Loss of Tumor Suppressor CYLD Expression Triggers Cisplatin Resistance in Oral Squamous Cell Carcinoma

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
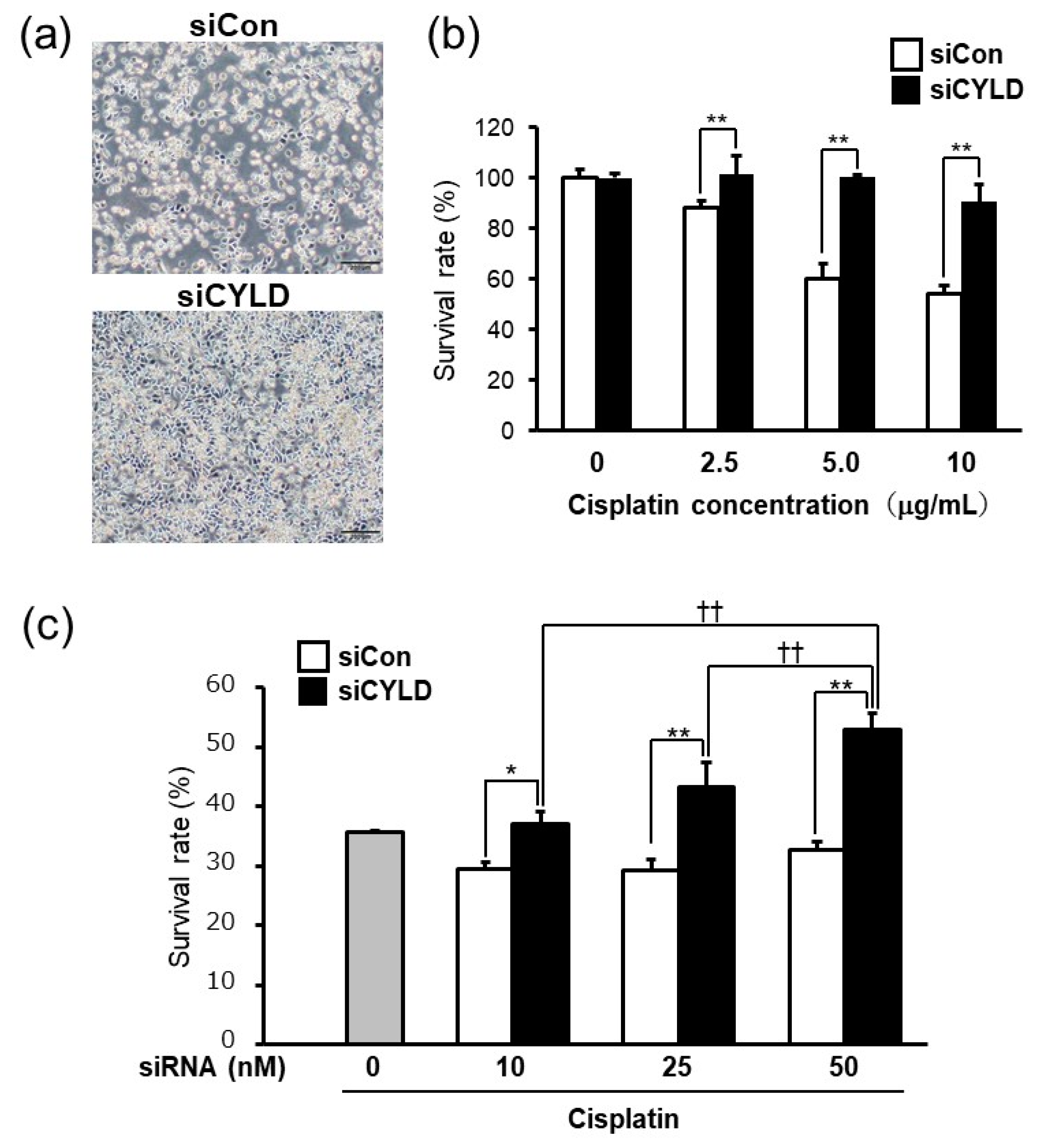
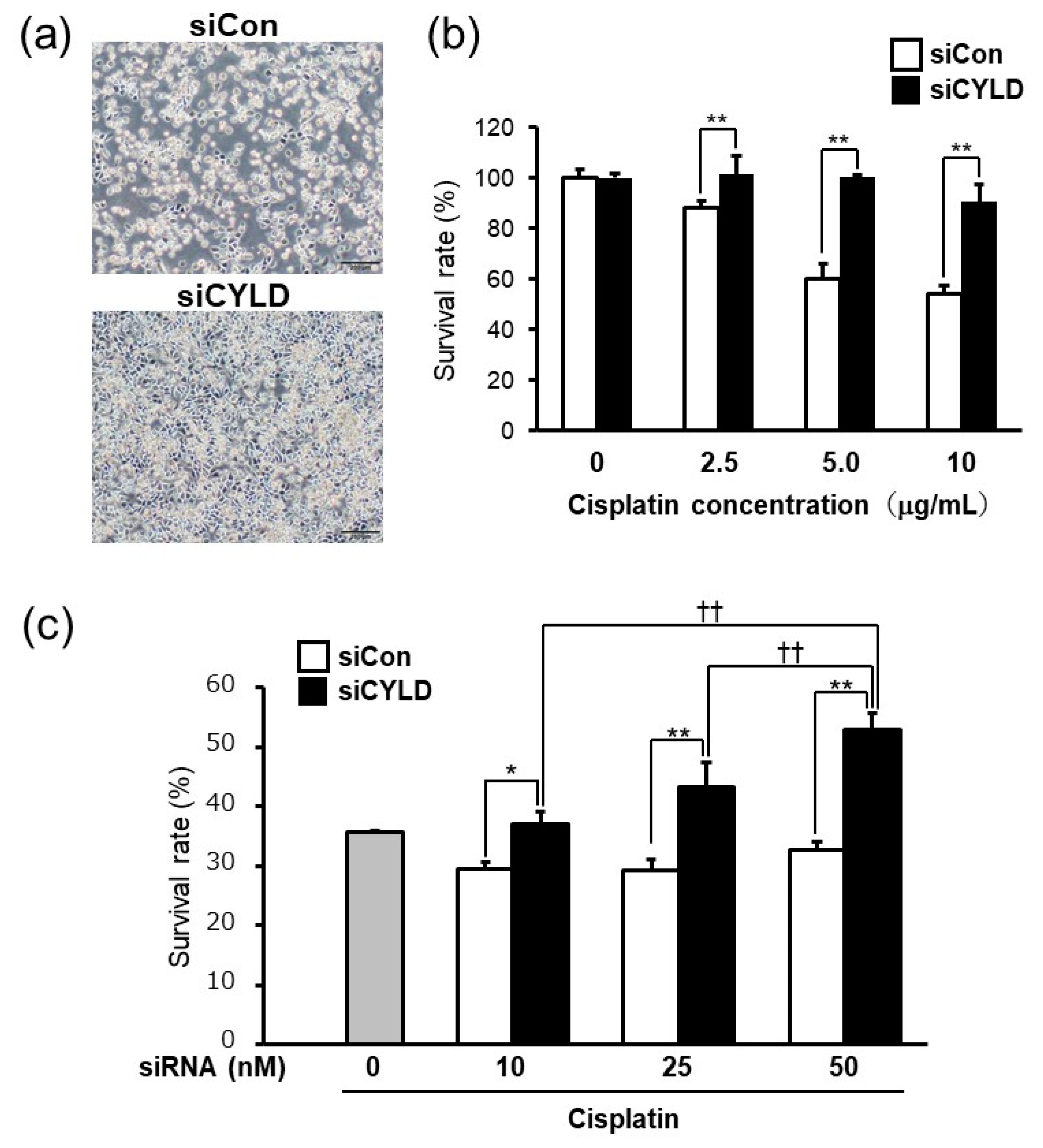
2.1. Effect of CYLD Down-Regulation on Cisplatin Sensitivity in OSCC Cells
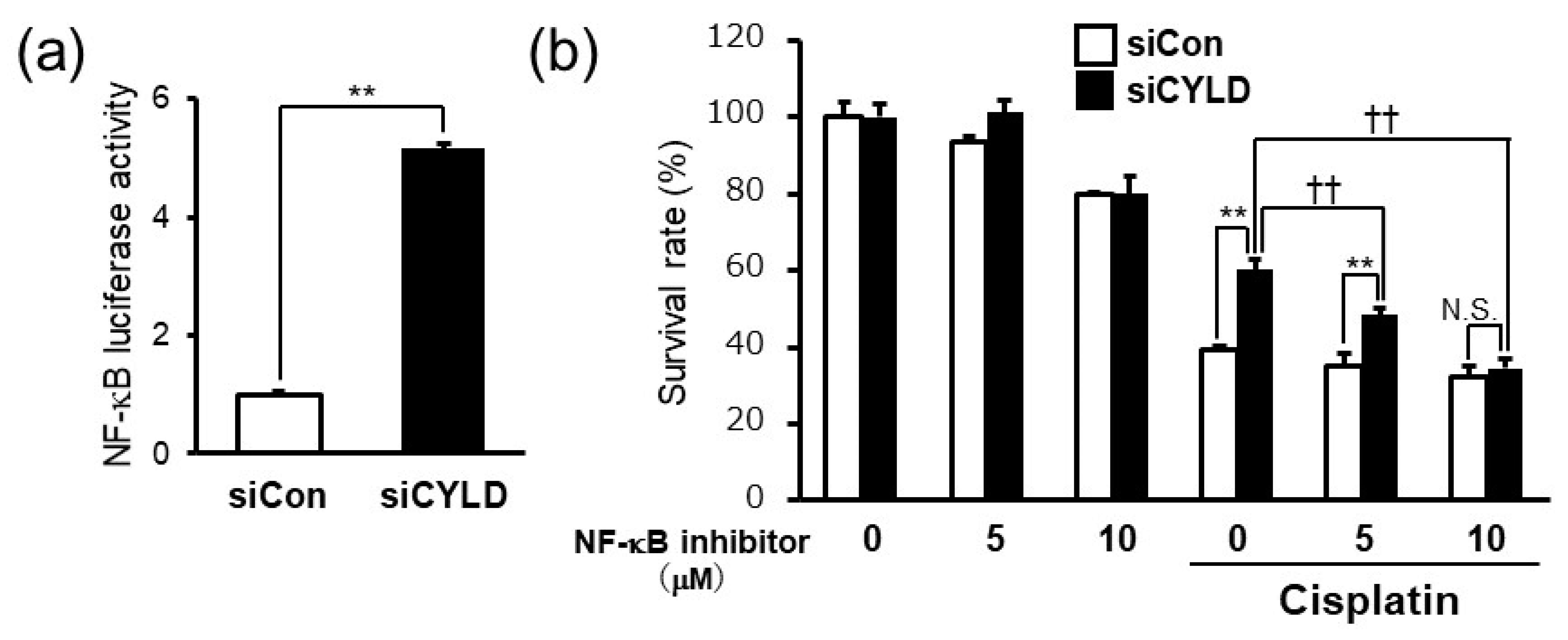
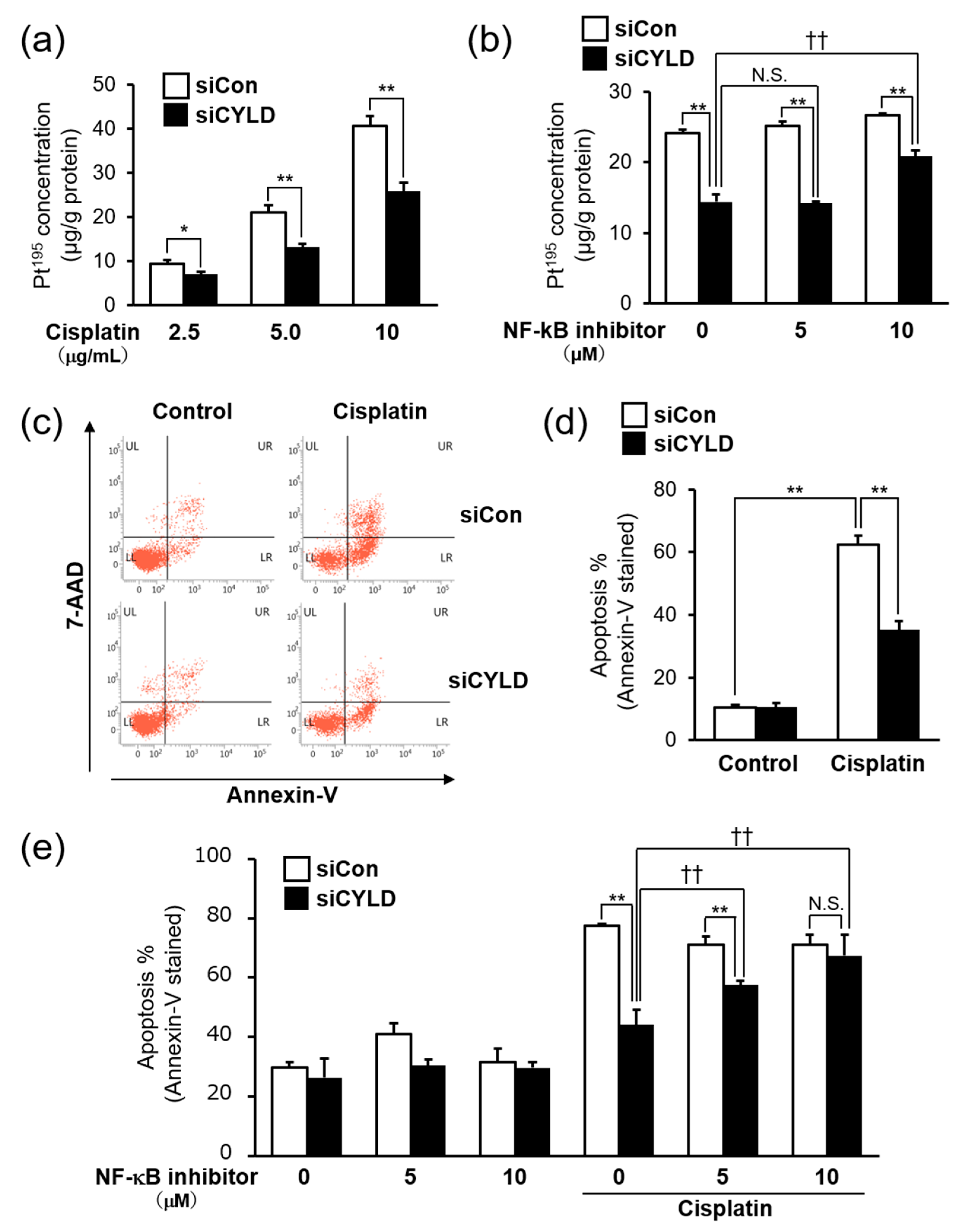
2.2. CYLD Down-Regulation Induced Cisplatin Resistance through NF-κB Hyperactivation
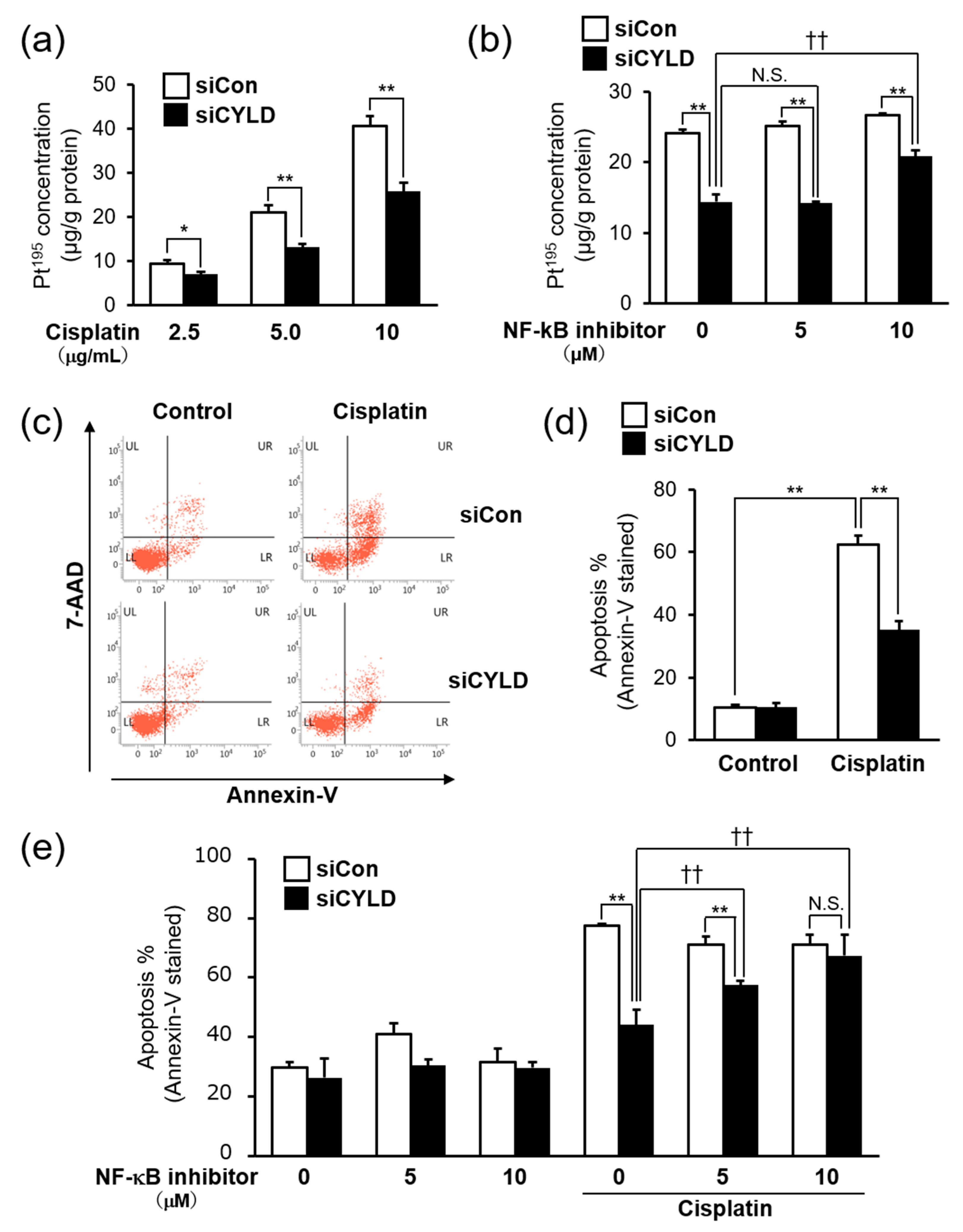
2.3. Effect of CYLD Down-Regulation on Intracellular Cisplatin Accumulation and Cisplatin-Induced Apoptosis in OSCC Cells
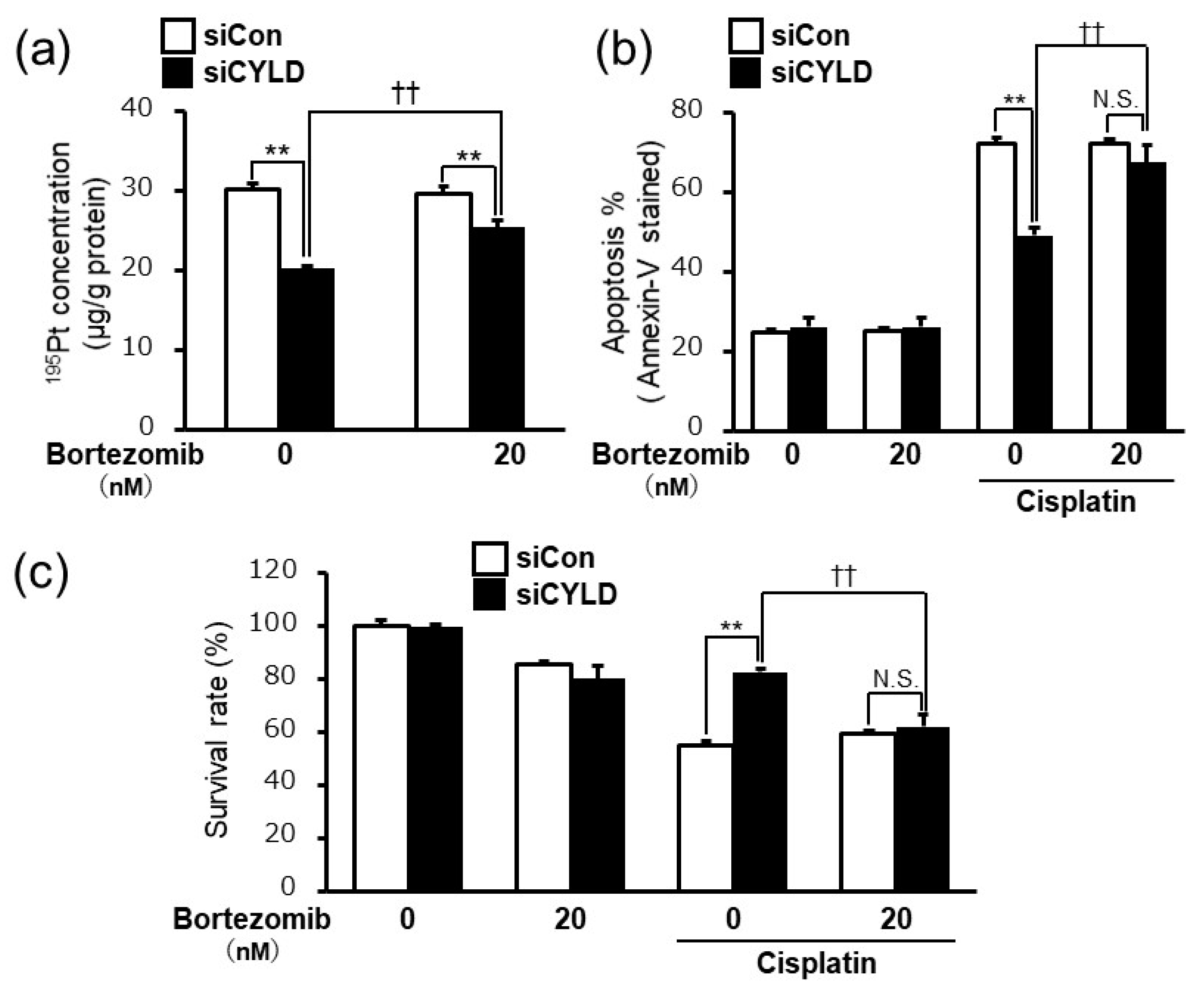
2.4. Bortezomib, a Proteasome Inhibitor, Released the Cisplatin Resistance Caused by CYLD Down-Regulation in OSCC Cells
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Lines and Cell Cultures
4.3. Transfection with siRNA
4.4. Measurement of Cell Survival Rate
4.5. NF-κB Reporter Assay
4.6. Inductively Coupled Plasma Mass Spectrometry (ICP-MS)
4.7. Apoptosis
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CYLD | Cylindromatosis |
| OSCC | Oral squamous cell carcinoma |
| NF-κB | Nuclear factor-κB |
| ICP-MS | Inductively coupled plasma mass spectrometry |
| siRNA | Small interfering RNA |
| 7-AAD | 7-amino-actinomycin D |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016: Cancer Statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Warnakulasuriya, S. Global epidemiology of oral and oropharyngeal cancer. Oral Oncol. 2009, 45, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Forastiere, A.A.; Goepfert, H.; Maor, M.; Pajak, T.F.; Weber, R.; Morrison, W.; Glisson, B.; Trotti, A.; Ridge, J.A.; Chao, C.; et al. Concurrent Chemotherapy and Radiotherapy for Organ Preservation in Advanced Laryngeal Cancer. N. Engl. J. Med. 2003, 349, 2091–2098. [Google Scholar] [CrossRef] [PubMed]
- Pignon, J.P.; Bourhis, J.; Domenge, C.; Designé, L. Chemotherapy added to locoregional treatment for head and neck squamous-cell carcinoma: Three meta-analyses of updated individual data. Lancet 2000, 355, 7. [Google Scholar] [CrossRef]
- Zheng, H.-C. The molecular mechanisms of chemoresistance in cancers. Oncotarget 2017, 8, 59950. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.-T.; Li, Z.-L.; He, Z.-X.; Qiu, J.-X.; Zhou, S.-F. Molecular mechanisms for tumour resistance to chemotherapy. Clin. Exp. Pharm. Physiol. 2016, 43, 723–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brummelkamp, T.R.; Nijman, S.M.B.; Dirac, A.M.G.; Bernards, R. Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF-κB. Nature 2003, 424, 797–801. [Google Scholar] [CrossRef]
- Kovalenko, A.; Chable-Bessia, C.; Cantarella, G. The tumour suppressor CYLD negatively regulates NF-kB signalling by deubiquitination. Nature 2003, 424, 801–805. [Google Scholar] [CrossRef]
- Trompouki, E.; Hatzivassiliou, E.; Tsichritzis, T.; Farmer, H.; Ashworth, A.; Mosialos, G. CYLD is a deubiquitinating enzyme that negatively regulates NF-kB activation by TNFR family members. Nature 2003, 424, 793–796. [Google Scholar] [CrossRef]
- Lork, M.; Verhelst, K.; Beyaert, R. CYLD, A20 and OTULIN deubiquitinases in NF-κB signaling and cell death: So similar, yet so different. Cell Death Differ. 2017, 24, 1172–1183. [Google Scholar] [CrossRef]
- Jono, H.; Lim, J.H.; Chen, L.-F.; Xu, H.; Trompouki, E.; Pan, Z.K.; Mosialos, G.; Li, J.-D. NF-κB Is Essential for Induction of CYLD, the Negative Regulator of NF-κB. J. Biol. Chem. 2004, 279, 36171–36174. [Google Scholar] [CrossRef] [PubMed]
- Tauriello, D.V.F.; Haegebarth, A.; Kuper, I.; Edelmann, M.J.; Henraat, M.; Canninga-van Dijk, M.R.; Kessler, B.M.; Clevers, H.; Maurice, M.M. Loss of the Tumor Suppressor CYLD Enhances Wnt/β-Catenin Signaling through K63-Linked Ubiquitination of Dvl. Mol. Cell 2010, 37, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Reiley, W.; Zhang, M.; Sun, S.-C. Negative Regulation of JNK Signaling by the tumor suppressor CYLD. J. Biol. Chem. 2004, 279, 55161–55167. [Google Scholar] [CrossRef] [PubMed]
- Tesio, M.; Tang, Y.; Müdder, K.; Saini, M.; von Paleske, L.; Macintyre, E.; Pasparakis, M.; Waisman, A.; Trumpp, A. Hematopoietic stem cell quiescence and function are controlled by the CYLD–TRAF2–p38MAPK pathway. J. Exp. Med. 2015, 212, 525–538. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, Z.; Wang, P.; Li, D.; Zhou, J.; Wu, S. CYLD negatively regulates Hippo signaling by limiting Hpo phosphorylation in Drosophila. Biochem. Biophys. Res. Commun. 2014, 452, 808–812. [Google Scholar] [CrossRef]
- Rajan, N.; Elliott, R.J.R.; Smith, A.; Sinclair, N.; Swift, S.; Lord, C.J.; Ashworth, A. The cylindromatosis gene product, CYLD, interacts with MIB2 to regulate notch signalling. Oncotarget 2014, 5, 12126. [Google Scholar] [CrossRef]
- Patel, M.; Horgan, P.G.; McMillan, D.C.; Edwards, J. NF-κB pathways in the development and progression of colorectal cancer. Transl. Res. 2018, 197, 43–56. [Google Scholar] [CrossRef]
- Xia, Y.; Shen, S.; Verma, I.M. NF-κB, an Active Player in Human Cancers. Cancer Immunol. Res. 2014, 2, 823–830. [Google Scholar] [CrossRef]
- Hayashi, M.; Jono, H.; Shinriki, S.; Nakamura, T.; Guo, J.; Sueta, A.; Tomiguchi, M.; Fujiwara, S.; Yamamoto-Ibusuki, M.; Murakami, K.; et al. Clinical significance of CYLD downregulation in breast cancer. Breast Cancer Res. Treat. 2014, 143, 447–457. [Google Scholar] [CrossRef]
- Guo, J.; Shinriki, S.; Su, Y.; Nakamura, T.; Hayashi, M.; Tsuda, Y.; Murakami, Y.; Tasaki, M.; Hide, T.; Takezaki, T.; et al. Hypoxia suppresses cylindromatosis (CYLD) expression to promote inflammation in glioblastoma possible link to acquired resistance to anti-VEGF therapy. Oncotarget 2014, 5, 6353–6364. [Google Scholar] [CrossRef]
- Kinoshita, H.; Okabe, H.; Beppu, T.; Chikamoto, A.; Hayashi, H.; Imai, K.; Mima, K.; Nakagawa, S.; Yokoyama, N.; Ishiko, T.; et al. CYLD downregulation is correlated with tumor development in patients with hepatocellular carcinoma. Mol. Clin. Oncol. 2013, 1, 309–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinriki, S.; Jono, H.; Maeshiro, M.; Nakamura, T.; Guo, J.; Li, J.-D.; Ueda, M.; Yoshida, R.; Shinohara, M.; Nakayama, H.; et al. Loss of CYLD promotes cell invasion via ALK5 stabilization in oral squamous cell carcinoma: Association of CYLD with OSCC-related invasion. J. Pathol. 2018, 244, 367–379. [Google Scholar] [CrossRef]
- Nariai, Y.; Mishima, K.; Yoshimura, Y.; Sekine, J. FAP-1 and NF-κB expressions in oral squamous cell carcinoma as potential markers for chemo-radio sensitivity and prognosis. Int. J. Oral Maxillofac. Surg. 2011, 40, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Bindhu, O.S.; Ramadas, K.; Sebastian, P.; Pillai, M.R. High expression levels of nuclear factor kappa B and gelatinases in the tumorigenesis of oral squamous cell carcinoma. Head Neck 2006, 28, 916–925. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-J.; Zhou, X.; Wang, W.; Tang, F.; Qi, C.-L.; Yang, X.; Wu, S.; Lin, Y.-Q.; Wang, J.-T.; Geng, J.-G. Andrographolide Inhibits Oral Squamous Cell Carcinogenesis through NF-κB Inactivation. J. Dent. Res. 2011, 90, 1246–1252. [Google Scholar] [CrossRef]
- Li, F.; Shanmugam, M.K.; Siveen, K.S.; Wang, F.; Ong, T.H.; Loo, S.Y.; Swamy, M.M.M.; Mandal, S.; Kumar, A.P.; Goh, B.C.; et al. Garcinol sensitizes human head and neck carcinoma to cisplatin in a xenograft mouse model despite downregulation of proliferative biomarkers. Oncotarget 2015, 6, 5147. [Google Scholar] [CrossRef]
- Nagata, M.; Nakayama, H.; Tanaka, T.; Yoshida, R.; Yoshitake, Y.; Fukuma, D.; Kawahara, K.; Nakagawa, Y.; Ota, K.; Hiraki, A.; et al. Overexpression of cIAP2 contributes to 5-FU resistance and a poor prognosis in oral squamous cell carcinoma. Br. J. Cancer 2011, 105, 1322–1330. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Michels, J.; Brenner, C.; Szabadkai, G.; Harel-Bellan, A.; Castedo, M.; Kroemer, G. Systems biology of cisplatin resistance: Past, present and future. Cell Death Dis. 2014, 5, e1257. [Google Scholar] [CrossRef]
- Siddik, Z.H. Cisplatin: Mode of cytotoxic action and molecular basis of resistance. Oncogene 2003, 22, 7265–7279. [Google Scholar] [CrossRef]
- Loh, S.; Mistry, P.; Kelland, L.; Abel, G.; Harrap, K. Reduced drug accumulation as a major mechanism of acquired resistance to cisplatin in a human ovarian carcinoma cell line: Circumvention studies using novel platinum (II) and (IV) ammine/amine complexes. Br. J. Cancer 1992, 66, 1109–1115. [Google Scholar] [CrossRef]
- Chou, A.J.; Gorlick, R. Chemotherapy resistance in osteosarcoma: Current challenges and future directions. Expert Rev. Anticancer Ther. 2006, 6, 1075–1085. [Google Scholar] [CrossRef] [PubMed]
- Zajączkowska, R.; Kocot-Kępska, M.; Leppert, W.; Wrzosek, A.; Mika, J.; Wordliczek, J. Mechanisms of Chemotherapy-Induced Peripheral Neuropathy. Int. J. Mol. Sci. 2019, 20, 1451. [Google Scholar] [CrossRef] [PubMed]
- Cengiz Seval, G.; Beksac, M. The safety of bortezomib for the treatment of multiple myeloma. Expert Opin. Drug Saf. 2018, 17, 953–962. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Frezza, M.; Schmitt, S.; Kanwar, J.; Dou, P.Q. Bortezomib as the First Proteasome Inhibitor Anticancer Drug: Current Status and Future Perspectives. Curr. Cancer Drug Targets 2011, 11, 239–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitsudo, K.; Shigetomi, T.; Fujimoto, Y.; Nishiguchi, H.; Yamamoto, N.; Furue, H.; Ueda, M.; Itoh, Y.; Fuwa, N.; Tohnai, I. Organ preservation with daily concurrent chemoradiotherapy using superselective intra-arterial infusion via a superficial temporal artery for T3 and T4 head and neck cancer. Int. J. Radiat. Oncol. 2011, 79, 1428–1435. [Google Scholar] [CrossRef] [PubMed]
- Konstantakou, E.; Voutsinas, G.; Karkoulis, P.; Aravantinos, G.; Margaritis, L.; Stravopodis, D. Human bladder cancer cells undergo cisplatin-induced apoptosis that is associated with p53-dependent and p53-independent responses. Int. J. Oncol. 2009, 35, 401–416. [Google Scholar] [PubMed]
- Schinkel, A.H.; Jonker, J.W. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: An overview. Adv. Drug Deliv. Rev. 2003, 55, 3–29. [Google Scholar] [CrossRef]
- Bentires-Alj, M.; Barbu, V.; Fillet, M.; Chariot, A.; Relic, B.; Jacobs, N.; Gielen, J.; Merville, M.-P.; Bours, V. NF-κB transcription factor induces drug resistance through MDR1 expression in cancer cells. Oncogene 2003, 22, 90–97. [Google Scholar] [CrossRef]
- Kang, M.H.; Reynolds, C.P. Bcl-2 Inhibitors: Targeting Mitochondrial Apoptotic Pathways in Cancer Therapy. Clin. Cancer Res. 2009, 15, 1126–1132. [Google Scholar] [CrossRef]
- Wang, L.-H.; Li, Y.; Yang, S.-N.; Wang, F.-Y.; Hou, Y.; Cui, W.; Chen, K.; Cao, Q.; Wang, S.; Zhang, T.-Y.; et al. Gambogic acid synergistically potentiates cisplatin-induced apoptosis in non-small-cell lung cancer through suppressing NF-κB and MAPK/HO-1 signalling. Br. J. Cancer 2014, 110, 341–352. [Google Scholar] [CrossRef]
- Solomon, L.A.; Ali, S.; Banerjee, S.; Munkarah, A.R.; Morris, R.T.; Sarkar, F.H. Sensitization of ovarian cancer cells to cisplatin by genistein: The role of NF-kappaB. J. Ovarian Res. 2008, 1, 9. [Google Scholar] [CrossRef] [PubMed]
- Asselin, E.; Mills, G.B.; Tsang, B.K. XIAP regulates Akt activity and caspase-3-dependent cleavage during cisplatin-induced apoptosis in human ovarian epithelial cancer cells. Cancer Res. 2001, 61, 1862–1868. [Google Scholar] [PubMed]
- Pérez-Sayáns, M.; Somoza-Martín, J.; Barros-Angueira, F.; Diz, P.G.; José Manuel Gándara, R.; Gándara, J.M. Multidrug resistance in oral squamous cell carcinoma: The role of vacuolar ATPases. Cancer Lett. 2010, 295, 135–143. [Google Scholar]
- Allen, C.T.; Conley, B.; Sunwoo, J.B.; Van Waes, C. CCR 20th Anniversary commentary: Preclinical study of proteasome inhibitor bortezomib in head and neck cancer. Clin. Cancer Res. 2015, 21, 942–943. [Google Scholar] [CrossRef]
- Van Waes, C.; Chang, A.A.; Lebowitz, P.F.; Druzgal, C.H.; Chen, Z.; Elsayed, Y.A.; Sunwoo, J.B.; Rudy, S.F.; Morris, J.C.; Mitchell, J.B.; et al. Inhibition of nuclear factor-κB and target genes during combined therapy with proteasome inhibitor bortezomib and reirradiation in patients with recurrent head-and-neck squamous cell carcinoma. Int. J. Radiat. Oncol. 2005, 63, 1400–1412. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suenaga, N.; Kuramitsu, M.; Komure, K.; Kanemaru, A.; Takano, K.; Ozeki, K.; Nishimura, Y.; Yoshida, R.; Nakayama, H.; Shinriki, S.; et al. Loss of Tumor Suppressor CYLD Expression Triggers Cisplatin Resistance in Oral Squamous Cell Carcinoma. Int. J. Mol. Sci. 2019, 20, 5194. https://doi.org/10.3390/ijms20205194
Suenaga N, Kuramitsu M, Komure K, Kanemaru A, Takano K, Ozeki K, Nishimura Y, Yoshida R, Nakayama H, Shinriki S, et al. Loss of Tumor Suppressor CYLD Expression Triggers Cisplatin Resistance in Oral Squamous Cell Carcinoma. International Journal of Molecular Sciences. 2019; 20(20):5194. https://doi.org/10.3390/ijms20205194
Chicago/Turabian StyleSuenaga, Naoki, Mimi Kuramitsu, Kanae Komure, Ayumi Kanemaru, Kanako Takano, Kazuya Ozeki, Yuka Nishimura, Ryoji Yoshida, Hideki Nakayama, Satoru Shinriki, and et al. 2019. "Loss of Tumor Suppressor CYLD Expression Triggers Cisplatin Resistance in Oral Squamous Cell Carcinoma" International Journal of Molecular Sciences 20, no. 20: 5194. https://doi.org/10.3390/ijms20205194