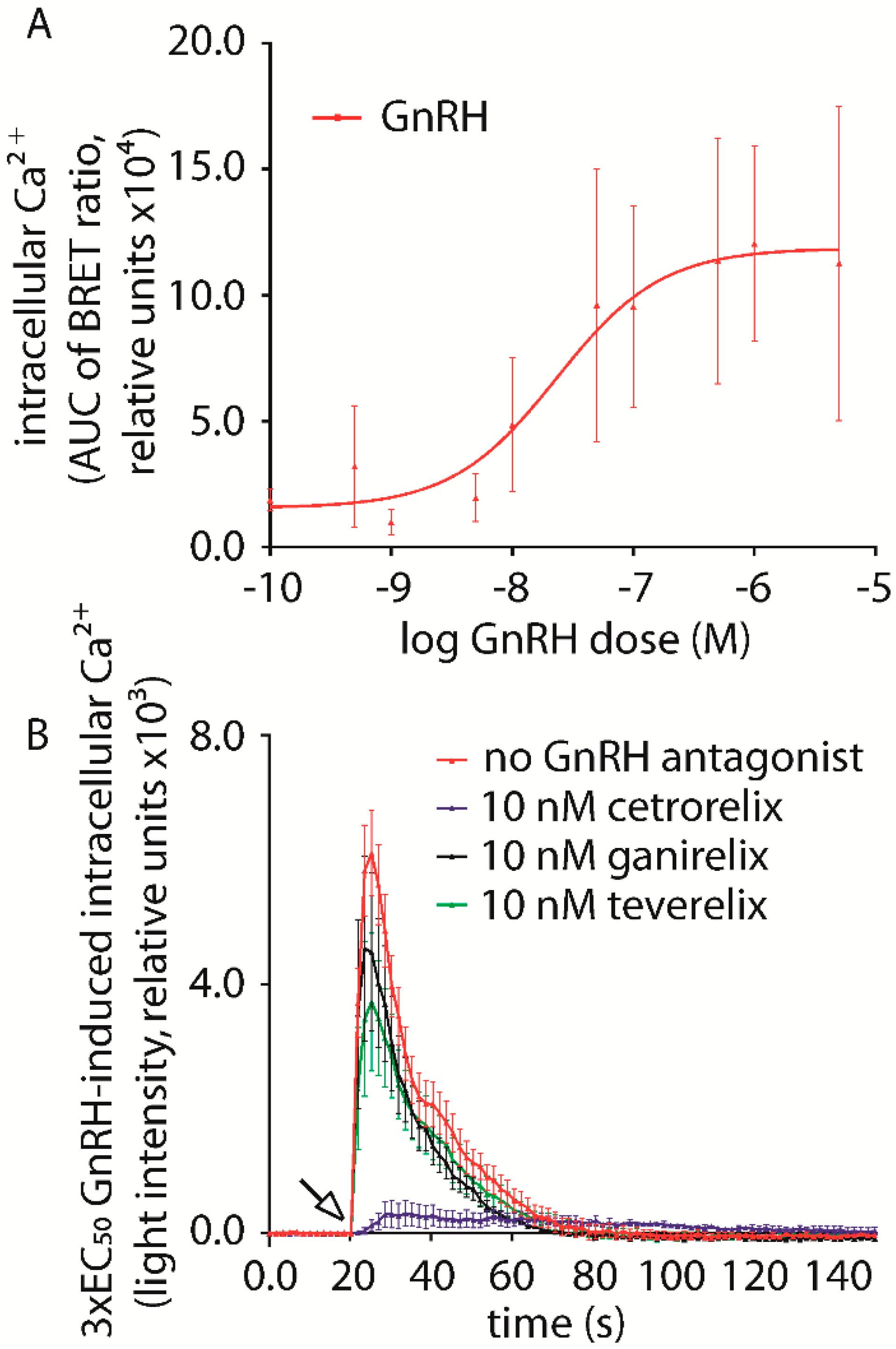
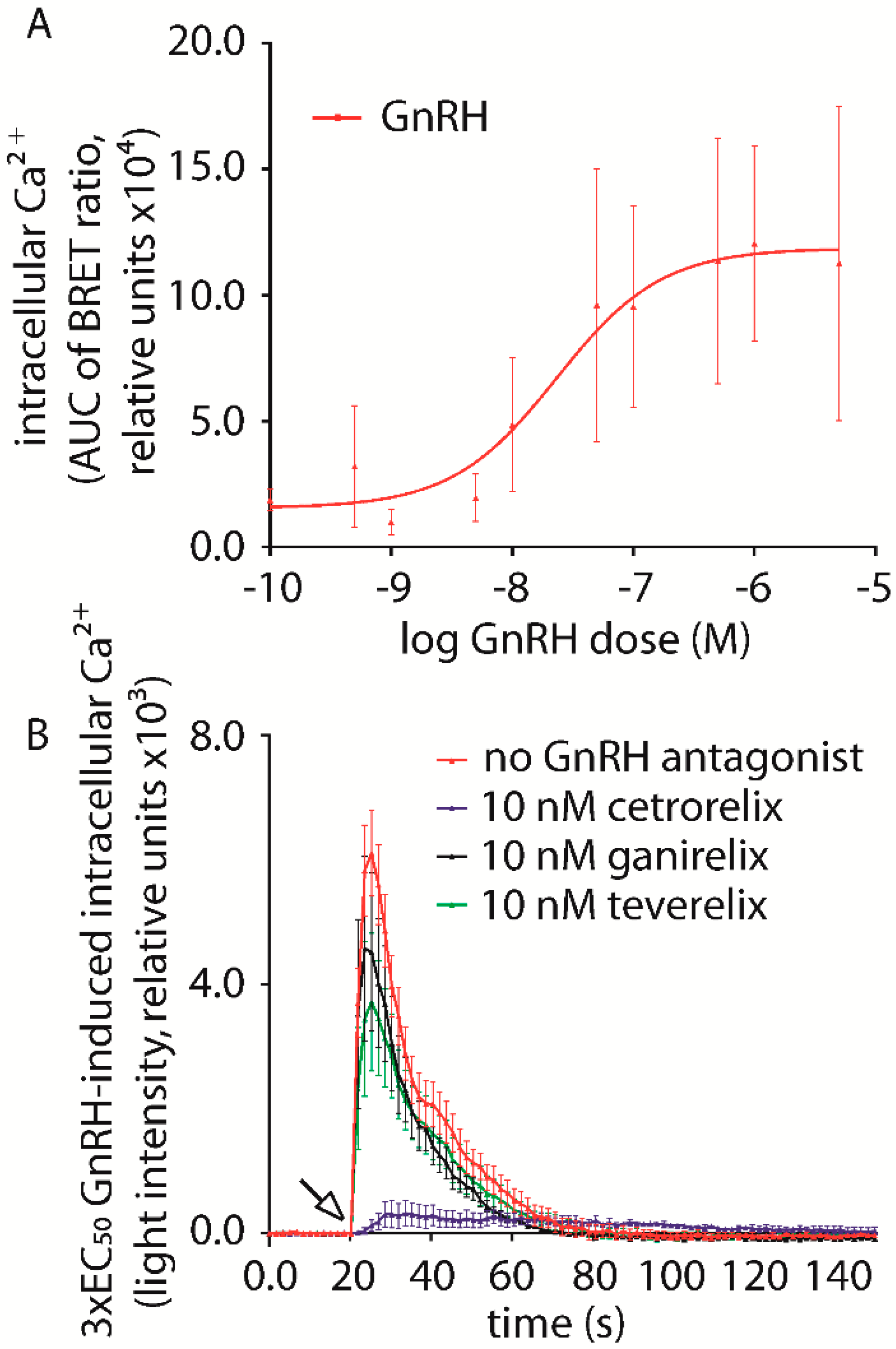
2.1. Gonadotropin Releasing Hormone (GnRH) Antagonist-Induced Inhibition of Intracellular Ca2+ Increase
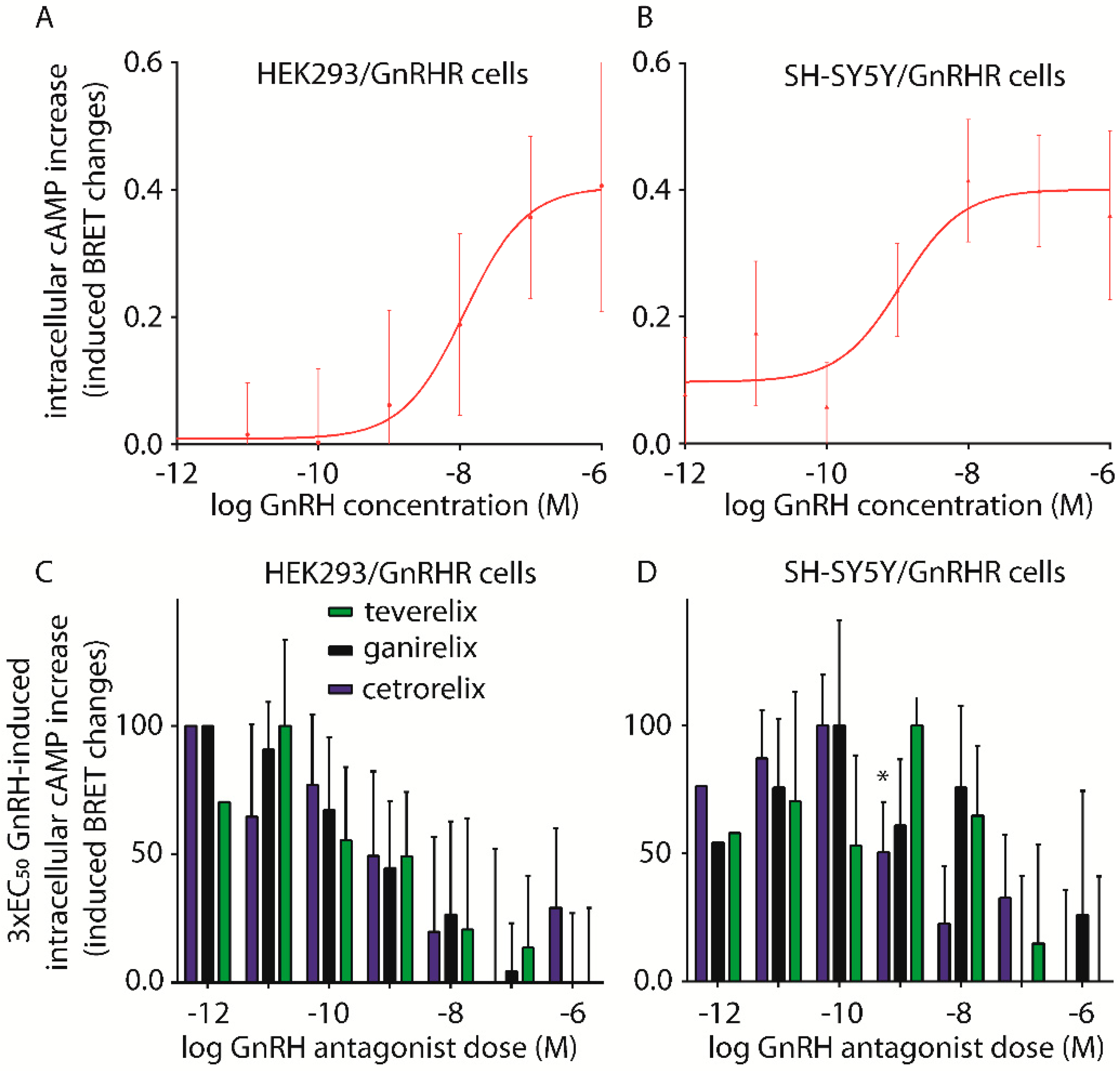
2.2. Assessment of cAMP Accumulation
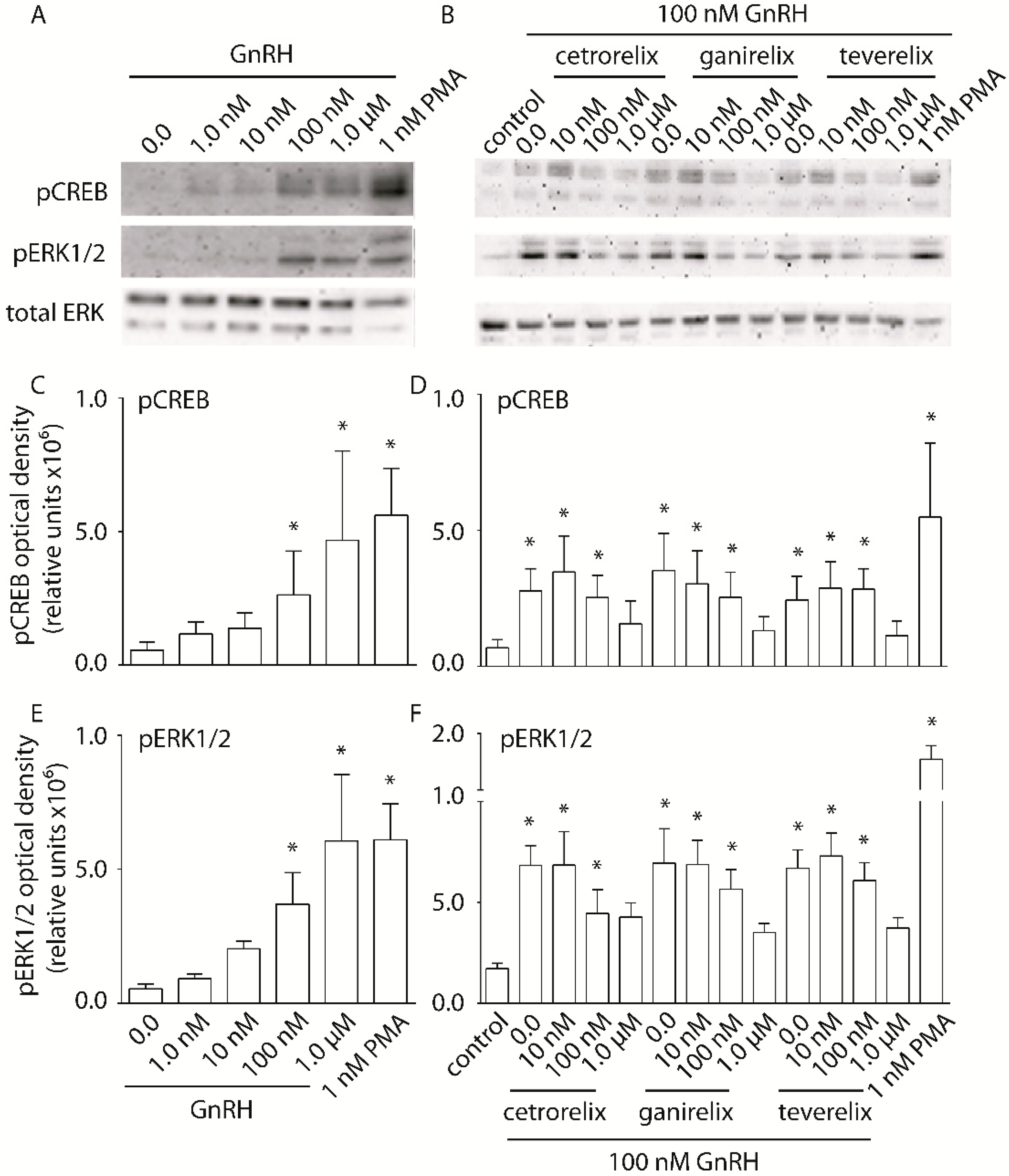
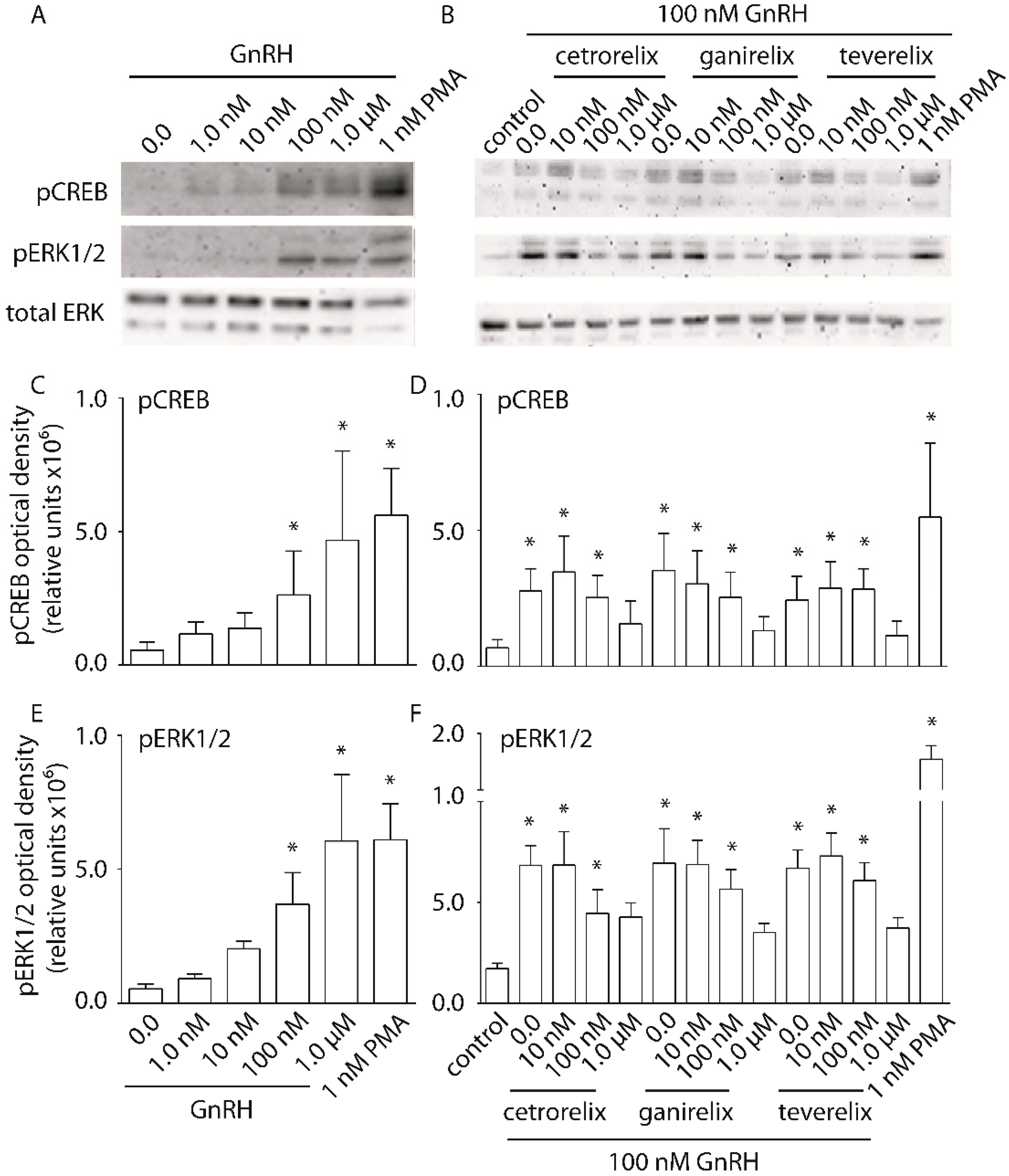
2.3. Evaluation of pERK1/2 and pCREB Activation
2.4. Analysis of β-Catenin Activation and Localization
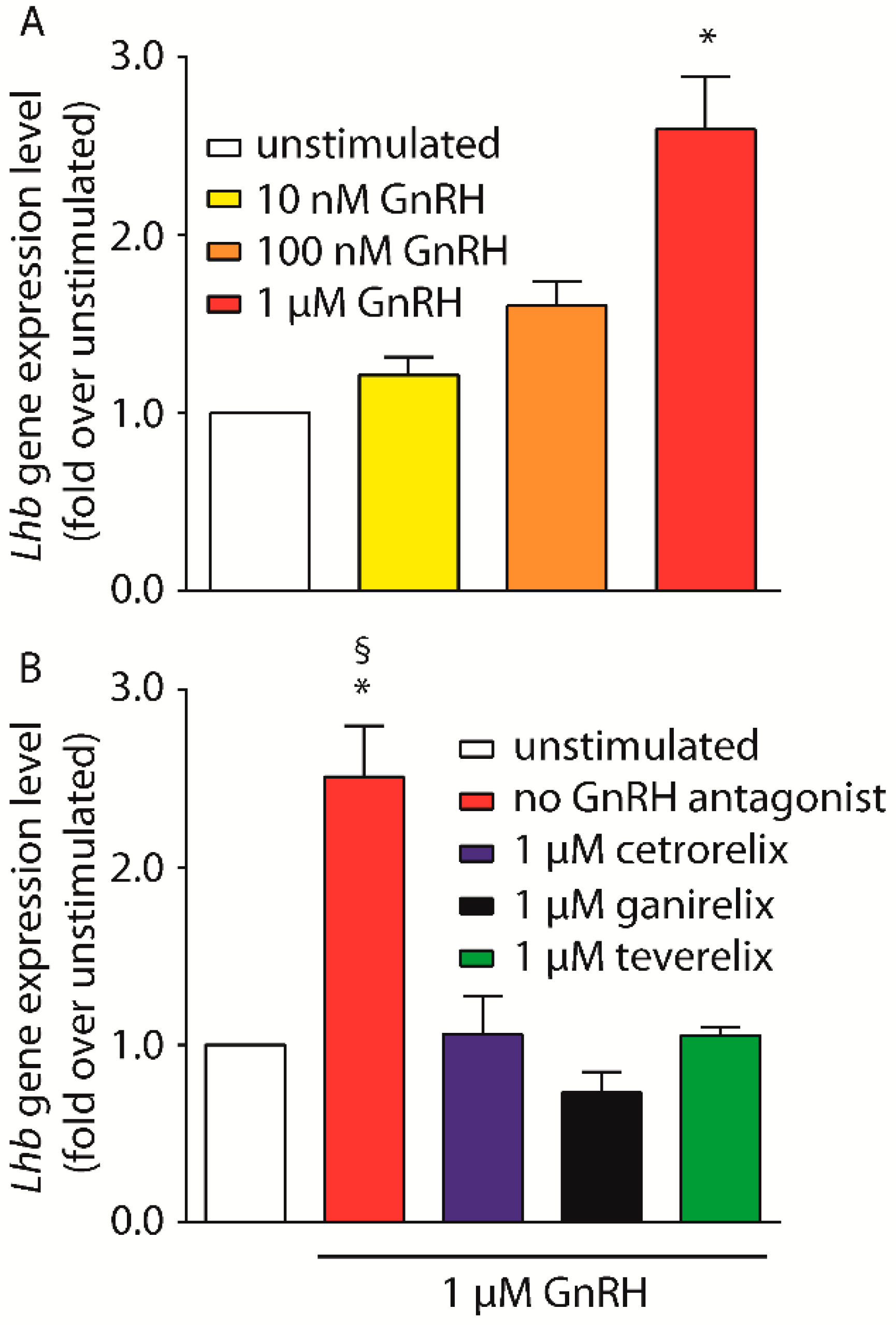
2.5. Analysis of GnRH-Induced Lhb Gene Expression
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Transfection Methods
4.4. BRET Measurement of Intracellular Ca2+ Increase and cAMP Production
4.5. Western Blotting Analysis
4.6. Immunofluorescence Analysis
4.7. Lhb Gene Expression
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- McArdle, C.A. Gonadotropin-releasing hormone receptor signaling: Biased and unbiased. Mini Rev. Med. Chem. 2012, 12, 841–850. [Google Scholar] [CrossRef]
- Naor, Z. Signaling by G-protein-coupled receptor (GPCR): Studies on the GnRH receptor. Front. Neuroendocr. 2009, 30, 10–29. [Google Scholar] [CrossRef]
- Pawson, A.J.; McNeilly, A.S. The pituitary effects of GnRH. Anim. Reprod. Sci. 2005, 88, 75–94. [Google Scholar] [CrossRef]
- Kaiser, U.B. Studies of Gonadotropin-Releasing Hormone (GnRH) Action Using GnRH Receptor-Expressing Pituitary Cell Lines. Endocr. Rev. 1997, 18, 46–70. [Google Scholar] [CrossRef] [PubMed]
- Millar, R.P.; Lu, Z.L.; Pawson, A.J.; Flanagan, C.A.; Morgan, K.; Maudsley, S.R. Gonadotropin-Releasing Hormone Receptors. Endocr. Rev. 2004, 25, 235–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grosse, R.; Schmid, A.; Schöneberg, T.; Herrlich, A.; Muhn, P.; Schultz, G.; Gudermann, T. Gonadotropin-releasing hormone receptor initiates multiple signaling pathways by exclusively coupling to G(q/11) proteins. J. Boil. Chem. 2000, 275, 9193–9200. [Google Scholar] [CrossRef] [PubMed]
- Mulvaney, J.M. Divergent Signaling Pathways Requiring Discrete Calcium Signals Mediate Concurrent Activation of Two Mitogen-activated Protein Kinases by Gonadotropin-releasing Hormone. J. Boil. Chem. 2000, 275, 14182–14189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobkin-Bekman, M.; Naidich, M.; Pawson, A.J.; Millar, R.P.; Seger, R.; Naor, Z. Activation of Mitogen-activated protein kinase (MAPK) by GnRH is cell-context dependent. Mol. Cell. Endocrinol. 2006, 252, 184–190. [Google Scholar] [CrossRef]
- Neves, S.R.; Ram, P.T.; Iyengar, R. G protein pathways. Science 2002, 296, 1636–1639. [Google Scholar] [CrossRef]
- Naor, Z.; Benard, O.; Seger, R. Activation of MAPK cascades by G-protein-coupled receptors: The case of gonadotropin-releasing hormone receptor. Trends Endocrinol. Metab. 2000, 11, 91–99. [Google Scholar] [CrossRef]
- Bonfil, D.; Chuderland, D.; Kraus, S.; Shahbazian, D.; Friedberg, I.; Seger, R.; Naor, Z. Extracellular signal-regulated kinase, Jun N-terminal kinase, p38, and c-Src are involved in gonadotropin-releasing hormone-stimulated activity of the glycoprotein hormone follicle-stimulating hormone beta-subunit promoter. Endocrinology 2004, 145, 2228–2244. [Google Scholar] [CrossRef] [PubMed]
- Tsutsumi, R.; Mistry, D.; Webster, N.J.G. Signaling Responses to Pulsatile Gonadotropin-releasing Hormone in LbetaT2 Gonadotrope Cells. J. Boil. Chem. 2010, 285, 20262–20272. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Usui, I.; Evans, L.G.; Austin, D.A.; Mellon, P.L.; Olefsky, J.M.; Webster, N.J.G. Involvement of Both G q/11 and G s Proteins in Gonadotropin-releasing Hormone Receptor-mediated Signaling in LβT2 Cells. J. Biol. Chem. 2002, 277, 32099–32108. [Google Scholar] [CrossRef]
- Perrett, R.M.; McArdle, C.A. Molecular Mechanisms of Gonadotropin-Releasing Hormone Signaling: Integrating Cyclic Nucleotides into the Network. Front. Endocrinol. 2013, 4, 4. [Google Scholar] [CrossRef]
- Maudsley, S.; Davidson, L.; Pawson, A.J.; Chan, R.; López de Maturana, R.; Millar, R.P. Gonadotropin-releasing hormone (GnRH) antagonists promote proapoptotic signaling in peripheral reproductive tumor cells by activating a Galphai-coupling state of the type I GnRH receptor. Cancer Res. 2004, 64, 7533–7544. [Google Scholar] [CrossRef]
- Naor, Z.; Huhtaniemi, I. Interactions of the GnRH receptor with heterotrimeric G proteins. Front. Neuroendocr. 2013, 34, 88–94. [Google Scholar] [CrossRef]
- Larivière, S.; Garrel, G.; Simon, V.; Soh, J.W.; Laverrière, J.N.; Counis, R.; Cohen-Tannoudji, J. Gonadotropin-releasing hormone couples to 3′,5′-cyclic adenosine-5′-monophosphate pathway through novel protein kinase Cdelta and -epsilon in LbetaT2 gonadotrope cells. Endocrinology 2007, 148, 1099–1107. [Google Scholar] [CrossRef]
- Thompson, I.R.; Kaiser, U.B. GnRH pulse frequency-dependent differential regulation of LH and FSH gene expression. Mol. Cell. Endocrinol. 2014, 385, 28–35. [Google Scholar] [CrossRef]
- Gardner, S.; Maudsley, S.; Millar, R.P.; Pawson, A.J. Nuclear stabilization of beta-catenin and inactivation of glycogen synthase kinase-3beta by gonadotropin-releasing hormone: Targeting Wnt signaling in the pituitary gonadotrope. Mol. Endocrinol. 2007, 21, 3028–3038. [Google Scholar] [CrossRef]
- Salisbury, T.B.; Binder, A.K.; Nilson, J.H. Welcoming beta-catenin to the gonadotropin-releasing hormone transcriptional network in gonadotropes. Mol. Endocrinol. 2008, 22, 1295–1303. [Google Scholar] [CrossRef]
- Brembeck, F.H.; Rosário, M.; Birchmeier, W. Balancing cell adhesion and Wnt signaling, the key role of beta-catenin. Curr. Opin. Genet. Dev. 2006, 16, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Nelson, W.J.; Nusse, R. Convergence of Wnt, beta-catenin, and cadherin pathways. Science 2004, 303, 1483–1487. [Google Scholar] [CrossRef] [PubMed]
- Moon, R.T.; Kohn, A.D.; De Ferrari, G.V.; Kaykas, A. WNT and beta-catenin signalling: Diseases and therapies. Nat. Rev. Genet. 2004, 5, 691–701. [Google Scholar] [CrossRef]
- Salisbury, T.B.; Binder, A.K.; Grammer, J.C.; Nilson, J.H. Maximal activity of the luteinizing hormone beta-subunit gene requires beta-catenin. Mol. Endocrinol. 2007, 21, 963–971. [Google Scholar] [CrossRef] [PubMed]
- Reissmann, T.; Felberbaum, R.; Diedrich, K.; Engel, J.; Comaru-Schally, A.M.; Schally, A.V. Endocrinology: Development and applications of luteinizing hormone-releasing hormone antagonists in the treatment of infertility: An overview. Hum. Reprod. 1995, 10, 1974–1981. [Google Scholar] [CrossRef]
- Millar, R.P.; Newton, C.L. Current and future applications of GnRH, kisspeptin and neurokinin B analogues. Nat. Rev. Endocrinol. 2013, 9, 451–466. [Google Scholar] [CrossRef] [Green Version]
- Engel, J.B.; Schally, A.V. Drug Insight: Clinical use of agonists and antagonists of luteinizing-hormone-releasing hormone. Nat. Clin. Pract. Endocrinol. Metab. 2007, 3, 157–167. [Google Scholar] [CrossRef]
- Emons, G.; Schally, A.V. The use of luteinizing hormone releasing hormone agonists and antagonists in gynaecological cancers. Hum. Reprod. 1994, 9, 1364–1379. [Google Scholar] [CrossRef]
- Wilcox, J.; Potter, D.; Moore, M.; Ferrande, L.; Kelly, E.; CAP IV Investigator Group. Prospective, randomized trial comparing cetrorelix acetate and ganirelix acetate in a programmed, flexible protocol for premature luteinizing hormone surge prevention in assisted reproductive technologies. Fertil. Steril. 2005, 84, 108–117. [Google Scholar] [CrossRef]
- Check, J.H.; Brasile, D.; Choe, J.K.; Amui, J.; Wilson, C. The effect of cetrorelix vs. ganirelix on pregnancy outcome using minimal gonadotropin stimulation in women with elevated day 3 serum follicle stimulating hormone levels. Clin. Exp. Obstet. Gynecol. 2009, 36, 148–149. [Google Scholar]
- Deghenghi, R.; Boutignon, F.; Wüthrich, P.; Lenaerts, V. Antarelix (EP 24332) a novel water soluble LHRH antagonist. Biomed. Pharmacother. 1993, 47, 107–110. [Google Scholar] [CrossRef]
- Erb, K.; Pechstein, B.; Schueler, A.; Engel, J.; Hermann, R. Pituitary and gonadal endocrine effects and pharmacokinetics of the novel luteinizing hormone-releasing hormone antagonist teverelix in healthy men-a first-dose-in-humans study. Clin. Pharmacol. Ther. 2000, 67, 660–669. [Google Scholar] [CrossRef] [PubMed]
- Gianotti, L.; Veldhuis, J.D.; Destefanis, S.; Lanfranco, F.; Ramunni, J.; Arvat, E.; Marzetto, M.; Boutignon, F.; Deghenghi, R.; Ghigo, E. Suppression and recovery of LH secretion by a potent and selective GnRH-receptor antagonist peptide in healthy early follicular-phase women are mediated via selective control of LH secretory burst mass. Clin. Endocrinol. 2003, 59, 526–532. [Google Scholar] [CrossRef]
- Millar, R.P.; Pawson, A.J.; Morgan, K.; Rissman, E.F.; Lu, Z.L. Diversity of actions of GnRHs mediated by ligand-induced selective signaling. Front. Neuroendocrinol. 2008, 29, 17–35. [Google Scholar] [CrossRef] [Green Version]
- Turgeon, J.L. Steroid and pulsatile gonadotropin-releasing hormone (GnRH) regulation of luteinizing hormone and GnRH receptor in a novel gonadotrope cell line. Mol. Endocrinol. 1996, 10, 439–450. [Google Scholar]
- Wilson, A.C.; Salamat, M.S.; Haasl, R.J.; Roche, K.M.; Karande, A.; Meethal, S.V.; Terasawa, E.; Bowen, R.L.; Atwood, C.S. Human neurons express type I GnRH receptor and respond to GnRH I by increasing luteinizing hormone expression. J. Endocrinol. 2004, 191, 651–663. [Google Scholar] [CrossRef]
- Huirne, J.A.; Lambalk, C.B. Gonadotropin-releasing-hormone-receptor antagonists. Lancet 2001, 358, 1793–1803. [Google Scholar] [CrossRef]
- Sealfon, S.C.; Weinstein, H.; Millar, R.P. Molecular Mechanisms of Ligand Interaction with the Gonadotropin-Releasing Hormone Receptor. Endocr. Rev. 1997, 18, 180–205. [Google Scholar] [CrossRef]
- Barran, P.E.; Roeske, R.W.; Pawson, A.J.; Sellar, R.; Bowers, M.T.; Morgan, K.; Lu, Z.L.; Tsuda, M.; Kusakabe, T.; Millar, R.P. Evolution of Constrained Gonadotropin-releasing Hormone Ligand Conformation and Receptor Selectivity. J. Boil. Chem. 2005, 280, 38569–38575. [Google Scholar] [CrossRef] [Green Version]
- Stojilkovic, S.S.; Bjelobaba, I.; Zemkova, H. Ion Channels of Pituitary Gonadotrophs and Their Roles in Signaling and Secretion. Front. Endocrinol. 2017, 8, 126. [Google Scholar] [CrossRef]
- Merelli, F.; Stojilkovic, S.S.; Iida, T.; Krsmanovic, L.Z.; Zheng, L.; Mellon, P.L.; Catt, K.J. Gonadotropin-releasing hormone-induced calcium signaling in clonal pituitary gonadotrophs. Endocrinology 1992, 131, 925–932. [Google Scholar] [PubMed]
- Durán-Pastén, M.L.; Fiordelisio, T. GnRH-Induced Ca2+ Signaling Patterns and Gonadotropin Secretion in Pituitary Gonadotrophs. Functional Adaptations to Both Ordinary and Extraordinary Physiological Demands. Front. Endocrinol. 2013, 4, 127. [Google Scholar] [CrossRef]
- Naor, Z.; Capponi, A.M.; Rossier, M.F.; Ayalon, D.; Limor, R. Gonadotropin-Releasing Hormone- Induced Rise in Cytosolic Free Ca2+ Levels: Mobilization of Cellular and Extracellular Ca2+ Pools and Relationship to Gonadotropin Secretion. Mol. Endocrinol. 1988, 2, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Kawaminami, M.; Uematsu, N.; Funahashi, K.; Kokubun, R.; Kurusu, S. Gonadotropin releasing hormone (GnRH) enhances annexin A5 mRNA expression through mitogen activated protein kinase (MAPK) in LbetaT2 pituitary gonadotrope cells. Endocr. J. 2008, 55, 1005–1014. [Google Scholar] [CrossRef] [PubMed]
- Miles, L.E.C.; Hanyaloglu, A.C.; Dromey, J.R.; Pfleger, K.D.G.; Eidne, K.A. Gonadotropin-releasing hormone receptor-mediated growth suppression of immortalized LbetaT2 gonadotrope and stable HEK293 cell lines. Endocrinology 2004, 145, 194–204. [Google Scholar] [CrossRef]
- Maiti, K.; Oh, D.Y.; Moon, J.S.; Acharjee, S.; Li, J.H.; Bai, D.G.; Park, H.S.; Lee, K.; Lee, Y.C.; Jung, N.C.; et al. Differential Effects of Gonadotropin-Releasing Hormone (GnRH)-I and GnRH-II on Prostate Cancer Cell Signaling and Death. J. Clin. Endocrinol. Metab. 2005, 90, 4287–4298. [Google Scholar] [CrossRef] [Green Version]
- Knollman, P.E.; Conn, P.M. Multiple G proteins compete for binding with the human gonadotropin releasing hormone receptor. Arch. Biochem. Biophys. 2008, 477, 92–97. [Google Scholar] [CrossRef] [Green Version]
- Oh, D.Y.; Song, J.A.; Moon, J.S.; Moon, M.J.; Kim, J.I.; Kim, K.; Seong, J.Y. Membrane-Proximal Region of the Carboxyl Terminus of the Gonadotropin-Releasing Hormone Receptor (GnRHR) Confers Differential Signal Transduction between Mammalian and Nonmammalian GnRHRs. Mol. Endocrinol. 2005, 19, 722–731. [Google Scholar] [CrossRef] [Green Version]
- Hanyaloglu, A.C.; Vrecl, M.; Kroeger, K.M.; Miles, L.E.C.; Qian, H.; Thomas, W.G.; Eidne, K.A. Casein Kinase II Sites in the Intracellular C-terminal Domain of the Thyrotropin-releasing Hormone Receptor and Chimeric Gonadotropin-releasing Hormone Receptors Contribute to β-Arrestin-dependent Internalization. J. Boil. Chem. 2001, 276, 18066–18074. [Google Scholar] [CrossRef]
- Avet, C.; Garrel, G.; Denoyelle, C.; Laverrière, J.N.; Counis, R.; Cohen-Tannoudji, J.; Simon, V. SET Protein Interacts with Intracellular Domains of the Gonadotropin-releasing Hormone Receptor and Differentially Regulates Receptor Signaling to cAMP and Calcium in Gonadotrope Cells. J. Biol. Chem. 2013, 288, 2641–2654. [Google Scholar] [CrossRef] [Green Version]
- Steen, A.; Larsen, O.; Thiele, S.; Rosenkilde, M.M. Biased and G Protein-Independent Signaling of Chemokine Receptors. Front. Immunol. 2014, 5, 277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, I.R.; Ciccone, N.A.; Zhou, Q.; Xu, S.; Khogeer, A.; Carroll, R.S.; Kaiser, U.B. GnRH Pulse Frequency Control of Fshb Gene Expression Is Mediated via ERK1/2 Regulation of ICER. Mol. Endocrinol. 2016, 30, 348–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer, S.I.; Dexheimer, V.; Nishida, E.; Kitajima, S.; Thiel, G. Expression of the Transcriptional Repressor ATF3 in Gonadotrophs Is Regulated by Egr-1, CREB, and ATF2 after Gonadotropin-Releasing Hormone Receptor Stimulation. Endocrinology 2008, 149, 6311–6325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, Y.L.; Ubuka, T.; Millar, R.P.; Kanasaki, H.; Tsutsui, K. Gonadotropin-Inhibitory Hormone Inhibits GnRH-Induced Gonadotropin Subunit Gene Transcriptions by Inhibiting AC/cAMP/PKA-Dependent ERK Pathway in LβT2 Cells. Endocrinology 2012, 153, 2332–2343. [Google Scholar] [CrossRef]
- Gardner, S.; Stavrou, E.; Rischitor, P.E.; Faccenda, E.; Pawson, A.J. Targeting mediators of Wnt signalling pathways by GnRH in gonadotropes. J. Mol. Endocrinol. 2010, 44, 195–201. [Google Scholar] [CrossRef] [Green Version]
- Riccetti, L.; Klett, D.; Ayoub, M.A.; Boulo, T.; Pignatti, E.; Tagliavini, S.; Varani, M.; Trenti, T.; Nicoli, A.; Capodanno, F.; et al. Heterogeneous hCG and hMG commercial preparations result in different intracellular signalling but induce a similar long-term progesterone response in vitro. Mol. Hum. Reprod. 2017, 23, 685–697. [Google Scholar] [CrossRef] [Green Version]
- Riccetti, L.; De Pascali, F.; Gilioli, L.; Potì, F.; Giva, L.B.; Marino, M.; Tagliavini, S.; Trenti, T.; Fanelli, F.; Mezzullo, M.; et al. Human LH and hCG stimulate differently the early signalling pathways but result in equal testosterone synthesis in mouse Leydig cells in vitro. Reprod. Boil. Endocrinol. 2017, 15, 2. [Google Scholar] [CrossRef]
- Tello, J.A.; Newton, C.L.; Bouligand, J.; Guiochon-Mantel, A.; Millar, R.P.; Young, J. Congenital Hypogonadotropic Hypogonadism Due to GNRH Receptor Mutations in Three Brothers Reveal Sites Affecting Conformation and Coupling. PLoS ONE 2012, 7, e38456. [Google Scholar] [CrossRef]
- Ayoub, M.A.; Landomiel, F.; Gallay, N.; Jégot, G.; Poupon, A.; Crépieux, P.; Reiter, E. Assessing Gonadotropin Receptor Function by Resonance Energy Transfer-Based Assays. Front. Endocrinol. 2015, 6, 130. [Google Scholar] [CrossRef] [Green Version]
- Riccetti, L.; Yvinec, R.; Klett, D.; Gallay, N.; Combarnous, Y.; Reiter, E.; Simoni, M.; Casarini, L.; Ayoub, M.A. Human Luteinizing Hormone and Chorionic Gonadotropin Display Biased Agonism at the LH and LH/CG Receptors. Sci. Rep. 2017, 7, 940. [Google Scholar] [CrossRef]
- Lazzaretti, C.; Riccetti, L.; Sperduti, S.; Anzivino, C.; Brigante, G.; De Pascali, F.; Potì, F.; Rovei, V.; Restagno, G.; Mari, C.; et al. Inferring biallelism of two FSH receptor mutations associated with spontaneous ovarian hyperstimulation syndrome by evaluating FSH, LH and HCG cross-activity. Reprod. Biomed. Online 2019, 38, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Casarini, L.; Riccetti, L.; Limoncella, S.; Lazzaretti, C.; Barbagallo, F.; Pacifico, S.; Guerrini, R.; Tagliavini, S.; Trenti, T.; Simoni, M.; et al. Probing the Effect of Sildenafil on Progesterone and Testosterone Production by an Intracellular FRET/BRET Combined Approach. Biochemistry 2019, 58, 799–808. [Google Scholar] [CrossRef] [PubMed]
- Brigante, G.; Riccetti, L.; Lazzaretti, C.; Rofrano, L.; Sperduti, S.; Potì, F.; Diazzi, C.; Prodam, F.; Guaraldi, G.; Lania, A.G.; et al. Abacavir, nevirapine, and ritonavir modulate intracellular calcium levels without affecting GHRH-mediated growth hormone secretion in somatotropic cells in vitro. Mol. Cell. Endocrinol. 2019, 482, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.I.; Collins, J.; Davis, R.; Lin, K.M.; DeCamp, D.; Roach, T.; Hsueh, R.; Rebres, R.A.; Ross, E.M.; Taussig, R.; et al. Use of a cAMP BRET sensor to characterize a novel regulation of cAMP by the sphingosine 1-phosphate/G13 pathway. J. Boil. Chem. 2007, 282, 10576–10584. [Google Scholar] [CrossRef]
- Casarini, L.; Reiter, E.; Simoni, M. β-arrestins regulate gonadotropin receptor-mediated cell proliferation and apoptosis by controlling different FSHR or LHCGR intracellular signaling in the hGL5 cell line. Mol. Cell. Endocrinol. 2016, 437, 11–21. [Google Scholar] [CrossRef]
- Casarini, L.; Riccetti, L.; De Pascali, F.; Gilioli, L.; Marino, M.; Vecchi, E.; Morini, D.; Nicoli, A.; La Sala, G.B.; Simoni, M. Estrogen Modulates Specific Life and Death Signals Induced by LH and hCG in Human Primary Granulosa Cells In Vitro. Int. J. Mol. Sci. 2017, 18, 926. [Google Scholar] [CrossRef]
- Collins, T.J. ImageJ for microscopy. Biotechniques 2007, 43, 25–30. [Google Scholar] [CrossRef]
- Bennett, C.N.; Ross, S.E.; Longo, K.A.; Bajnok, L.; Hemati, N.; Johnson, K.W.; Harrison, S.D.; MacDougald, O. Regulation of Wnt Signaling during Adipogenesis. J. Boil. Chem. 2002, 277, 30998–31004. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real- Time Quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
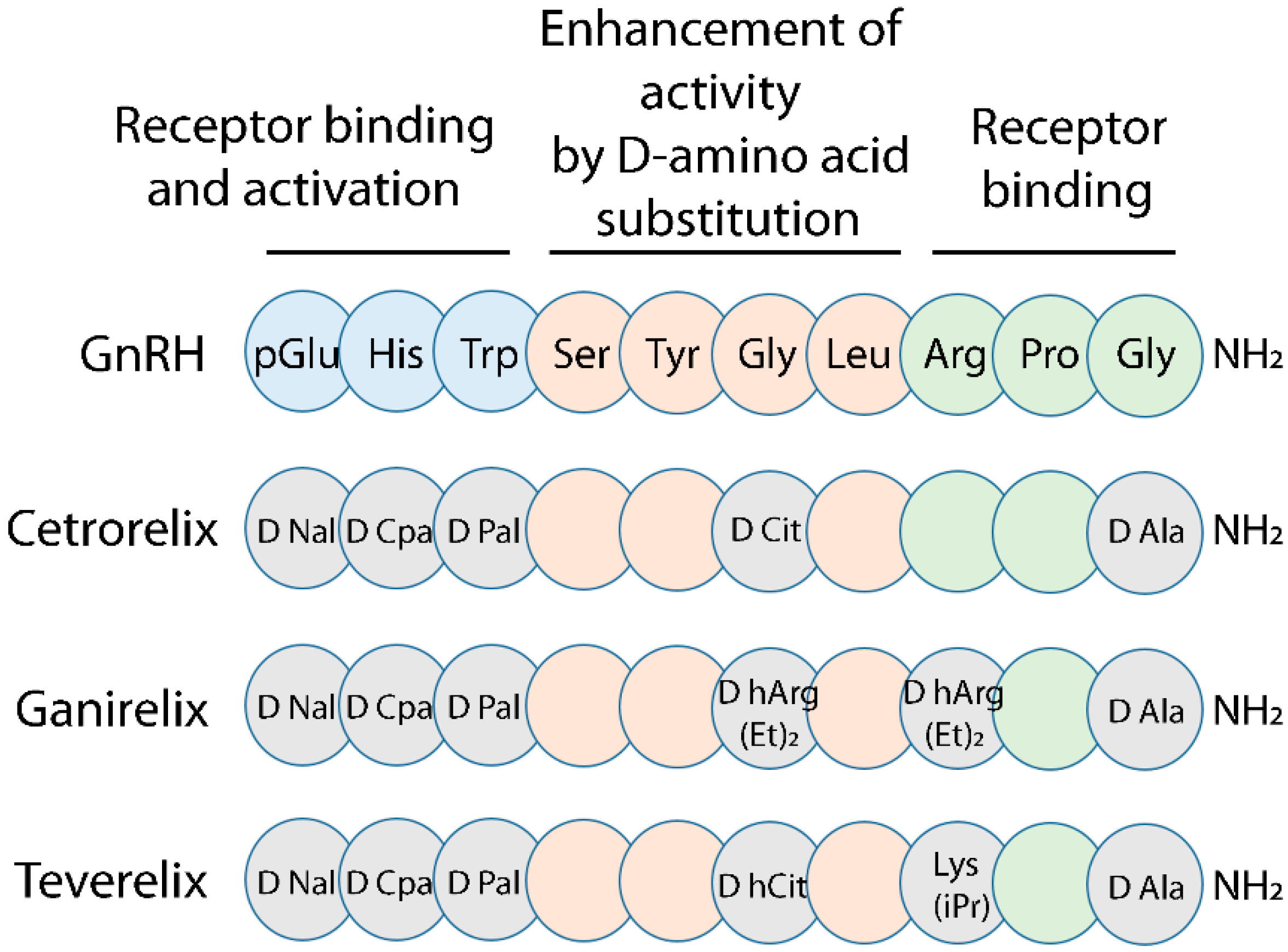




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No Antagonist | 109,340 ± 13,866 | ||
|---|---|---|---|
| Cetrorelix | Ganirelix | Teverelix | |
| 1 pM | 78,113 ± 29,979 | 76,189 ± 18,564 | 85,301 ± 21,321 |
| 10 pM | 124,981 ± 33,684 | 91,896 ± 21,766 | 98,939 ± 27,373 |
| 100 pM | 126,243 ± 34,982 | 101,943 ± 27,388 | 58,864 ± 27,101 |
| 1 nM | 71,127 ± 25,268 | 66,665 ± 16,035 | 81,702 ± 18,920 |
| 10 nM | 21,482 ± 6718 * | 73,164 ± 16,237 | 74,321 ± 17,569 |
| 75 nM | 18,632 ± 4423 * | 43,870 ± 4193 * | 42,401 ± 8974 * |
| 100 nM | 8946 ± 3296 * | 34,734 ± 13,731 * | 29,931 ± 8731 * |
| 1 µM | 3264 ± 3024 * | 3346 ± 2977 * | 4107 ± 3804 * |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sperduti, S.; Limoncella, S.; Lazzaretti, C.; Paradiso, E.; Riccetti, L.; Turchi, S.; Ferrigno, I.; Bertacchini, J.; Palumbo, C.; Potì, F.; et al. GnRH Antagonists Produce Differential Modulation of the Signaling Pathways Mediated by GnRH Receptors. Int. J. Mol. Sci. 2019, 20, 5548. https://doi.org/10.3390/ijms20225548
Sperduti S, Limoncella S, Lazzaretti C, Paradiso E, Riccetti L, Turchi S, Ferrigno I, Bertacchini J, Palumbo C, Potì F, et al. GnRH Antagonists Produce Differential Modulation of the Signaling Pathways Mediated by GnRH Receptors. International Journal of Molecular Sciences. 2019; 20(22):5548. https://doi.org/10.3390/ijms20225548
Chicago/Turabian StyleSperduti, Samantha, Silvia Limoncella, Clara Lazzaretti, Elia Paradiso, Laura Riccetti, Sara Turchi, Ilaria Ferrigno, Jessika Bertacchini, Carla Palumbo, Francesco Potì, and et al. 2019. "GnRH Antagonists Produce Differential Modulation of the Signaling Pathways Mediated by GnRH Receptors" International Journal of Molecular Sciences 20, no. 22: 5548. https://doi.org/10.3390/ijms20225548