The Crosstalk of miRNA and Oxidative Stress in the Liver: From Physiology to Pathology and Clinical Implications

 ,
,
Abstract
:1. Introduction
2. Physiology and Metabolism
3. Alcoholic Liver Disease (ALD) and Alcoholic Steatohepatitis (ASH)
4. Nonalcoholic Fatty Liver Disease (NAFLD) and Nonalcoholic Steatohepatitis (NASH)
5. Viral Hepatitis
6. Hepatocellular Carcinoma (HCC)
7. Clinical Implications/Studies
7.1. ALD and ASH
7.2. NAFLD/NASH
7.3. HCC
8. Summary
Author Contributions
Funding
Conflicts of Interest
References
- Cichoz-Lach, H.; Michalak, A. Oxidative stress as a crucial factor in liver diseases. World J. Gastroenterol. 2014, 20, 8082–8091. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S.; Maltepe, E.; Goldwasser, E.; Mathieu, C.E.; Simon, M.C.; Schumacker, P.T. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. USA 1998, 95, 11715–11720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Nemoto, S.; Takeda, K.; Yu, Z.X.; Ferrans, V.J.; Finkel, T. Role for mitochondrial oxidants as regulators of cellular metabolism. Mol. Cell. Biol. 2000, 20, 7311–7318. [Google Scholar] [CrossRef] [PubMed]
- Lo, Y.Y.; Cruz, T.F. Involvement of reactive oxygen species in cytokine and growth factor induction of c-fos expression in chondrocytes. J. Biol. Chem. 1995, 270, 11727–11730. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.S.; Kang, S.W.; Seo, M.S.; Baines, I.C.; Tekle, E.; Chock, P.B.; Rhee, S.G. Epidermal growth factor (EGF)-induced generation of hydrogen peroxide. Role in EGF receptor-mediated tyrosine phosphorylation. J. Biol. Chem. 1997, 272, 217–221. [Google Scholar] [CrossRef]
- Urao, N.; Ushio-Fukai, M. Redox regulation of stem/progenitor cells and bone marrow niche. Free Radic. Biol. Med. 2013, 54, 26–39. [Google Scholar] [CrossRef]
- The Alpha-Tocopherol Beta Carotene Cancer Prevention Study Group. The effect of vitamin E and beta carotene on the incidence of lung cancer and other cancers in male smokers. N. Engl. J. Med. 1994, 330, 1029–1035. [Google Scholar] [CrossRef]
- Watson, J. Oxidants, antioxidants and the current incurability of metastatic cancers. Open Biol. 2013, 3, 120144. [Google Scholar] [CrossRef] [Green Version]
- Anastasiou, D.; Poulogiannis, G.; Asara, J.M.; Boxer, M.B.; Jiang, J.K.; Shen, M.; Bellinger, G.; Sasaki, A.T.; Locasale, J.W.; Auld, D.S.; et al. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science 2011, 334, 1278–1283. [Google Scholar] [CrossRef]
- Mathew, R.; White, E. Autophagy, stress, and cancer metabolism: What doesn’t kill you makes you stronger. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Holmstrom, K.M.; Finkel, T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell. Biol. 2014, 15, 411–421. [Google Scholar] [CrossRef]
- Calin, G.A.; Croce, C.M. MicroRNA signatures in human cancers. Nat. Rev. Cancer 2006, 6, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Pichler, M.; Calin, G.A. MicroRNAs in cancer: From developmental genes in worms to their clinical application in patients. Br. J. Cancer 2015, 113, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Kinser, H.E.; Pincus, Z. MicroRNAs as modulators of longevity and the aging process. In Human Genetics; Springer: Berlin, Germany, 2019. [Google Scholar]
- Greco, S.; Gaetano, C.; Martelli, F. Long Noncoding Competing Endogenous RNA Networks in Age-Associated Cardiovascular Diseases. Int. J. Mol. Sci 2019, 20, 3079. [Google Scholar] [CrossRef] [PubMed]
- Piontek, K.; Selaru, F.M. MicroRNAs in the biology and diagnosis of cholangiocarcinoma. Semin. Liver Dis. 2015, 35, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Swierczynski, S.; Klieser, E.; Illig, R.; Alinger-Scharinger, B.; Kiesslich, T.; Neureiter, D. Histone deacetylation meets miRNA: Epigenetics and post-transcriptional regulation in cancer and chronic diseases. Expert Opin. Biol. Ther. 2015, 15, 651–664. [Google Scholar] [CrossRef]
- Gjorgjieva, M.; Sobolewski, C.; Dolicka, D.; Correia de Sousa, M.; Foti, M. miRNAs and NAFLD: From pathophysiology to therapy. Gut 2019, 68, 2065–2079. [Google Scholar] [CrossRef]
- Collin, F. Chemical Basis of Reactive Oxygen Species Reactivity and Involvement in Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 2407. [Google Scholar] [CrossRef]
- Glasauer, A.; Chandel, N.S. Ros. Curr. Biol. 2013, 23, R100–R102. [Google Scholar] [CrossRef]
- Parola, M.; Robino, G. Oxidative stress-related molecules and liver fibrosis. J. Hepatol. 2001, 35, 297–306. [Google Scholar] [CrossRef]
- Fierro-Fernandez, M.; Miguel, V.; Lamas, S. Role of redoximiRs in fibrogenesis. Redox Biol. 2016, 7, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. On the history of oxidative stress: Concept and some aspects of current development. Curr. Opin. Toxicol. 2018, 7, 122–126. [Google Scholar] [CrossRef]
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15. [Google Scholar] [CrossRef] [Green Version]
- Dickinson, B.C.; Chang, C.J. Chemistry and biology of reactive oxygen species in signaling or stress responses. Nat. Chem. Biol. 2011, 7, 504–511. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Wu, H.; Collier-Hyams, L.S.; Hansen, J.M.; Li, T.; Yamoah, K.; Pan, Z.Q.; Jones, D.P.; Neish, A.S. Commensal bacteria modulate cullin-dependent signaling via generation of reactive oxygen species. EMBO J. 2007, 26, 4457–4466. [Google Scholar] [CrossRef] [Green Version]
- Ha, E.M.; Oh, C.T.; Bae, Y.S.; Lee, W.J. A direct role for dual oxidase in Drosophila gut immunity. Science 2005, 310, 847–850. [Google Scholar] [CrossRef]
- Jones, R.M.; Luo, L.; Ardita, C.S.; Richardson, A.N.; Kwon, Y.M.; Mercante, J.W.; Alam, A.; Gates, C.L.; Wu, H.; Swanson, P.A.; et al. Symbiotic lactobacilli stimulate gut epithelial proliferation via Nox-mediated generation of reactive oxygen species. EMBO J. 2013, 32, 3017–3028. [Google Scholar] [CrossRef]
- Ceni, E.; Mello, T.; Galli, A. Pathogenesis of alcoholic liver disease: Role of oxidative metabolism. World J. Gastroenterol. 2014, 20, 17756–17772. [Google Scholar] [CrossRef]
- Li, S.; Tan, H.Y.; Wang, N.; Zhang, Z.J.; Lao, L.; Wong, C.W.; Feng, Y. The Role of Oxidative Stress and Antioxidants in Liver Diseases. Int. J. Mol. Sci. 2015, 16, 26087–26124. [Google Scholar] [CrossRef] [Green Version]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Francoz, C.; Belghiti, J.; Durand, F. Indications of liver transplantation in patients with complications of cirrhosis. Best Pract. Res. Clin. Gastroenterol. 2007, 21, 175–190. [Google Scholar] [CrossRef] [PubMed]
- Poli, G. Pathogenesis of liver fibrosis: Role of oxidative stress. Mol. Asp. Med. 2000, 21, 49–98. [Google Scholar] [CrossRef]
- Richter, K.; Kietzmann, T. Reactive oxygen species and fibrosis: Further evidence of a significant liaison. Cell Tissue Res. 2016, 365, 591–605. [Google Scholar] [CrossRef]
- Meroni, M.; Longo, M.; Rametta, R.; Dongiovanni, P. Genetic and Epigenetic Modifiers of Alcoholic Liver Disease. Int. J. Mol. Sci. 2018, 19, 3857. [Google Scholar] [CrossRef]
- Bala, S.; Marcos, M.; Kodys, K.; Csak, T.; Catalano, D.; Mandrekar, P.; Szabo, G. Up-regulation of microRNA-155 in macrophages contributes to increased tumor necrosis factor α (TNFα) production via increased mRNA half-life in alcoholic liver disease. J. Biol. Chem. 2011, 286, 1436–1444. [Google Scholar] [CrossRef]
- Hritz, I.; Mandrekar, P.; Velayudham, A.; Catalano, D.; Dolganiuc, A.; Kodys, K.; Kurt-Jones, E.; Szabo, G. The critical role of toll-like receptor (TLR) 4 in alcoholic liver disease is independent of the common TLR adapter MyD88. Hepatology 2008, 48, 1224–1231. [Google Scholar] [CrossRef]
- Bala, S.; Csak, T.; Saha, B.; Zatsiorsky, J.; Kodys, K.; Catalano, D.; Satishchandran, A.; Szabo, G. The pro-inflammatory effects of miR-155 promote liver fibrosis and alcohol-induced steatohepatitis. J. Hepatol. 2016, 64, 1378–1387. [Google Scholar] [CrossRef] [Green Version]
- Saikia, P.; Bellos, D.; McMullen, M.R.; Pollard, K.A.; de la Motte, C.; Nagy, L.E. MicroRNA 181b-3p and its target importin alpha5 regulate toll-like receptor 4 signaling in Kupffer cells and liver injury in mice in response to ethanol. Hepatology 2017, 66, 602–615. [Google Scholar] [CrossRef]
- Saikia, P.; Roychowdhury, S.; Bellos, D.; Pollard, K.A.; McMullen, M.R.; McCullough, R.L.; McCullough, A.J.; Gholam, P.; de la Motte, C.; Nagy, L.E. Hyaluronic acid 35 normalizes TLR4 signaling in Kupffer cells from ethanol-fed rats via regulation of microRNA291b and its target Tollip. Sci. Rep. 2017, 7, 15671. [Google Scholar] [CrossRef] [PubMed]
- Raver-Shapira, N.; Marciano, E.; Meiri, E.; Spector, Y.; Rosenfeld, N.; Moskovits, N.; Bentwich, Z.; Oren, M. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol. Cell 2007, 26, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Padhye, A.; Sharma, A.; Song, G.; Miao, J.; Mo, Y.Y.; Wang, L.; Kemper, J.K. A pathway involving farnesoid X receptor and small heterodimer partner positively regulates hepatic sirtuin 1 levels via microRNA-34a inhibition. J. Biol. Chem. 2010, 285, 12604–12611. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Liang, X.; Jogasuria, A.; Davidson, N.O.; You, M. miR-217 regulates ethanol-induced hepatic inflammation by disrupting sirtuin 1-lipin-1 signaling. Am. J. Pathol. 2015, 185, 1286–1296. [Google Scholar] [CrossRef]
- Satishchandran, A.; Ambade, A.; Rao, S.; Hsueh, Y.C.; Iracheta-Vellve, A.; Tornai, D.; Lowe, P.; Gyongyosi, B.; Li, J.; Catalano, D.; et al. MicroRNA 122, Regulated by GRLH2, Protects Livers of Mice and Patients from Ethanol-Induced Liver Disease. Gastroenterology 2018, 154, 238–252. [Google Scholar] [CrossRef]
- Li, F.; Duan, K.; Wang, C.; McClain, C.; Feng, W. Probiotics and Alcoholic Liver Disease: Treatment and Potential Mechanisms. Gastroenterol. Res. Pract. 2016, 2016, 5491465. [Google Scholar] [CrossRef]
- Szabo, G.; Bala, S.; Petrasek, J.; Gattu, A. Gut-liver axis and sensing microbes. Dig. Dis. 2010, 28, 737–744. [Google Scholar] [CrossRef]
- Ng, R.; Song, G.; Roll, G.R.; Frandsen, N.M.; Willenbring, H. A microRNA-21 surge facilitates rapid cyclin D1 translation and cell cycle progression in mouse liver regeneration. J. Clin. Investig. 2012, 122, 1097–1108. [Google Scholar] [CrossRef]
- Francis, H.; McDaniel, K.; Han, Y.; Liu, X.; Kennedy, L.; Yang, F.; McCarra, J.; Zhou, T.; Glaser, S.; Venter, J.; et al. Regulation of the extrinsic apoptotic pathway by microRNA-21 in alcoholic liver injury. J. Biol. Chem. 2014, 289, 27526–27539. [Google Scholar] [CrossRef]
- Xu, T.; Li, L.; Hu, H.Q.; Meng, X.M.; Huang, C.; Zhang, L.; Qin, J.; Li, J. MicroRNAs in alcoholic liver disease: Recent advances and future applications. J. Cell. Physiol. 2018, 234, 382–394. [Google Scholar] [CrossRef] [Green Version]
- Yeligar, S.; Tsukamoto, H.; Kalra, V.K. Ethanol-induced expression of ET-1 and ET-BR in liver sinusoidal endothelial cells and human endothelial cells involves hypoxia-inducible factor-1alpha and microrNA-199. J. Immunol. 2009, 183, 5232–5243. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Liu, H.; Chen, F.; Li, D.; Zhao, Y. MiR-214 promotes the alcohol-induced oxidative stress via down-regulation of glutathione reductase and cytochrome P450 oxidoreductase in liver cells. Alcohol. Clin. Exp. Res. 2014, 38, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yu, D.; Tolleson, W.H.; Yu, L.R.; Green, B.; Zeng, L.; Chen, Y.; Chen, S.; Ren, Z.; Guo, L.; et al. A systematic evaluation of microRNAs in regulating human hepatic CYP2E1. Biochem. Pharmacol. 2017, 138, 174–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araujo, A.R.; Rosso, N.; Bedogni, G.; Tiribelli, C.; Bellentani, S. Global epidemiology of non-alcoholic fatty liver disease/non-alcoholic steatohepatitis: What we need in the future. Liver Int. 2018, 38, 47–51. [Google Scholar] [CrossRef] [Green Version]
- Maher, J.J.; Leon, P.; Ryan, J.C. Beyond insulin resistance: Innate immunity in nonalcoholic steatohepatitis. Hepatology 2008, 48, 670–678. [Google Scholar] [CrossRef] [Green Version]
- Wruck, W.; Graffmann, N.; Kawala, M.A.; Adjaye, J. Concise Review: Current Status and Future Directions on Research Related to Nonalcoholic Fatty Liver Disease. Stem Cells 2017, 35, 89–96. [Google Scholar] [CrossRef]
- Manne, V.; Handa, P.; Kowdley, K.V. Pathophysiology of Nonalcoholic Fatty Liver Disease/Nonalcoholic Steatohepatitis. Clin. Liver Dis. 2018, 22, 23–37. [Google Scholar] [CrossRef]
- Magee, N.; Zou, A.; Zhang, Y. Pathogenesis of Nonalcoholic Steatohepatitis: Interactions between Liver Parenchymal and Nonparenchymal Cells. Biomed. Res. Int. 2016, 2016, 5170402. [Google Scholar] [CrossRef]
- Begriche, K.; Massart, J.; Robin, M.A.; Bonnet, F.; Fromenty, B. Mitochondrial adaptations and dysfunctions in nonalcoholic fatty liver disease. Hepatology 2013, 58, 1497–1507. [Google Scholar] [CrossRef]
- Hu, F.; Liu, F. Mitochondrial stress: A bridge between mitochondrial dysfunction and metabolic diseases? Cell. Signal. 2011, 23, 1528–1533. [Google Scholar] [CrossRef] [Green Version]
- Koliaki, C.; Szendroedi, J.; Kaul, K.; Jelenik, T.; Nowotny, P.; Jankowiak, F.; Herder, C.; Carstensen, M.; Krausch, M.; Knoefel, W.T.; et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 2015, 21, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, N.U.; Sheikh, T.A. Endoplasmic reticulum stress and Oxidative stress in the pathogenesis of Non-alcoholic fatty liver disease. Free Radic. Res. 2015, 49, 1405–1418. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, R.J. Stress signaling from the lumen of the endoplasmic reticulum: Coordination of gene transcriptional and translational controls. Genes Dev. 1999, 13, 1211–1233. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, Y.; Mauer, A.S.; Kumar, S.; Mott, J.L.; Malhi, H. Mmu-miR-615-3p regulates lipoapoptosis by inhibiting C/EBP homologous protein. PLoS ONE 2014, 9, e109637. [Google Scholar] [CrossRef]
- Park, S.W.; Zhou, Y.; Lee, J.; Lee, J.; Ozcan, U. Sarco(endo)plasmic reticulum Ca2+-ATPase 2b is a major regulator of endoplasmic reticulum stress and glucose homeostasis in obesity. Proc. Natl. Acad. Sci. USA 2010, 107, 19320–19325. [Google Scholar] [CrossRef]
- Zhang, J.; Li, Y.; Jiang, S.; Yu, H.; An, W. Enhanced endoplasmic reticulum SERCA activity by overexpression of hepatic stimulator substance gene prevents hepatic cells from ER stress-induced apoptosis. Am. J. Physiol. Cell Physiol. 2014, 306, C279–C290. [Google Scholar] [CrossRef] [Green Version]
- Chowdhry, S.; Nazmy, M.H.; Meakin, P.J.; Dinkova-Kostova, A.T.; Walsh, S.V.; Tsujita, T.; Dillon, J.F.; Ashford, M.L.; Hayes, J.D. Loss of Nrf2 markedly exacerbates nonalcoholic steatohepatitis. Free Radic. Biol. Med. 2010, 48, 357–371. [Google Scholar] [CrossRef]
- Sugimoto, H.; Okada, K.; Shoda, J.; Warabi, E.; Ishige, K.; Ueda, T.; Taguchi, K.; Yanagawa, T.; Nakahara, A.; Hyodo, I.; et al. Deletion of nuclear factor-E2-related factor-2 leads to rapid onset and progression of nutritional steatohepatitis in mice. Am. J. Physiol. Gastrointest Liver Physiol. 2010, 298, G283–G294. [Google Scholar] [CrossRef] [PubMed]
- Zhan, S.S.; Jiang, J.X.; Wu, J.; Halsted, C.; Friedman, S.L.; Zern, M.A.; Torok, N.J. Phagocytosis of apoptotic bodies by hepatic stellate cells induces NADPH oxidase and is associated with liver fibrosis in vivo. Hepatology 2006, 43, 435–443. [Google Scholar] [CrossRef]
- Gallego-Duran, R.; Romero-Gomez, M. Epigenetic mechanisms in non-alcoholic fatty liver disease: An emerging field. World J. Hepatol. 2015, 7, 2497–2502. [Google Scholar] [CrossRef]
- Dattaroy, D.; Pourhoseini, S.; Das, S.; Alhasson, F.; Seth, R.K.; Nagarkatti, M.; Michelotti, G.A.; Diehl, A.M.; Chatterjee, S. Micro-RNA 21 inhibition of SMAD7 enhances fibrogenesis via leptin-mediated NADPH oxidase in experimental and human nonalcoholic steatohepatitis. Am. J. Physiol. Gastrointest Liver Physiol. 2015, 308, G298–G312. [Google Scholar] [CrossRef] [PubMed]
- Vinciguerra, M.; Sgroi, A.; Veyrat-Durebex, C.; Rubbia-Brandt, L.; Buhler, L.H.; Foti, M. Unsaturated fatty acids inhibit the expression of tumor suppressor phosphatase and tensin homolog (PTEN) via microRNA-21 up-regulation in hepatocytes. Hepatology 2009, 49, 1176–1184. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, P.M.; Afonso, M.B.; Simao, A.L.; Carvalho, C.C.; Trindade, A.; Duarte, A.; Borralho, P.M.; Machado, M.V.; Cortez-Pinto, H.; Rodrigues, C.M.; et al. miR-21 ablation and obeticholic acid ameliorate nonalcoholic steatohepatitis in mice. Cell Death Dis. 2017, 8, e2748. [Google Scholar] [CrossRef] [PubMed]
- Loyer, X.; Paradis, V.; Henique, C.; Vion, A.C.; Colnot, N.; Guerin, C.L.; Devue, C.; On, S.; Scetbun, J.; Romain, M.; et al. Liver microRNA-21 is overexpressed in non-alcoholic steatohepatitis and contributes to the disease in experimental models by inhibiting PPARalpha expression. Gut 2016, 65, 1882–1894. [Google Scholar] [CrossRef]
- Sulaiman, S.A.; Muhsin, N.I.A.; Jamal, R. Regulatory Non-coding RNAs Network in Non-alcoholic Fatty Liver Disease. Front. Physiol. 2019, 10, 279. [Google Scholar] [CrossRef]
- Brandt, S.; Roos, J.; Inzaghi, E.; Kotnik, P.; Kovac, J.; Battelino, T.; Cianfarani, S.; Nobili, V.; Colajacomo, M.; Kratzer, W.; et al. Circulating levels of miR-122 and nonalcoholic fatty liver disease in pre-pubertal obese children. Pediatr. Obes. 2018, 13, 175–182. [Google Scholar] [CrossRef]
- Latorre, J.; Moreno-Navarrete, J.M.; Mercader, J.M.; Sabater, M.; Rovira, O.; Girones, J.; Ricart, W.; Fernandez-Real, J.M.; Ortega, F.J. Decreased lipid metabolism but increased FA biosynthesis are coupled with changes in liver microRNAs in obese subjects with NAFLD. Int. J. Obes. (Lond.) 2017, 41, 620–630. [Google Scholar] [CrossRef]
- Su, Q.; Kumar, V.; Sud, N.; Mahato, R.I. MicroRNAs in the pathogenesis and treatment of progressive liver injury in NAFLD and liver fibrosis. Adv. Drug Deliv. Rev. 2018, 129, 54–63. [Google Scholar] [CrossRef]
- Cermelli, S.; Ruggieri, A.; Marrero, J.A.; Ioannou, G.N.; Beretta, L. Circulating microRNAs in patients with chronic hepatitis C and non-alcoholic fatty liver disease. PLoS ONE 2011, 6, e23937. [Google Scholar] [CrossRef]
- Miyaaki, H.; Ichikawa, T.; Kamo, Y.; Taura, N.; Honda, T.; Shibata, H.; Milazzo, M.; Fornari, F.; Gramantieri, L.; Bolondi, L.; et al. Significance of serum and hepatic microRNA-122 levels in patients with non-alcoholic fatty liver disease. Liver Int. 2014, 34, e302–e307. [Google Scholar] [CrossRef]
- Yu, F.; Jiang, Z.; Chen, B.; Dong, P.; Zheng, J. NEAT1 accelerates the progression of liver fibrosis via regulation of microRNA-122 and Kruppel-like factor 6. J. Mol. Med. (Berl.) 2017, 95, 1191–1202. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.H.; Wang, B.; Kota, J.; Yu, J.; Costinean, S.; Kutay, H.; Yu, L.; Bai, S.; La Perle, K.; Chivukula, R.R.; et al. Essential metabolic, anti-inflammatory, and anti-tumorigenic functions of miR-122 in liver. J. Clin. Investig. 2012, 122, 2871–2883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salvoza, N.C.; Klinzing, D.C.; Gopez-Cervantes, J.; Baclig, M.O. Association of Circulating Serum miR-34a and miR-122 with Dyslipidemia among Patients with Non-Alcoholic Fatty Liver Disease. PLoS ONE 2016, 11, e0153497. [Google Scholar] [CrossRef] [PubMed]
- Yamada, H.; Suzuki, K.; Ichino, N.; Ando, Y.; Sawada, A.; Osakabe, K.; Sugimoto, K.; Ohashi, K.; Teradaira, R.; Inoue, T.; et al. Associations between circulating microRNAs (miR-21, miR-34a, miR-122 and miR-451) and non-alcoholic fatty liver. Clin. Chim. Acta 2013, 424, 99–103. [Google Scholar] [CrossRef]
- Chang, T.C.; Wentzel, E.A.; Kent, O.A.; Ramachandran, K.; Mullendore, M.; Lee, K.H.; Feldmann, G.; Yamakuchi, M.; Ferlito, M.; Lowenstein, C.J.; et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol. Cell 2007, 26, 745–752. [Google Scholar] [CrossRef]
- Ding, J.; Li, M.; Wan, X.; Jin, X.; Chen, S.; Yu, C.; Li, Y. Effect of miR-34a in regulating steatosis by targeting PPARalpha expression in nonalcoholic fatty liver disease. Sci. Rep. 2015, 5, 13729. [Google Scholar] [CrossRef]
- Shan, W.; Gao, L.; Zeng, W.; Hu, Y.; Wang, G.; Li, M.; Zhou, J.; Ma, X.; Tian, X.; Yao, J. Activation of the SIRT1/p66shc antiapoptosis pathway via carnosic acid-induced inhibition of miR-34a protects rats against nonalcoholic fatty liver disease. Cell Death Dis. 2015, 6, e1833. [Google Scholar] [CrossRef]
- Xu, Y.; Zalzala, M.; Xu, J.; Li, Y.; Yin, L.; Zhang, Y. A metabolic stress-inducible miR-34a-HNF4alpha pathway regulates lipid and lipoprotein metabolism. Nat. Commun. 2015, 6, 7466. [Google Scholar] [CrossRef]
- Castro, R.E.; Ferreira, D.M.; Afonso, M.B.; Borralho, P.M.; Machado, M.V.; Cortez-Pinto, H.; Rodrigues, C.M. miR-34a/SIRT1/p53 is suppressed by ursodeoxycholic acid in the rat liver and activated by disease severity in human non-alcoholic fatty liver disease. J. Hepatol. 2013, 58, 119–125. [Google Scholar] [CrossRef]
- Derdak, Z.; Villegas, K.A.; Harb, R.; Wu, A.M.; Sousa, A.; Wands, J.R. Inhibition of p53 attenuates steatosis and liver injury in a mouse model of non-alcoholic fatty liver disease. J. Hepatol. 2013, 58, 785–791. [Google Scholar] [CrossRef]
- Braza-Boils, A.; Mari-Alexandre, J.; Molina, P.; Arnau, M.A.; Barcelo-Molina, M.; Domingo, D.; Girbes, J.; Giner, J.; Martinez-Dolz, L.; Zorio, E. Deregulated hepatic microRNAs underlie the association between non-alcoholic fatty liver disease and coronary artery disease. Liver Int. 2016, 36, 1221–1229. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Benz, F.; Luedde, T.; Roderburg, C. The role of miRNAs in the regulation of inflammatory processes during hepatofibrogenesis. Hepatobiliary Surg. Nutr. 2015, 4, 24–33. [Google Scholar] [PubMed]
- Liu, M.X.; Gao, M.; Li, C.Z.; Yu, C.Z.; Yan, H.; Peng, C.; Li, Y.; Li, C.G.; Ma, Z.L.; Zhao, Y.; et al. Dicer1/miR-29/HMGCR axis contributes to hepatic free cholesterol accumulation in mouse non-alcoholic steatohepatitis. Acta Pharmacol. Sin. 2017, 38, 660–671. [Google Scholar] [CrossRef] [PubMed]
- Mattis, A.N.; Song, G.; Hitchner, K.; Kim, R.Y.; Lee, A.Y.; Sharma, A.D.; Malato, Y.; McManus, M.T.; Esau, C.C.; Koller, E.; et al. A screen in mice uncovers repression of lipoprotein lipase by microRNA-29a as a mechanism for lipid distribution away from the liver. Hepatology 2015, 61, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, C.L.; Fannin, E.E.; Toth, C.L.; Pearson, D.S.; Vickers, K.C.; Sethupathy, P. Inhibition of miR-29 has a significant lipid-lowering benefit through suppression of lipogenic programs in liver. Sci. Rep. 2015, 5, 12911. [Google Scholar] [CrossRef] [Green Version]
- Bandyopadhyay, S.; Friedman, R.C.; Marquez, R.T.; Keck, K.; Kong, B.; Icardi, M.S.; Brown, K.E.; Burge, C.B.; Schmidt, W.N.; Wang, Y.; et al. Hepatitis C virus infection and hepatic stellate cell activation downregulate miR-29: miR-29 overexpression reduces hepatitis C viral abundance in culture. J. Infect. Dis. 2011, 203, 1753–1762. [Google Scholar] [CrossRef]
- Pogribny, I.P.; Starlard-Davenport, A.; Tryndyak, V.P.; Han, T.; Ross, S.A.; Rusyn, I.; Beland, F.A. Difference in expression of hepatic microRNAs miR-29c, miR-34a, miR-155, and miR-200b is associated with strain-specific susceptibility to dietary nonalcoholic steatohepatitis in mice. Lab. Investig. 2010, 90, 1437–1446. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Ren, T.; Zhu, Z.; Cheng, M.; Mou, Q.; Mu, M.; Liu, Y.; Yao, Y.; Cheng, Y.; Zhang, B.; et al. Thymosin-beta4 Mediates Hepatic Stellate Cell Activation by Interfering with CircRNA-0067835/miR-155/FoxO3 Signaling Pathway. Cell. Physiol. Biochem. 2018, 51, 1389–1398. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, N.; Wang, Z.; Ai, D.M.; Cao, Z.Y.; Pan, H.P. Decreased MiR-155 Level in the Peripheral Blood of Non-Alcoholic Fatty Liver Disease Patients may Serve as a Biomarker and may Influence LXR Activity. Cell. Physiol. Biochem. 2016, 39, 2239–2248. [Google Scholar] [CrossRef] [Green Version]
- Csak, T.; Bala, S.; Lippai, D.; Satishchandran, A.; Catalano, D.; Kodys, K.; Szabo, G. microRNA-122 regulates hypoxia-inducible factor-1 and vimentin in hepatocytes and correlates with fibrosis in diet-induced steatohepatitis. Liver Int. 2015, 35, 532–541. [Google Scholar] [CrossRef]
- Csak, T.; Bala, S.; Lippai, D.; Kodys, K.; Catalano, D.; Iracheta-Vellve, A.; Szabo, G. MicroRNA-155 Deficiency Attenuates Liver Steatosis and Fibrosis without Reducing Inflammation in a Mouse Model of Steatohepatitis. PLoS ONE 2015, 10, e0129251. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Koda, M.; Okamoto, T.; Onoyama, T.; Miyoshi, K.; Kishina, M.; Kato, J.; Tokunaga, S.; Sugihara, T.A.; Hara, Y.; et al. A Series of microRNA in the Chromosome 14q32.2 Maternally Imprinted Region Related to Progression of Non-Alcoholic Fatty Liver Disease in a Mouse Model. PLoS ONE 2016, 11, e0154676. [Google Scholar] [CrossRef] [PubMed]
- Behrman, S.; Acosta-Alvear, D.; Walter, P. A CHOP-regulated microRNA controls rhodopsin expression. J. Cell Biol. 2011, 192, 919–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chitnis, N.S.; Pytel, D.; Bobrovnikova-Marjon, E.; Pant, D.; Zheng, H.; Maas, N.L.; Frederick, B.; Kushner, J.A.; Chodosh, L.A.; Koumenis, C.; et al. miR-211 is a prosurvival microRNA that regulates chop expression in a PERK-dependent manner. Mol. Cell 2012, 48, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.G.; Chen, L.; Dong, Q.; He, J.; Zhao, H.D.; Li, F.L.; Li, H. Mmu-miR-702 functions as an anti-apoptotic mirtron by mediating ATF6 inhibition in mice. Gene 2013, 531, 235–242. [Google Scholar] [CrossRef]
- Belmont, P.J.; Chen, W.J.; Thuerauf, D.J.; Glembotski, C.C. Regulation of microRNA expression in the heart by the ATF6 branch of the ER stress response. J. Mol. Cell Cardiol. 2012, 52, 1176–1182. [Google Scholar] [CrossRef] [Green Version]
- Malhi, H. Micrornas in er stress: Divergent roles in cell fate decisions. Curr. Pathobiol. Rep. 2014, 2, 117–122. [Google Scholar] [CrossRef]
- WHO. Hepatitis C. Available online: https://www.who.int/news-room/fact-sheets/detail/hepatitis-c (accessed on 29 September 2019).
- WHO. Hepatitis B. Available online: https://www.who.int/news-room/fact-sheets/detail/hepatitis-b (accessed on 29 September 2019).
- Choi, J.; Ou, J.H. Mechanisms of liver injury. III. Oxidative stress in the pathogenesis of hepatitis C virus. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G847–G851. [Google Scholar] [CrossRef]
- Muriel, P. Role of free radicals in liver diseases. Hepatol. Int. 2009, 3, 526–536. [Google Scholar] [CrossRef] [Green Version]
- Farinati, F.; Cardin, R.; De Maria, N.; Della Libera, G.; Marafin, C.; Lecis, E.; Burra, P.; Floreani, A.; Cecchetto, A.; Naccarato, R. Iron storage, lipid peroxidation and glutathione turnover in chronic anti-HCV positive hepatitis. J. Hepatol. 1995, 22, 449–456. [Google Scholar] [CrossRef]
- Hou, W.; Tian, Q.; Zheng, J.; Bonkovsky, H.L. MicroRNA-196 represses Bach1 protein and hepatitis C virus gene expression in human hepatoma cells expressing hepatitis C viral proteins. Hepatology 2010, 51, 1494–1504. [Google Scholar] [CrossRef] [PubMed]
- Jopling, C.L.; Yi, M.; Lancaster, A.M.; Lemon, S.M.; Sarnow, P. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science 2005, 309, 1577–1581. [Google Scholar] [CrossRef]
- Sarnow, P.; Sagan, S.M. Unraveling the Mysterious Interactions Between Hepatitis C Virus RNA and Liver-Specific MicroRNA-122. Annu. Rev. Virol. 2016, 3, 309–332. [Google Scholar] [CrossRef] [PubMed]
- Lowey, B.; Hertz, L.; Chiu, S.; Valdez, K.; Li, Q.; Liang, T.J. Hepatitis C Virus Infection Induces Hepatic Expression of NF-kappaB-Inducing Kinase and Lipogenesis by Downregulating miR-122. MBio 2019, 10, e01617-19. [Google Scholar] [CrossRef]
- Goh, G.Y.S.; Winter, J.J.; Bhanshali, F.; Doering, K.R.S.; Lai, R.; Lee, K.; Veal, E.A.; Taubert, S. NHR-49/HNF4 integrates regulation of fatty acid metabolism with a protective transcriptional response to oxidative stress and fasting. Aging Cell 2018, 17, e12743. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Pene, V.; Krishnamurthy, S.; Cha, H.; Liang, T.J. Hepatitis C virus infection activates an innate pathway involving IKK-alpha in lipogenesis and viral assembly. Nat. Med. 2013, 19, 722–729. [Google Scholar] [CrossRef]
- Wang, S.; Qiu, L.; Yan, X.; Jin, W.; Wang, Y.; Chen, L.; Wu, E.; Ye, X.; Gao, G.F.; Wang, F.; et al. Loss of microRNA 122 expression in patients with hepatitis B enhances hepatitis B virus replication through cyclin G(1) -modulated P53 activity. Hepatology 2012, 55, 730–741. [Google Scholar] [CrossRef]
- Wojcik, K.; Piekarska, A.; Szymanska, B.; Jablonowska, E. Hepatic expression of miR-122 and antioxidant genes in patients with chronic hepatitis B. Acta Biochim. Pol. 2016, 63, 527–531. [Google Scholar] [CrossRef]
- Varnholt, H.; Drebber, U.; Schulze, F.; Wedemeyer, I.; Schirmacher, P.; Dienes, H.P.; Odenthal, M. MicroRNA gene expression profile of hepatitis C virus-associated hepatocellular carcinoma. Hepatology 2008, 47, 1223–1232. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Petrick, J.L.; McGlynn, K.A. The changing epidemiology of primary liver cancer. Curr. Epidemiol. Rep. 2019, 6, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Reeves, H.L.; Kotsiliti, E.; Govaere, O.; Heikenwalder, M. From NASH to HCC: Current concepts and future challenges. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 411–428. [Google Scholar] [CrossRef] [PubMed]
- Neureiter, D.; Stintzing, S.; Kiesslich, T.; Ocker, M. Hepatocellular carcinoma: Therapeutic advances in signaling, epigenetic and immune targets. World J. Gastroenterol. 2019, 25, 3136–3150. [Google Scholar] [CrossRef] [PubMed]
- Engedal, N.; Zerovnik, E.; Rudov, A.; Galli, F.; Olivieri, F.; Procopio, A.D.; Rippo, M.R.; Monsurro, V.; Betti, M.; Albertini, M.C. From Oxidative Stress Damage to Pathways, Networks, and Autophagy via MicroRNAs. Oxid. Med. Cell. Longev. 2018, 2018, 4968321. [Google Scholar] [CrossRef]
- Mansouri, A.; Gattolliat, C.H.; Asselah, T. Mitochondrial Dysfunction and Signaling in Chronic Liver Diseases. Gastroenterology 2018, 155, 629–647. [Google Scholar] [CrossRef] [Green Version]
- Masarone, M.; Rosato, V.; Dallio, M.; Gravina, A.G.; Aglitti, A.; Loguercio, C.; Federico, A.; Persico, M. Role of Oxidative Stress in Pathophysiology of Nonalcoholic Fatty Liver Disease. Oxid. Med. Cell. Longev. 2018, 2018, 9547613. [Google Scholar] [CrossRef]
- He, Z.; Yu, Y.; Nong, Y.; Du, L.; Liu, C.; Cao, Y.; Bai, L.; Tang, H. Hepatitis B virus X protein promotes hepatocellular carcinoma invasion and metastasis via upregulating thioredoxin interacting protein. Oncol. Lett. 2017, 14, 1323–1332. [Google Scholar] [CrossRef]
- Ivanov, A.V.; Bartosch, B.; Smirnova, O.A.; Isaguliants, M.G.; Kochetkov, S.N. HCV and oxidative stress in the liver. Viruses 2013, 5, 439–469. [Google Scholar] [CrossRef]
- Medvedev, R.; Ploen, D.; Hildt, E. HCV and Oxidative Stress: Implications for HCV Life Cycle and HCV-Associated Pathogenesis. Oxid. Med. Cell. Longev. 2016, 2016, 9012580. [Google Scholar] [CrossRef]
- Severi, T.; Vander Borght, S.; Libbrecht, L.; VanAelst, L.; Nevens, F.; Roskams, T.; Cassiman, D.; Fevery, J.; Verslype, C.; van Pelt, J.F. HBx or HCV core gene expression in HepG2 human liver cells results in a survival benefit against oxidative stress with possible implications for HCC development. Chem. Biol. Interact. 2007, 168, 128–134. [Google Scholar] [CrossRef]
- Zhong, L.; Shu, W.; Dai, W.; Gao, B.; Xiong, S. Reactive Oxygen Species-Mediated c-Jun NH2-Terminal Kinase Activation Contributes to Hepatitis B Virus X Protein-Induced Autophagy via Regulation of the Beclin-1/Bcl-2 Interaction. J. Virol. 2017, 91, e00001-17. [Google Scholar] [CrossRef] [PubMed]
- Marra, M.; Sordelli, I.M.; Lombardi, A.; Lamberti, M.; Tarantino, L.; Giudice, A.; Stiuso, P.; Abbruzzese, A.; Sperlongano, R.; Accardo, M.; et al. Molecular targets and oxidative stress biomarkers in hepatocellular carcinoma: An overview. J. Transl. Med. 2011, 9, 171. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Fujita, N.; Sugimoto, R.; Urawa, N.; Horiike, S.; Kobayashi, Y.; Iwasa, M.; Ma, N.; Kawanishi, S.; Watanabe, S.; et al. Hepatic oxidative DNA damage is associated with increased risk for hepatocellular carcinoma in chronic hepatitis C. Br. J. Cancer 2008, 98, 580–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Wang, X.; Wu, Y.; Zhang, H.; Zhang, L.; Wang, C.; Zhang, R.; Guo, Z. 8-Hydroxy-2’-deoxyguanosine expression predicts hepatocellular carcinoma outcome. Oncol. Lett. 2012, 3, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Zhuang, Z.; Wang, W.; He, L.; Wu, H.; Cao, Y.; Pan, F.; Zhao, J.; Hu, Z.; Sekhar, C.; et al. OGG1 is essential in oxidative stress induced DNA demethylation. Cell. Signal. 2016, 28, 1163–1171. [Google Scholar] [CrossRef]
- Sartorius, K.; Sartorius, B.; Winkler, C.; Chuturgoon, A.; Makarova, J. The biological and diagnostic role of miRNA’s in hepatocellular carcinoma. Front. Biosci. 2018, 23, 1701–1720. [Google Scholar] [CrossRef]
- Ji, J.; Shi, J.; Budhu, A.; Yu, Z.; Forgues, M.; Roessler, S.; Ambs, S.; Chen, Y.; Meltzer, P.S.; Croce, C.M.; et al. MicroRNA expression, survival, and response to interferon in liver cancer. N. Engl. J. Med. 2009, 361, 1437–1447. [Google Scholar] [CrossRef]
- Wong, C.M.; Tsang, F.H.; Ng, I.O. Non-coding RNAs in hepatocellular carcinoma: Molecular functions and pathological implications. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 137–151. [Google Scholar] [CrossRef]
- Deng, M.; Zhang, R.; He, Z.; Qiu, Q.; Lu, X.; Yin, J.; Liu, H.; Jia, X.; He, Z. TET-Mediated Sequestration of miR-26 Drives EZH2 Expression and Gastric Carcinogenesis. Cancer Res. 2017, 77, 6069–6082. [Google Scholar] [CrossRef]
- Lu, J.; He, M.L.; Wang, L.; Chen, Y.; Liu, X.; Dong, Q.; Chen, Y.C.; Peng, Y.; Yao, K.T.; Kung, H.F.; et al. MiR-26a inhibits cell growth and tumorigenesis of nasopharyngeal carcinoma through repression of EZH2. Cancer Res. 2011, 71, 225–233. [Google Scholar] [CrossRef]
- Lin, L.L.; Wang, W.; Hu, Z.; Wang, L.W.; Chang, J.; Qian, H. Erratum to: Negative feedback of miR-29 family TET1 involves in hepatocellular cancer. Med. Oncol. 2015, 32, 39. [Google Scholar] [CrossRef] [Green Version]
- Chuang, K.H.; Whitney-Miller, C.L.; Chu, C.Y.; Zhou, Z.; Dokus, M.K.; Schmit, S.; Barry, C.T. MicroRNA-494 is a master epigenetic regulator of multiple invasion-suppressor microRNAs by targeting ten eleven translocation 1 in invasive human hepatocellular carcinoma tumors. Hepatology 2015, 62, 466–480. [Google Scholar] [CrossRef]
- Zhang, W.; Lu, Z.; Gao, Y.; Ye, L.; Song, T.; Zhang, X. MiR-520b suppresses proliferation of hepatoma cells through targeting ten-eleven translocation 1 (TET1) mRNA. Biochem. Biophys. Res. Commun. 2015, 460, 793–798. [Google Scholar] [CrossRef]
- Cardin, R.; Piciocchi, M.; Sinigaglia, A.; Lavezzo, E.; Bortolami, M.; Kotsafti, A.; Cillo, U.; Zanus, G.; Mescoli, C.; Rugge, M.; et al. Oxidative DNA damage correlates with cell immortalization and mir-92 expression in hepatocellular carcinoma. BMC Cancer 2012, 12, 177. [Google Scholar] [CrossRef]
- Henrici, A.; Montalbano, R.; Neureiter, D.; Krause, M.; Stiewe, T.; Slater, E.P.; Quint, K.; Ocker, M.; Di Fazio, P. The pan-deacetylase inhibitor panobinostat suppresses the expression of oncogenic miRNAs in hepatocellular carcinoma cell lines. Mol. Carcinog. 2015, 54, 585–597. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, Y.; Han, N.; Chen, X.; Yu, W.; Zhang, W.; Zou, F. Profiles of oxidative stress-related microRNA and mRNA expression in auditory cells. Brain Res. 2010, 1346, 14–25. [Google Scholar] [CrossRef]
- Lei, P.P.; Zhang, Z.J.; Shen, L.J.; Li, J.Y.; Zou, Q.; Zhang, H.X. Expression and hypermethylation of p27 kip1 in hepatocarcinogenesis. World J. Gastroenterol. 2005, 11, 4587–4591. [Google Scholar] [CrossRef]
- Di Fazio, P.; Montalbano, R.; Neureiter, D.; Alinger, B.; Schmidt, A.; Merkel, A.L.; Quint, K.; Ocker, M. Downregulation of HMGA2 by the pan-deacetylase inhibitor panobinostat is dependent on hsa-let-7b expression in liver cancer cell lines. Exp. Cell Res. 2012, 318, 1832–1843. [Google Scholar] [CrossRef]
- Pillon Barcelos, R.; Freire Royes, L.F.; Gonzalez-Gallego, J.; Bresciani, G. Oxidative stress and inflammation: Liver responses and adaptations to acute and regular exercise. Free Radic. Res. 2017, 51, 222–236. [Google Scholar] [CrossRef]
- Farias, M.S.; Budni, P.; Ribeiro, C.M.; Parisotto, E.B.; Santos, C.E.; Dias, J.F.; Dalmarco, E.M.; Frode, T.S.; Pedrosa, R.C.; Wilhelm Filho, D. Antioxidant supplementation attenuates oxidative stress in chronic hepatitis C patients. Gastroenterol. Hepatol. 2012, 35, 386–394. [Google Scholar] [CrossRef]
- Chakraborty, C.; Sharma, A.R.; Sharma, G.; Doss, C.G.P.; Lee, S.S. Therapeutic miRNA and siRNA: Moving from Bench to Clinic as Next Generation Medicine. Mol. Ther. Nucleic Acids 2017, 8, 132–143. [Google Scholar] [CrossRef] [Green Version]
- Vienberg, S.; Geiger, J.; Madsen, S.; Dalgaard, L.T. MicroRNAs in metabolism. Acta Physiol. (Oxf.) 2017, 219, 346–361. [Google Scholar] [CrossRef] [PubMed]
- Elmen, J.; Lindow, M.; Schutz, S.; Lawrence, M.; Petri, A.; Obad, S.; Lindholm, M.; Hedtjarn, M.; Hansen, H.F.; Berger, U.; et al. LNA-mediated microRNA silencing in non-human primates. Nature 2008, 452, 896–899. [Google Scholar] [CrossRef] [PubMed]
- Petri, A.; Lindow, M.; Kauppinen, S. MicroRNA silencing in primates: Towards development of novel therapeutics. Cancer Res. 2009, 69, 393–395. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y. Preclinical and Clinical Advances of GalNAc-Decorated Nucleic Acid Therapeutics. Mol. Ther. Nucleic Acids 2017, 6, 116–132. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Le, Q.T.; Giaccia, A.J. MiR-210--micromanager of the hypoxia pathway. Trends Mol. Med. 2010, 16, 230–237. [Google Scholar] [CrossRef]
- Tsuchiya, S.; Fujiwara, T.; Sato, F.; Shimada, Y.; Tanaka, E.; Sakai, Y.; Shimizu, K.; Tsujimoto, G. MicroRNA-210 regulates cancer cell proliferation through targeting fibroblast growth factor receptor-like 1 (FGFRL1). J. Biol. Chem. 2011, 286, 420–428. [Google Scholar] [CrossRef]
- Puissegur, M.P.; Mazure, N.M.; Bertero, T.; Pradelli, L.; Grosso, S.; Robbe-Sermesant, K.; Maurin, T.; Lebrigand, K.; Cardinaud, B.; Hofman, V.; et al. miR-210 is overexpressed in late stages of lung cancer and mediates mitochondrial alterations associated with modulation of HIF-1 activity. Cell Death Differ. 2011, 18, 465–478. [Google Scholar] [CrossRef]
- Devlin, C.; Greco, S.; Martelli, F.; Ivan, M. miR-210: More than a silent player in hypoxia. IUBMB Life 2011, 63, 94–100. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Ding, L.; Bennewith, K.L.; Tong, R.T.; Welford, S.M.; Ang, K.K.; Story, M.; Le, Q.T.; Giaccia, A.J. Hypoxia-inducible mir-210 regulates normoxic gene expression involved in tumor initiation. Mol. Cell 2009, 35, 856–867. [Google Scholar] [CrossRef]
- Momen-Heravi, F.; Saha, B.; Kodys, K.; Catalano, D.; Satishchandran, A.; Szabo, G. Increased number of circulating exosomes and their microRNA cargos are potential novel biomarkers in alcoholic hepatitis. J. Transl. Med. 2015, 13, 261. [Google Scholar] [CrossRef]
- Trajkovski, M.; Hausser, J.; Soutschek, J.; Bhat, B.; Akin, A.; Zavolan, M.; Heim, M.H.; Stoffel, M. MicroRNAs 103 and 107 regulate insulin sensitivity. Nature 2011, 474, 649–653. [Google Scholar] [CrossRef] [Green Version]
- Bala, S.; Petrasek, J.; Mundkur, S.; Catalano, D.; Levin, I.; Ward, J.; Alao, H.; Kodys, K.; Szabo, G. Circulating microRNAs in exosomes indicate hepatocyte injury and inflammation in alcoholic, drug-induced, and inflammatory liver diseases. Hepatology 2012, 56, 1946–1957. [Google Scholar] [CrossRef] [Green Version]
- Janssen, H.L.; Reesink, H.W.; Lawitz, E.J.; Zeuzem, S.; Rodriguez-Torres, M.; Patel, K.; van der Meer, A.J.; Patick, A.K.; Chen, A.; Zhou, Y.; et al. Treatment of HCV infection by targeting microRNA. N. Engl. J. Med. 2013, 368, 1685–1694. [Google Scholar] [CrossRef]
- Gebert, L.F.; Rebhan, M.A.; Crivelli, S.E.; Denzler, R.; Stoffel, M.; Hall, J. Miravirsen (SPC3649) can inhibit the biogenesis of miR-122. Nucleic Acids Res. 2014, 42, 609–621. [Google Scholar] [CrossRef]
- Streba, L.A.; Vere, C.C.; Rogoveanu, I.; Streba, C.T. Nonalcoholic fatty liver disease, metabolic risk factors, and hepatocellular carcinoma: An open question. World J. Gastroenterol. 2015, 21, 4103–4110. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver; European Association for the Study of Diabetes. EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 1388–1402. [Google Scholar] [CrossRef]
- Ardekani, A.M.; Naeini, M.M. The Role of MicroRNAs in Human Diseases. Avicenna J. Med. Biotechnol. 2010, 2, 161–179. [Google Scholar]
- Liu, X.; Huang, K.; Niu, Z.; Mei, D.; Zhang, B. Protective effect of isochlorogenic acid B on liver fibrosis in non-alcoholic steatohepatitis of mice. Basic Clin. Pharmacol. Toxicol. 2019, 124, 144–153. [Google Scholar] [CrossRef]
- Kumar, V.; Mondal, G.; Dutta, R.; Mahato, R.I. Co-delivery of small molecule hedgehog inhibitor and miRNA for treating liver fibrosis. Biomaterials 2016, 76, 144–156. [Google Scholar] [CrossRef]
- Maurer, B.; Stanczyk, J.; Jungel, A.; Akhmetshina, A.; Trenkmann, M.; Brock, M.; Kowal-Bielecka, O.; Gay, R.E.; Michel, B.A.; Distler, J.H.; et al. MicroRNA-29, a key regulator of collagen expression in systemic sclerosis. Arthritis Rheum. 2010, 62, 1733–1743. [Google Scholar] [CrossRef] [PubMed]
- Okada, H.; Honda, M.; Campbell, J.S.; Takegoshi, K.; Sakai, Y.; Yamashita, T.; Shirasaki, T.; Takabatake, R.; Nakamura, M.; Tanaka, T.; et al. Inhibition of microRNA-214 ameliorates hepatic fibrosis and tumor incidence in platelet-derived growth factor C transgenic mice. Cancer Sci. 2015, 106, 1143–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, L.L.; Li, S.D.; Ma, Y.C.; Tang, J.R.; Lv, J.Y.; Zhang, Y.Q.; Miao, Y.L.; Ma, Y.Q.; Li, C.M.; Chu, Y.Y.; et al. MicroRNA-30b regulates insulin sensitivity by targeting SERCA2b in non-alcoholic fatty liver disease. Liver Int. 2019, 39, 1504–1513. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cheng, Z.J.; Liu, Y.; Yan, Z.L.; Wang, K.; Wu, D.; Wan, X.Y.; Xia, Y.; Lau, W.Y.; Wu, M.C.; et al. Serum thioredoxin is a diagnostic marker for hepatocellular carcinoma. Oncotarget 2015, 6, 9551–9563. [Google Scholar] [CrossRef]
- Mollbrink, A.; Jawad, R.; Vlamis-Gardikas, A.; Edenvik, P.; Isaksson, B.; Danielsson, O.; Stal, P.; Fernandes, A.P. Expression of thioredoxins and glutaredoxins in human hepatocellular carcinoma: Correlation to cell proliferation, tumor size and metabolic syndrome. Int. J. Immunopathol. Pharmacol. 2014, 27, 169–183. [Google Scholar] [CrossRef]
- Tamai, T.; Uto, H.; Takami, Y.; Oda, K.; Saishoji, A.; Hashiguchi, M.; Kumagai, K.; Kure, T.; Mawatari, S.; Moriuchi, A.; et al. Serum manganese superoxide dismutase and thioredoxin are potential prognostic markers for hepatitis C virus-related hepatocellular carcinoma. World J. Gastroenterol. 2011, 17, 4890–4898. [Google Scholar] [CrossRef]
- Valavanidis, A.; Vlachogianni, T.; Fiotakis, K.; Loridas, S. Pulmonary oxidative stress, inflammation and cancer: Respirable particulate matter, fibrous dusts and ozone as major causes of lung carcinogenesis through reactive oxygen species mechanisms. Int. J. Environ. Res. Public Health 2013, 10, 3886–3907. [Google Scholar] [CrossRef]
- Chuma, M.; Hige, S.; Nakanishi, M.; Ogawa, K.; Natsuizaka, M.; Yamamoto, Y.; Asaka, M. 8-Hydroxy-2’-deoxy-guanosine is a risk factor for development of hepatocellular carcinoma in patients with chronic hepatitis C virus infection. J. Gastroenterol. Hepatol. 2008, 23, 1431–1436. [Google Scholar] [CrossRef]
- Ichiba, M.; Maeta, Y.; Mukoyama, T.; Saeki, T.; Yasui, S.; Kanbe, T.; Okano, J.; Tanabe, Y.; Hirooka, Y.; Yamada, S.; et al. Expression of 8-hydroxy-2’-deoxyguanosine in chronic liver disease and hepatocellular carcinoma. Liver Int. 2003, 23, 338–345. [Google Scholar] [CrossRef]
- Ma-On, C.; Sanpavat, A.; Whongsiri, P.; Suwannasin, S.; Hirankarn, N.; Tangkijvanich, P.; Boonla, C. Oxidative stress indicated by elevated expression of Nrf2 and 8-OHdG promotes hepatocellular carcinoma progression. Med. Oncol. 2017, 34, 57. [Google Scholar] [CrossRef]
- Tanaka, S.; Miyanishi, K.; Kobune, M.; Kawano, Y.; Hoki, T.; Kubo, T.; Hayashi, T.; Sato, T.; Sato, Y.; Takimoto, R.; et al. Increased hepatic oxidative DNA damage in patients with nonalcoholic steatohepatitis who develop hepatocellular carcinoma. J. Gastroenterol. 2013, 48, 1249–1258. [Google Scholar] [CrossRef] [PubMed]
- Jopling, C. Liver-specific microRNA-122: Biogenesis and function. RNA Biol. 2012, 9, 137–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilic, M.; Kasperczyk, H.; Fulda, S.; Debatin, K.M. Role of hypoxia inducible factor-1 alpha in modulation of apoptosis resistance. Oncogene 2007, 26, 2027–2038. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Zhang, X.; Wang, Q.; Sun, Y.; Liu, L.; Huang, L.; Ding, L.; Shen, M.; Zhang, L.; Duan, Y. Simple and rational design of a polymer nano-platform for high performance of HCV related miR-122 reduction in the liver. Biomater Sci. 2018, 6, 2667–2680. [Google Scholar] [CrossRef]
- Fu, B.; Meng, W.; Zeng, X.; Zhao, H.; Liu, W.; Zhang, T. TXNRD1 Is an Unfavorable Prognostic Factor for Patients with Hepatocellular Carcinoma. Biomed. Res. Int. 2017, 2017, 4698167. [Google Scholar] [CrossRef]
- Lee, D.; Xu, I.M.; Chiu, D.K.; Leibold, J.; Tse, A.P.; Bao, M.H.; Yuen, V.W.; Chan, C.Y.; Lai, R.K.; Chin, D.W.; et al. Induction of Oxidative Stress Through Inhibition of Thioredoxin Reductase 1 Is an Effective Therapeutic Approach for Hepatocellular Carcinoma. Hepatology 2019, 69, 1768–1786. [Google Scholar] [CrossRef]
- Liu, L.; Cao, Y.; Chen, C.; Zhang, X.; McNabola, A.; Wilkie, D.; Wilhelm, S.; Lynch, M.; Carter, C. Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res. 2006, 66, 11851–11858. [Google Scholar] [CrossRef]
- Wilhelm, S.M.; Carter, C.; Tang, L.; Wilkie, D.; McNabola, A.; Rong, H.; Chen, C.; Zhang, X.; Vincent, P.; McHugh, M.; et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004, 64, 7099–7109. [Google Scholar] [CrossRef]
- Coriat, R.; Nicco, C.; Chereau, C.; Mir, O.; Alexandre, J.; Ropert, S.; Weill, B.; Chaussade, S.; Goldwasser, F.; Batteux, F. Sorafenib-induced hepatocellular carcinoma cell death depends on reactive oxygen species production in vitro and in vivo. Mol. Cancer Ther. 2012, 11, 2284–2293. [Google Scholar] [CrossRef]
- Paech, F.; Mingard, C.; Grunig, D.; Abegg, V.F.; Bouitbir, J.; Krahenbuhl, S. Mechanisms of mitochondrial toxicity of the kinase inhibitors ponatinib, regorafenib and sorafenib in human hepatic HepG2 cells. Toxicology 2018, 395, 34–44. [Google Scholar] [CrossRef]
- Bull, V.H.; Rajalingam, K.; Thiede, B. Sorafenib-induced mitochondrial complex I inactivation and cell death in human neuroblastoma cells. J. Proteome Res. 2012, 11, 1609–1620. [Google Scholar] [CrossRef] [PubMed]
- Awan, F.M.; Naz, A.; Obaid, A.; Ikram, A.; Ali, A.; Ahmad, J.; Naveed, A.K.; Janjua, H.A. MicroRNA pharmacogenomics based integrated model of miR-17-92 cluster in sorafenib resistant HCC cells reveals a strategy to forestall drug resistance. Sci. Rep. 2017, 7, 11448. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
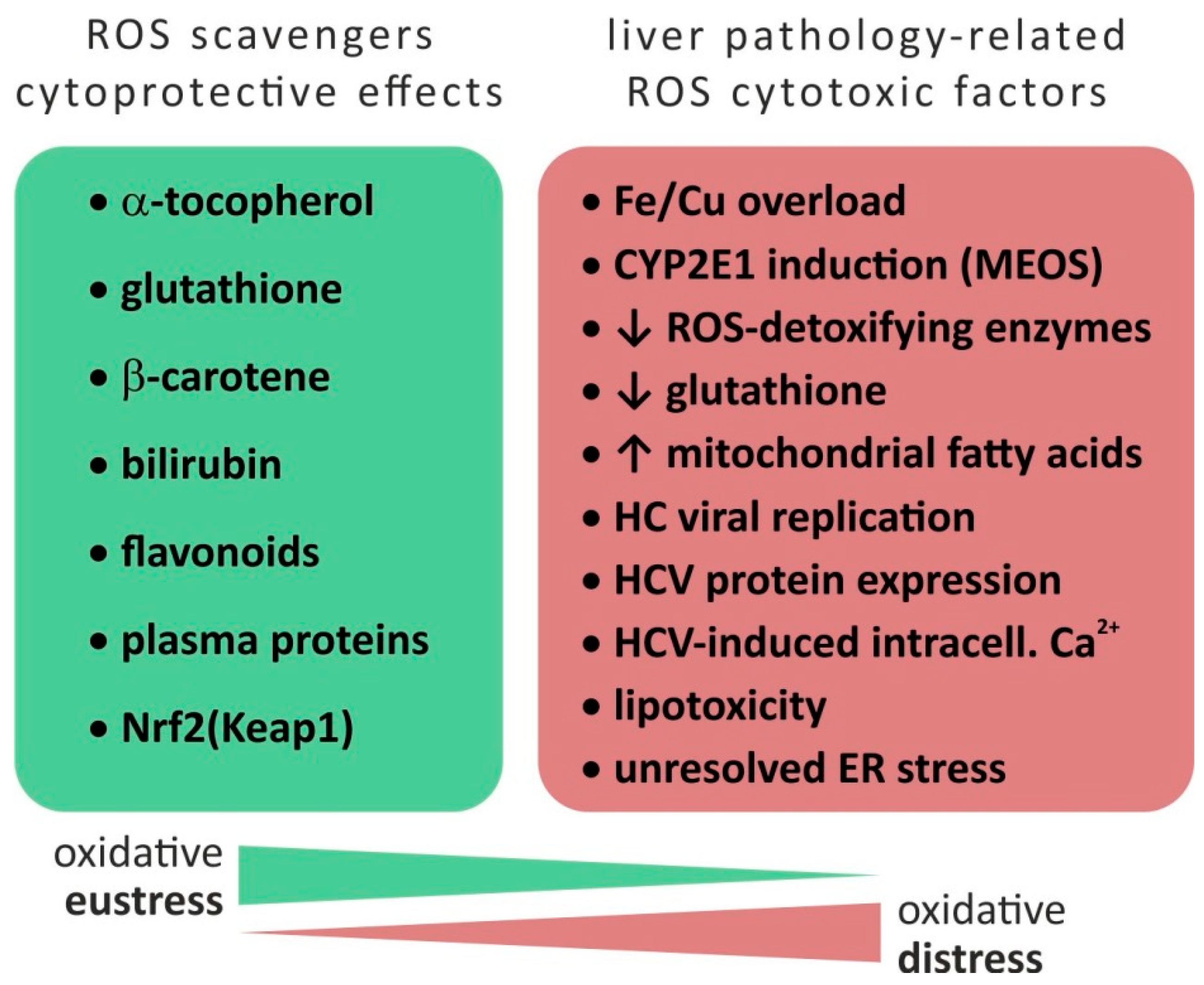
| Liver Disease | ROS-Production by | (Patho)Mechanism |
|---|---|---|
| Hemochromatosis, Wilson’s disease | Iron/copper overload | Presence of metal catalyst for ROS production |
| Alcoholic liver disease (ALD) | CYP2E1 induction (MEOS) | High NADPH oxidase activity of CYP2E1 associated with production of O2− and H2O2 |
| Reduced expression of ROS-detoxifying enzymes | Alcohol-induced reduction of PPARγ coactivator 1α | |
| Nonalcoholic steatohepatitis (NASH) | Increased concentration and metabolisms of fatty acids in mitochondria | Saturation of mitochondrial β-oxidation and H2O2 production through peroxisomal β-oxidation |
| CYP2E1 (CYP4A) induction | See above | |
| HCV infection | Reduction of ROS detoxification | Reduced levels of glutathione and its regeneration as well as ROS-detoxifying enzymes |
| Increased mitochondrial ROS production due to viral replication or virus protein expression | Inhibition of mitochondrial electron transport chain | |
| Increased NADPH oxidase triggered by calcium | Virus-induced redistribution of cellular calcium |
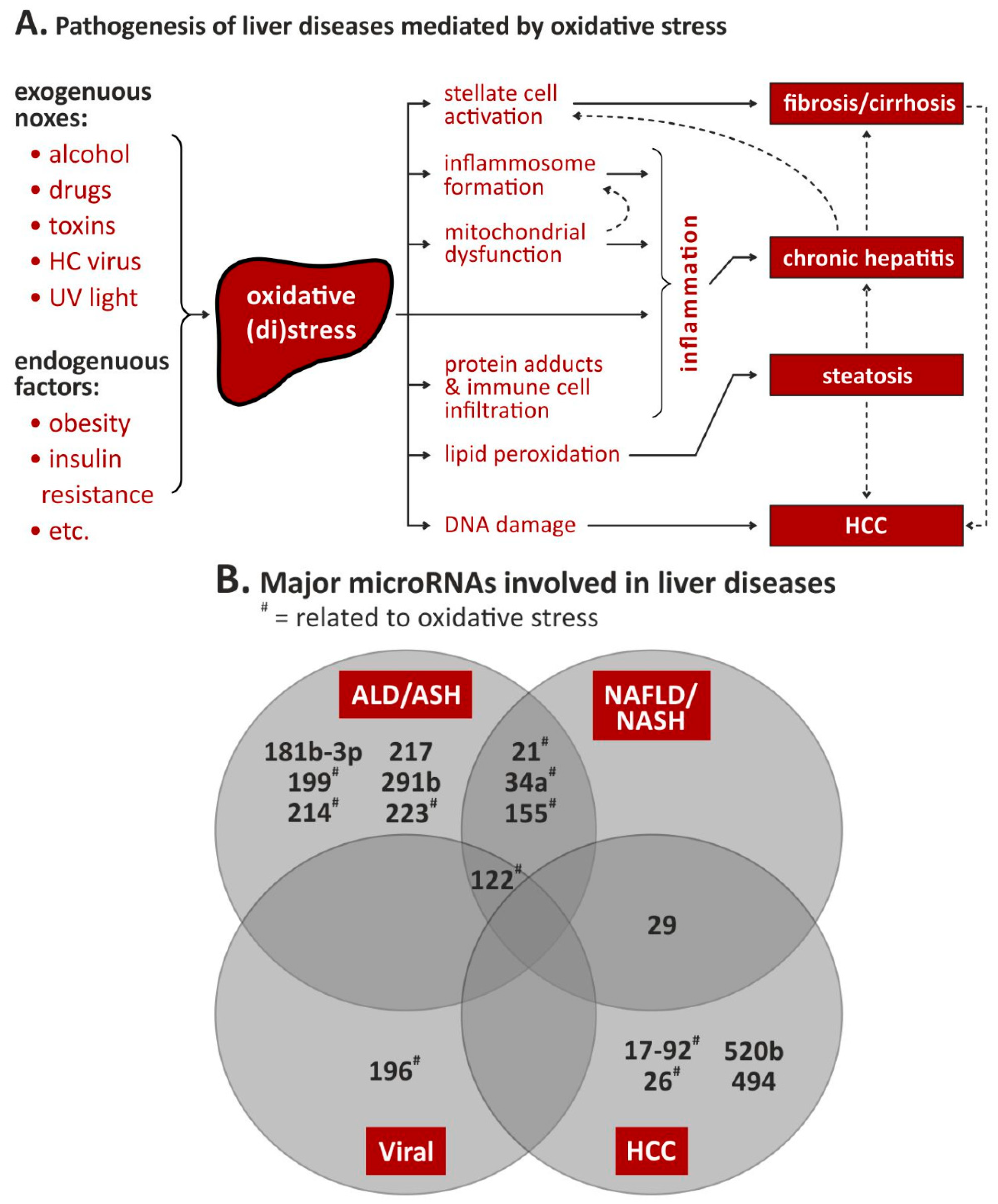
| miRNA | Evidence | Target Gene/Pathway | (Patho)Mechanism | References | |||
|---|---|---|---|---|---|---|---|
| In Vitro | In Vivo | In Situ | In Silico | ||||
| 155 ↑1 | ✓ | ✓ | ✓ | TNFα ↑ PPARα ↓ | LPS mediates the activation of NFκB. Increase of miR-155; release of TNFα, ROS and oxidative stress in Kupffer cells and hepatic stellate cells via suppression of PPARα causing overexpression of FABP4, ACC1 and LDLR | [38,39,40] | |
| 181b-3p ↑2 | ✓ | ✓ | TLR4 ↑ NFκB ↑ | Alterations in glucose and lipid homeostasis; activation of Kupffer cells | [41] | ||
| 291b ↑2 | ✓ | ✓ | Tollip ↓ | Loss of downregulation of TLR4/NFκβ in Kupffer cells | [42] | ||
| 34a ↑2 | ✓ | ✓ | SIRT1 ↓ | Inhibition of SIRT1 protein coding | [44] | ||
| 217 ↑2 | ✓ | ✓ | SIRT1 ↓ | Alcohol-associated inflammation | [45] | ||
| 122 ↓2 | ✓ | ✓ | HIF1α ↑ | miR-122 loss (deletion) or down-regulation (due to alcohol diet via GRHL2) leads to steathosis at birth, following fibrosis; miR-122 down-regulation enables HIF1α expression in ALD | [46,47] | ||
| 21 ↑2 | ✓ | ✓ | FASLG ↓ DR5 ↓ | Reduced ethanol induced cell death in hepatocytes; stellate cells dysregulation via miR-21 in ethanol-induced altered extrinsic apoptotic signaling and its progression to ALD | [50] | ||
| 223 ↑1 | IL-6 ↑ p47phox ↑ | Peripheral neutrophils activation and liver infiltration induced by ethanol; triggering NADPH oxidase → ROS | [51] | ||||
| 199 ↓1 | ✓ | ✓ | HIF1α ↑ ET-1 ↑ | Leading to steatohepatitis in cirrhosis patients | [52] | ||
| 214 ↑1 | ✓ | ✓ | ✓ | CypP450 ↓ GSR ↓ | Affecting alcohol metabolism and causing oxidative stress | [53,54] | |
| miRNA | Evidence | Target Gene/Pathway | (Patho)Mechanism | References | |||
|---|---|---|---|---|---|---|---|
| In Vitro | In Vivo | In Situ | In Silico | ||||
| 21 ↑3 | ✓ | PPARα ↓ | Liver injury, inflammation and fibrosis | [75] | |||
| 21 ↑3 | ✓ | ✓ | ✓ | PTEN ↓ | Development of steatosis | [73] | |
| 21 ↑1 | ✓ | ✓ | TGFβ ↑ | Induced collagen production and extracellular matrix formation fibrogenesis via increase of Col1α1 and α-SMA expression | [72] | ||
| 122 ↑2 | ✓ | KLF6 ↑ | Activation of hepatic stellate cells and progression of liver fibrosis | [82] | |||
| 34a ↑2 | ✓ | ✓ | HNF4α ↓ | Inhibition of very low-density lipoprotein secretion and promotion of liver steatosis and hypolipidemia | [89] | ||
| 34a ↑1 | ✓ | ✓ | PPARα ↓ | Loss of regulation genes encoding fatty acid metabolizing enzymes and mitochondrial fatty acid oxidation activity | [87] | ||
| 34a ↑1 | ✓ | SIRT1 ↓ | Increase of hepatic cholesterol synthesis and activation of pro-apoptotic genes (p53, p66shc) | [88] | |||
| 29a and c ↓2 | ✓ | ✓ | SIRT1 ↓ | Increased levels of free cholesterol | [94,95] | ||
| 29 ↑2 | ✓ | ✓ | Col1α1 ↓ | Downregulation in activated hepatic stellate cells and therefore loss of interaction with Col1α1 → decreased collagen production | [97] | ||
| 155 ↑1 | ✓ | ✓ | AKT/ FOXO3a ↑ | Regulates proliferation of hepatic stellate cells promotes liver fibrosis; FOXO3a proteins maintain intracellular redox balance and survival | [99] | ||
| 155 ↑2 | ✓ | ✓ | LXRα ↓ | Decreased SREBP1 and FAS resulting in an increased intracellular lipid content | [100] | ||
| 155 ↑2 | ✓ | ✓ | HIF1α and vimentin ↑ | NASH-induced liver fibrosis | [101] | ||
| miRNA | Evidence | Target Gene/Pathway | (Patho)Mechanism | References | |||
|---|---|---|---|---|---|---|---|
| In Vitro | In Vivo | In Situ | In Silico | ||||
| 196↓1, C | ✓ | Bach1/ HMOX1 ↓ | Down-regulation of Bach1 gene expression, up-regulation of HMOX1 gene expression, a key cytoprotective enzyme | [114] | |||
| 196↓2, C | ✓ | HCV NS5A gene ↓ | miR-196 perfectly matches coding region of the HCV NS5A gene down-regulatory effect of miR-196 on HCV expression in the HCV J6/JFH1 cell culture system | [114] | |||
| 122↓2, C | ✓ | HCV viral genome ↑ | Enhances viral RNA replication | [115,116] | |||
| 122↓1, C | ✓ | ✓ | ✓ | NIK ↑ and HNF4α ↑ | Disturbance of the NIK mediated lipid metabolism → lipogenesis and lipid droplet formation → promotion of oxidative stress | [117] | |
| 122↓2, B | ✓ | cyclin G1-p53 complex ↑ | Inhibits the effects of p53 on HBV replication | [120] | |||
| 122↓1, B | ✓ | NQO1 ↑ and HO1 ↓ | miR-122 affects balance between the pro-oxidants and antioxidants | [121] | |||
| miRNA | Evidence | Target Gene/Pathway | (Patho)Mechanism | References | |||
|---|---|---|---|---|---|---|---|
| In Vitro | In Vivo | In Situ | In Silico | ||||
| 26↓1 | ✓ | ✓ | EZH2 ↑ | Sequestration of miR-26 from its target EZH2, which released the suppression on EZH2, and thereby led to EZH2 overexpression in gastric cancer | [142] | ||
| 29b↓2 | ✓ | ✓ | TET1 ↓ | Feedback of miRNA-29-TET1 downregulation in HCC development suggesting a potential target in identification of the prognosis and application of cancer therapy for HCC patients | [144] | ||
| 494↑2 | ✓ | ✓ | TET1 ↓ | miR-494 inhibition or enforced TET1 expression is able to restore invasion-suppressor miRNAs and inhibit miR-494-mediated HCC cell invasion | [145] | ||
| 520b↓2 | ✓ | TET1 ↓ | Depresses proliferation of liver cancer cells through targeting 3’UTR of TET1 mRNA | [146] | |||
| 17-92 cluster↓1 | ✓ | E2F family ↑ | ROS-mediated oxidative DNA damage correlates with over-expression of miR-92–playing a role in both the apoptotic process and in cellular proliferation pathways | [147] | |||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klieser, E.; Mayr, C.; Kiesslich, T.; Wissniowski, T.; Di Fazio, P.; Neureiter, D.; Ocker, M. The Crosstalk of miRNA and Oxidative Stress in the Liver: From Physiology to Pathology and Clinical Implications. Int. J. Mol. Sci. 2019, 20, 5266. https://doi.org/10.3390/ijms20215266
Klieser E, Mayr C, Kiesslich T, Wissniowski T, Di Fazio P, Neureiter D, Ocker M. The Crosstalk of miRNA and Oxidative Stress in the Liver: From Physiology to Pathology and Clinical Implications. International Journal of Molecular Sciences. 2019; 20(21):5266. https://doi.org/10.3390/ijms20215266
Chicago/Turabian StyleKlieser, Eckhard, Christian Mayr, Tobias Kiesslich, Till Wissniowski, Pietro Di Fazio, Daniel Neureiter, and Matthias Ocker. 2019. "The Crosstalk of miRNA and Oxidative Stress in the Liver: From Physiology to Pathology and Clinical Implications" International Journal of Molecular Sciences 20, no. 21: 5266. https://doi.org/10.3390/ijms20215266