The Effect of Dexamethasone, Adrenergic and Cholinergic Receptor Agonists on Phospholipid Metabolism in Human Osteoarthritic Synoviocytes

 , ,
, ,
Abstract
:1. Introduction
2. Results
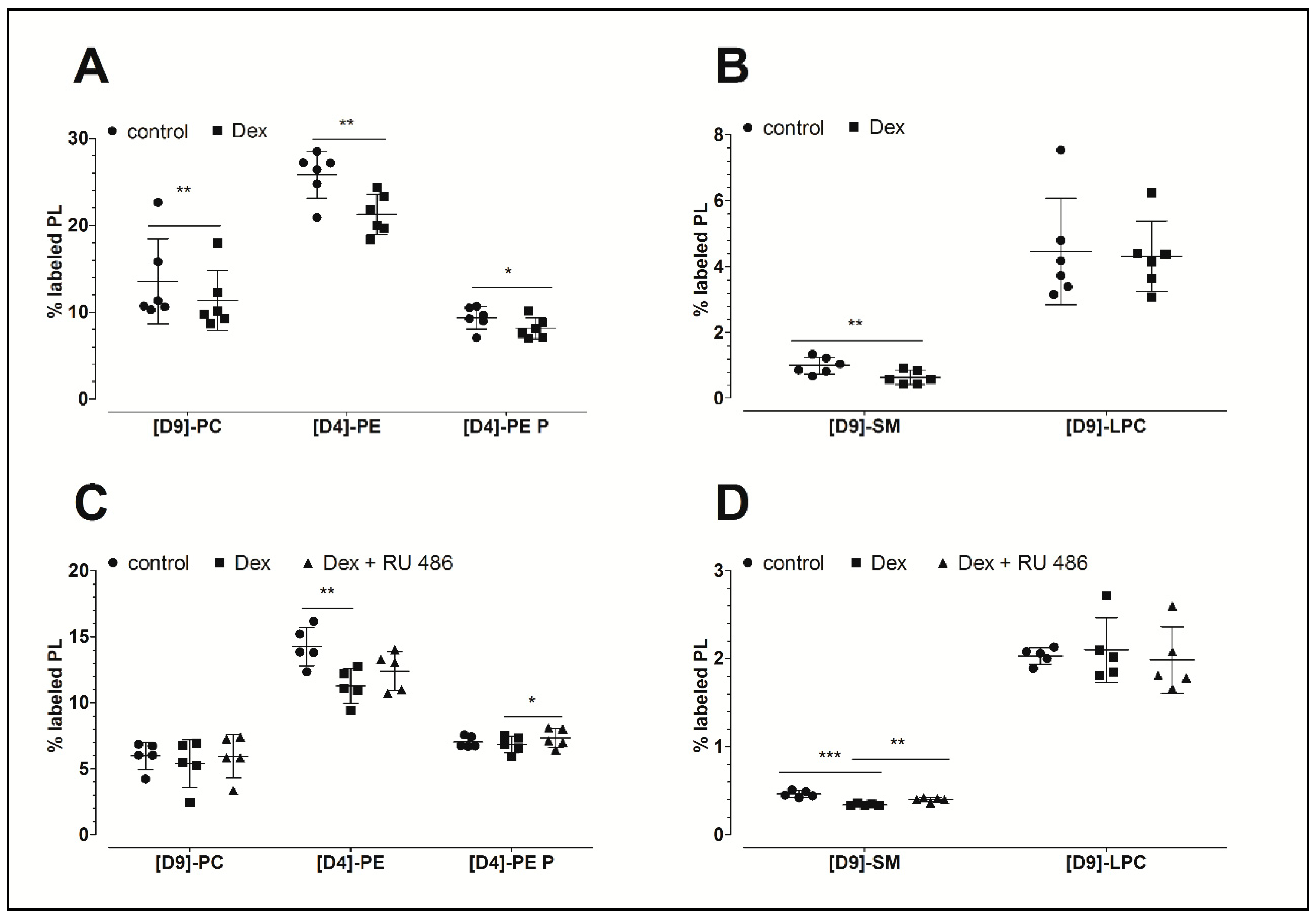
2.1. The Effect of Dexamethasone on the Biosynthesis of PL Classes
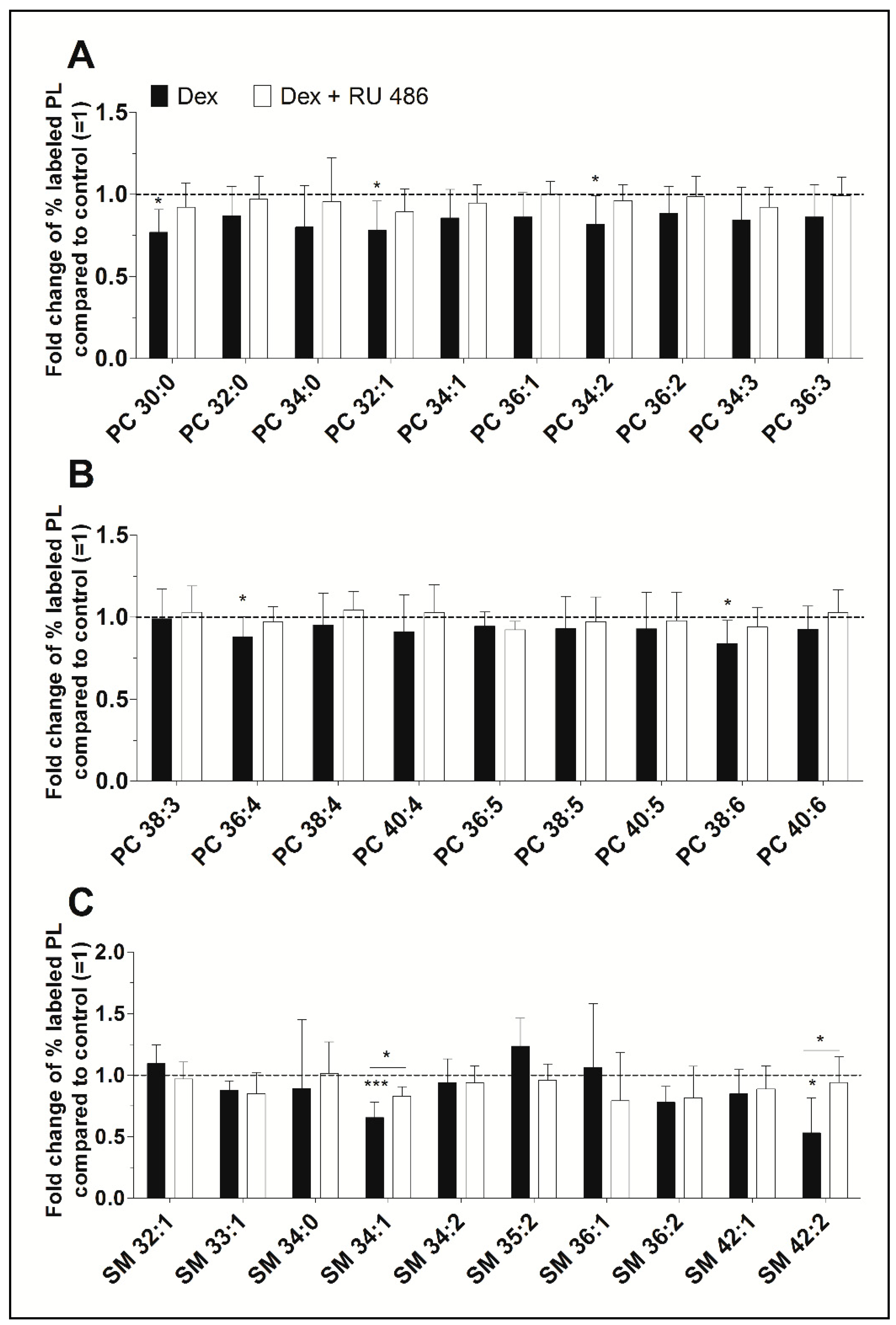
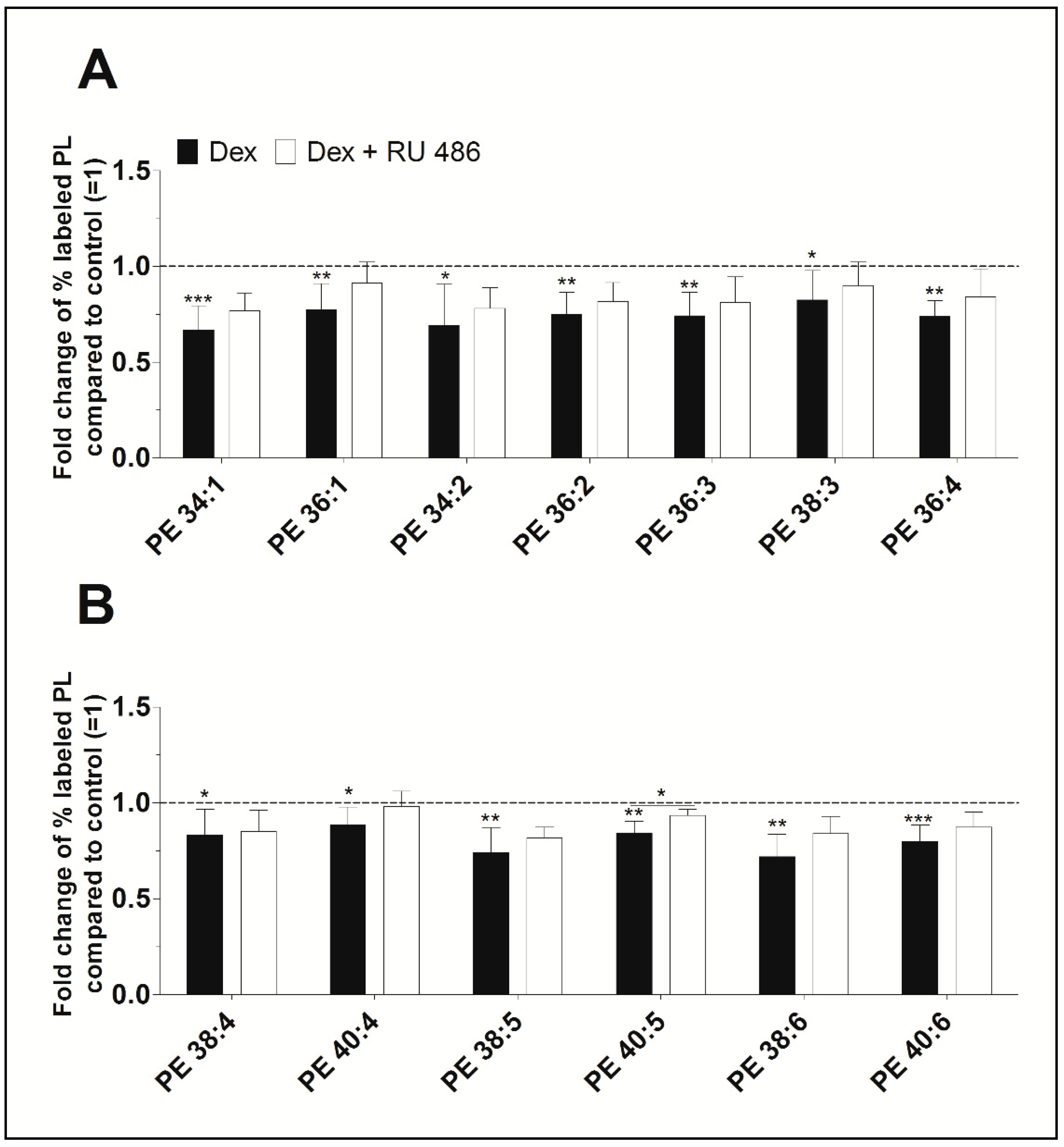
2.2. The Effect of the Glucocorticoid Receptor Antagonist RU 486
2.3. A Detailed Analysis of the Dexamethasone Effect on PL Species
2.4. The Effect of Adrenergic and Muscarinic Receptor Agonists on PL Biosynthesis
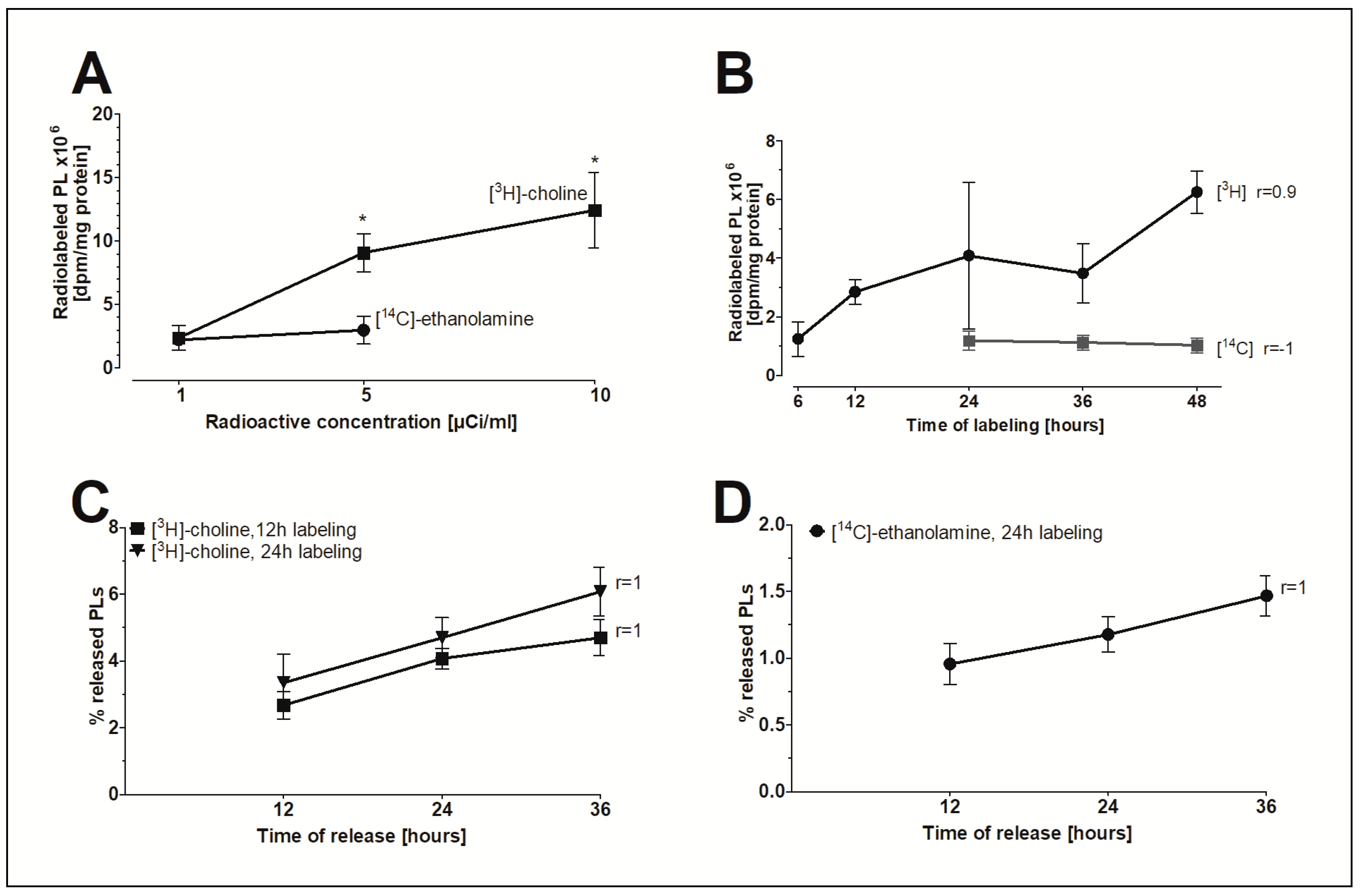
2.5. The In Vitro Model of FLS to Study PL Release
2.6. The Release of PLs from FLS as Modulated by Agonists
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Isolation of Fibroblast-Like Synoviocytes (FLS)
4.3. Cell Culture Procedure
4.4. FACS Analysis
4.5. The Effect of Dexamethasone, Cholinergic and Adrenergic Agonists on the Biosynthesis of PLs
4.6. Release Model
4.7. The Effect of Dexamethasone, Cholinergic and Adrenergic Agonists on PL Release
4.8. Lipid Extraction
4.9. PL Analysis by Mass Spectrometry
4.10. The Release of Radioactive PLs
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| COX | Cyclooxygenase |
| DPPC | Dipalmitoyl-Phosphatidylcholine |
| FLS | Fibroblast-like Synoviocytes |
| IL-1ß | Interleukin-1ß |
| LPC | Lysphosphatidylcholine |
| MS | Mass Spectrometry |
| OA | Osteoarthritis |
| PC | Phosphatidylcholine |
| PE | Phosphatidylethanolamine |
| PE P | Phosphatidylethanolamine-based plasmalogen |
| PL | Phospholipid |
| RA | Rheumatoid Arthritis |
| SM | Sphingomyelin |
| TNFα | Tumor necrosis factor α |
References
- Jahn, S.; Seror, J.; Klein, J. Lubrication of articular cartilage. Annu. Rev. Biomed. Eng. 2016, 18, 235–258. [Google Scholar] [CrossRef] [PubMed]
- Hills, B.A. Surface-active phospholipid: A pandora’s box of clinical applications. Part II. Barrier and lubricating properties. Intern. Med. J. 2002, 32, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Vance, J.E. Phospholipid synthesis and transport in mammalian cells. Traffic 2015, 16, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Bernhard, W. Lung surfactant: Function and composition in the context of development and respiratory physiology. Ann. Anat. 2016, 208, 146–150. [Google Scholar] [CrossRef]
- Mulugeta, S.; Nureki, S.; Beers, M.F. Lost after translation: Insights from pulmonary surfactant for understanding the role of alveolar epithelial dysfunction and cellular quality control in fibrotic lung disease. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 309, L507–L525. [Google Scholar] [CrossRef]
- Agassandian, M.; Mallampalli, R.K. Surfactant phospholipid metabolism. Biochim. Biophys. Acta 2013, 1831, 612–625. [Google Scholar] [CrossRef] [Green Version]
- Hills, B.A.; Crawford, R.W. Normal and prosthetic synovial joints are lubricated by surface-active phospholipid: A hypothesis. J. Arthroplasty 2003, 18, 499–505. [Google Scholar] [CrossRef]
- Crockett, R. Boundary lubrication in natural articular joints. Tribol. Lett. 2016, 35, 77–84. [Google Scholar] [CrossRef]
- Clark, R.B.; Schmidt, T.; Sachse, F.B.; Boyle, D.; Firestein, G.S.; Giles, W.R. Cellular electrophysiological principles that modulate secretion from synovial fibroblasts. J. Physiol. 2017, 595, 635–665. [Google Scholar] [CrossRef]
- Nagaoka, A.; Yoshida, H.; Nakamura, S.; Morikawa, T.; Kawabata, K.; Kobayashi, M.; Sakai, S.; Takahashi, Y.; Okada, Y.; Inoue, S. Regulation of hyaluronan (ha) metabolism mediated by hybid (hyaluronan-binding protein involved in ha depolymerization, kiaa1199) and ha synthases in growth factor-stimulated fibroblasts. J. Biol. Chem. 2015, 290, 30910–30923. [Google Scholar] [CrossRef]
- Kosinska, M.K.; Ludwig, T.E.; Liebisch, G.; Zhang, R.; Siebert, H.C.; Wilhelm, J.; Kaesser, U.; Dettmeyer, R.B.; Klein, H.; Ishaque, B.; et al. Articular joint lubricants during osteoarthritis and rheumatoid arthritis display altered levels and molecular species. PLoS ONE 2015, 10, e0125192. [Google Scholar] [CrossRef] [PubMed]
- Kosinska, M.K.; Liebisch, G.; Lochnit, G.; Wilhelm, J.; Klein, H.; Kaesser, U.; Lasczkowski, G.; Rickert, M.; Schmitz, G.; Steinmeyer, J. A lipidomic study of phospholipid classes and species in human synovial fluid. Arthritis Rheum. 2013, 65, 2323–2333. [Google Scholar] [CrossRef] [PubMed]
- King, G.; Damas, J.E.; Cake, M.H.; Berryman, D.; Maker, G.L. Influence of glucocorticoids, neuregulin-1beta, and sex on surfactant phospholipid secretion from type II cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 306, L292–L298. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, L.C.; Orgeig, S. Dexamethasone and epinephrine stimulate surfactant secretion in type II cells of embryonic chickens. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2001, 281, R770–R777. [Google Scholar] [CrossRef] [PubMed]
- Postle, A.D.; Gonzales, L.W.; Bernhard, W.; Clark, G.T.; Godinez, M.H.; Godinez, R.I.; Ballard, P.L. Lipidomics of cellular and secreted phospholipids from differentiated human fetal type II alveolar epithelial cells. J. Lipid Res. 2006, 47, 1322–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, D.J.; Williams, T.C.; Beck, S.L. Foamy macrophage responses in the rat lung following exposure to inhaled pharmaceuticals: A simple, pragmatic approach for inhaled drug development. J. Appl. Toxicol. 2014, 34, 319–331. [Google Scholar] [CrossRef]
- Enhorning, G. Surfactant in airway disease. Chest 2008, 133, 975–980. [Google Scholar] [CrossRef]
- McAuley, D.F.; Matthay, M.A. Is there a role for beta-adrenoceptor agonists in the management of acute lung injury and the acute respiratory distress syndrome? Treat. Respir. Med. 2005, 4, 297–307. [Google Scholar] [CrossRef]
- Gojnic, M.; Pervulov, M. Artificial fetal lung maturation--prevention of antenatal complications in premature deliveries. Clin. Exp. Obstet. Gynecol. 2005, 32, 61–64. [Google Scholar]
- Rooney, S.A. Lung surfactant. Environ. Health Perspect. 1984, 55, 205–226. [Google Scholar] [CrossRef]
- Jenei-Lanzl, Z.; Grassel, S.; Pongratz, G.; Kees, F.; Miosge, N.; Angele, P.; Straub, R.H. Norepinephrine inhibition of mesenchymal stem cell and chondrogenic progenitor cell chondrogenesis and acceleration of chondrogenic hypertrophy. Arthritis Rheumatol. 2014, 66, 2472–2481. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, J.; Schafer, N.; Bauer, R.; Jenei-Lanzl, Z.; Springorum, R.H.; Grassel, S. Norepinephrine modulates osteoarthritic chondrocyte metabolism and inflammatory responses. Osteoarthritis Cartilage 2016, 24, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Grassel, S.G. The role of peripheral nerve fibers and their neurotransmitters in cartilage and bone physiology and pathophysiology. Arthritis Res. Ther. 2014, 16, 485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grassel, S.; Muschter, D. Peripheral nerve fibers and their neurotransmitters in osteoarthritis pathology. Int. J. Mol. Sci. 2017, 18, 931. [Google Scholar] [CrossRef] [PubMed]
- Beckmann, J.; Schubert, J.; Morhenn, H.G.; Grau, V.; Schnettler, R.; Lips, K.S. Expression of choline and acetylcholine transporters in synovial tissue and cartilage of patients with rheumatoid arthritis and osteoarthritis. Cell Tissue Res. 2015, 359, 465–477. [Google Scholar] [CrossRef]
- Schubert, J.; Beckmann, J.; Hartmann, S.; Morhenn, H.G.; Szalay, G.; Heiss, C.; Schnettler, R.; Lips, K.S. Expression of the non-neuronal cholinergic system in human knee synovial tissue from patients with rheumatoid arthritis and osteoarthritis. Life Sci. 2012, 91, 1048–1052. [Google Scholar] [CrossRef]
- Beckmann, J.; Dittmann, N.; Schutz, I.; Klein, J.; Lips, K.S. Effect of M3 muscarinic acetylcholine receptor deficiency on collagen antibody-induced arthritis. Arthritis Res. Ther. 2016, 18, 17. [Google Scholar] [CrossRef]
- Gu, Q.; Li, D.; Wei, B.; Guo, Y.; Yan, J.; Mao, F.; Zhang, X.; Wang, L. Effects of nicotine on a rat model of early stage osteoarthritis. Int. J. Clin. Exp. Pathol. 2015, 8, 3602–3612. [Google Scholar]
- del Rincon, I.; Battafarano, D.F.; Restrepo, J.F.; Erikson, J.M.; Escalante, A. Glucocorticoid dose thresholds associated with all-cause and cardiovascular mortality in rheumatoid arthritis. Arthritis Rheumatol. 2014, 66, 264–272. [Google Scholar] [CrossRef]
- McAlindon, T.E.; Bannuru, R.R.; Sullivan, M.C.; Arden, N.K.; Berenbaum, F.; Bierma-Zeinstra, S.M.; Hawker, G.A.; Henrotin, Y.; Hunter, D.J.; Kawaguchi, H.; et al. OARSI guidelines for the non-surgical management of knee osteoarthritis. Osteoarthritis Cartilage 2014, 22, 363–388. [Google Scholar] [CrossRef] [Green Version]
- Richardson, D.W.; Dodge, G.R. Dose-dependent effects of corticosteroids on the expression of matrix-related genes in normal and cytokine-treated articular chondrocytes. Inflamm. Res. 2003, 52, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Shimpo, H.; Sakai, T.; Kondo, S.; Mishima, S.; Yoda, M.; Hiraiwa, H.; Ishiguro, N. Regulation of prostaglandin E(2) synthesis in cells derived from chondrocytes of patients with osteoarthritis. J. Orthop. Sci. 2009, 14, 611–617. [Google Scholar] [CrossRef] [PubMed]
- Huo, Y.; Rangarajan, P.; Ling, E.A.; Dheen, S.T. Dexamethasone inhibits the Nox-dependent ROS production via suppression of MKP-1-dependent MAPK pathways in activated microglia. BMC Neurosci. 2011, 12, 49. [Google Scholar] [CrossRef] [PubMed]
- Garvican, E.R.; Vaughan-Thomas, A.; Redmond, C.; Gabriel, N.; Clegg, P.D. MMP-mediated collagen breakdown induced by activated protein c in equine cartilage is reduced by corticosteroids. J. Orthop. Res. 2010, 28, 370–378. [Google Scholar] [CrossRef] [PubMed]
- Huebner, K.D.; Shrive, N.G.; Frank, C.B. Dexamethasone inhibits inflammation and cartilage damage in a new model of post-traumatic osteoarthritis. J. Orthop. Res. 2014, 32, 566–572. [Google Scholar] [CrossRef]
- Heard, B.J.; Barton, K.I.; Chung, M.; Achari, Y.; Shrive, N.G.; Frank, C.B.; Hart, D.A. Single intra-articular dexamethasone injection immediately post-surgery in a rabbit model mitigates early inflammatory responses and post-traumatic osteoarthritis-like alterations. J. Orthop. Res. 2015, 33, 1826–1834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huhtakangas, J.A.; Veijola, J.; Turunen, S.; Karjalainen, A.; Valkealahti, M.; Nousiainen, T.; Yli-Luukko, S.; Vuolteenaho, O.; Lehenkari, P. 1,25(oh)2d3 and calcipotriol, its hypocalcemic analog, exert a long-lasting anti-inflammatory and anti-proliferative effect in synoviocytes cultured from patients with rheumatoid arthritis and osteoarthritis. J. Steroid Biochem. Mol. Biol. 2017, 173, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Huhtakangas, J.A.; Veijola, J.; Turunen, S.; Karjalainen, A.; Valkealahti, M.; Nousiainen, T.; Yli-Luukko, S.; Vuolteenaho, O.; Lehenkari, P. Cytokine data obtained from synovial stromal cells of patients with rheumatoid arthritis or osteoarthritis. Data Brief 2017, 12, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Sampey, A.V.; Hutchinson, P.; Morand, E.F. Annexin I and dexamethasone effects on phospholipase and cyclooxygenase activity in human synoviocytes. Mediators Inflamm. 2000, 9, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Utomo, L.; van Osch, G.J.; Bayon, Y.; Verhaar, J.A.; Bastiaansen-Jenniskens, Y.M. Guiding synovial inflammation by macrophage phenotype modulation: An in vitro study towards a therapy for osteoarthritis. Osteoarthritis Cartilage 2016, 24, 1629–1638. [Google Scholar] [CrossRef]
- Gossye, V.; Elewaut, D.; Bougarne, N.; Bracke, D.; Van Calenbergh, S.; Haegeman, G.; De Bosscher, K. Differential mechanism of nf-kappab inhibition by two glucocorticoid receptor modulators in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 2009, 60, 3241–3250. [Google Scholar] [CrossRef] [PubMed]
- Hills, B.A.; Ethell, M.T.; Hodgson, D.R. Release of lubricating synovial surfactant by intra-articular steroid. Br. J. Rheumatol. 1998, 37, 649–652. [Google Scholar] [CrossRef] [PubMed]
- Gibellini, F.; Smith, T.K. The Kennedy pathway--de novo synthesis of phosphatidylethanolamine and phosphatidylcholine. IUBMB Life 2010, 62, 414–428. [Google Scholar] [CrossRef] [PubMed]
- Momchilova, A.; Markovska, T. Phosphatidylethanolamine and phosphatidylcholine are sources of diacylglycerol in ras-transformed nih 3t3 fibroblasts. Int. J. Biochem. Cell Biol. 1999, 31, 311–318. [Google Scholar] [CrossRef]
- Okamoto, Y.; Morishita, J.; Tsuboi, K.; Tonai, T.; Ueda, N. Molecular characterization of a phospholipase D generating anandamide and its congeners. J. Biol. Chem. 2004, 279, 5298–5305. [Google Scholar] [CrossRef] [PubMed]
- Carver, K.A.; Yang, D. N-acetylcysteine amide protects against oxidative stress-induced microparticle release from human retinal pigment epithelial cells. Invest. Ophthalmol. Vis. Sci. 2016, 57, 360–371. [Google Scholar] [CrossRef]
- Emoto, K.; Toyama-Sorimachi, N.; Karasuyama, H.; Inoue, K.; Umeda, M. Exposure of phosphatidylethanolamine on the surface of apoptotic cells. Exp. Cell Res. 1997, 232, 430–434. [Google Scholar] [CrossRef] [PubMed]
- Ichinose, Y.; Eguchi, K.; Migita, K.; Kawabe, Y.; Tsukada, T.; Koji, T.; Abe, K.; Aoyagi, T.; Nakamura, H.; Nagataki, S. Apoptosis induction in synovial fibroblasts by ceramide: In vitro and in vivo effects. J. Lab. Clin. Med. 1998, 131, 410–416. [Google Scholar] [CrossRef]
- Cutler, R.G.; Mattson, M.P. Sphingomyelin and ceramide as regulators of development and lifespan. Mech. Ageing Dev. 2001, 122, 895–908. [Google Scholar] [CrossRef]
- Zhao, J.; Wenzel, S. Interactions of RKIP with inflammatory signaling pathways. Crit. Rev. Oncog. 2014, 19, 497–504. [Google Scholar] [CrossRef]
- Guo, L.; Davies, S.S. Bioactive aldehyde-modified phosphatidylethanolamines. Biochimie 2013, 95, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Sluzalska, K.D.; Liebisch, G.; Lochnit, G.; Ishaque, B.; Hackstein, H.; Schmitz, G.; Rickert, M.; Steinmeyer, J. Interleukin-1beta affects the phospholipid biosynthesis of fibroblast-like synoviocytes from human osteoarthritic knee joints. Osteoarthritis Cartilage 2017, 25, 1890–1899. [Google Scholar] [CrossRef] [PubMed]
- John, K.; Marino, J.S.; Sanchez, E.R.; Hinds, T.D. The glucocorticoid receptor: Cause of or cure for obesity? Am. J. Physiol. Endocrinol. Metab. 2016, 310, E249–E257. [Google Scholar] [CrossRef] [PubMed]
- Löwenberg, M.; Stahn, C.; Hommes, D.W.; Buttgereit, F. Novel insights into mechanisms of glucocorticoid action and the development of new glucocorticoid receptor ligands. Steroids 2008, 73, 1025–1029. [Google Scholar]
- Urbach, V.; Verriere, V.; Grumbach, Y.; Bousquet, J.; Harvey, B.J. Rapid anti-secretory effects of glucocorticoids in human airway epithelium. Steroids 2006, 71, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Griese, M.; Gobran, L.I.; Rooney, S.A. Ontogeny of surfactant secretion in type II pneumocytes from fetal, newborn, and adult rats. Am. J. Physiol. 1992, 262, L337–L343. [Google Scholar] [CrossRef]
- Massaro, D.; Clerch, L.; Massaro, G.D. Surfactant secretion: Evidence that cholinergic stimulation of secretion is indirect. Am. J. Physiol. 1982, 243, C39–C45. [Google Scholar] [CrossRef]
- Oyarzun, M.J.; Clements, J.A.; Baritussio, A. Ventilation enhances pulmonary alveolar clearance of radioactive dipalmitoyl phosphatidylcholine in liposomes. Am. Rev. Respir. Dis. 1980, 121, 709–721. [Google Scholar]
- Wood, P.G.; Lopatko, O.V.; Orgeig, S.; Joss, J.M.; Smits, A.W.; Daniels, C.B. Control of pulmonary surfactant secretion: An evolutionary perspective. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000, 278, R611–R619. [Google Scholar] [CrossRef]
- Neumann, E.; Riepl, B.; Knedla, A.; Lefevre, S.; Tarner, I.H.; Grifka, J.; Steinmeyer, J.; Scholmerich, J.; Gay, S.; Muller-Ladner, U. Cell culture and passaging alters gene expression pattern and proliferation rate in rheumatoid arthritis synovial fibroblasts. Arthritis Res. Ther. 2010, 12, R83. [Google Scholar] [CrossRef] [Green Version]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Binder, M.; Liebisch, G.; Langmann, T.; Schmitz, G. Metabolic profiling of glycerophospholipid synthesis in fibroblasts loaded with free cholesterol and modified low density lipoproteins. J. Biol. Chem. 2006, 281, 21869–21877. [Google Scholar] [CrossRef] [PubMed]
- Liebisch, G.; Binder, M.; Schifferer, R.; Langmann, T.; Schulz, B.; Schmitz, G. High throughput quantification of cholesterol and cholesteryl ester by electrospray ionization tandem mass spectrometry (ESI-MS/MS). Biochim. Biophys. Acta 2006, 1761, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Liebisch, G.; Vizcaino, J.A.; Kofeler, H.; Trotzmuller, M.; Griffiths, W.J.; Schmitz, G.; Spener, F.; Wakelam, M.J. Shorthand notation for lipid structures derived from mass spectrometry. J. Lipid Res. 2013, 54, 1523–1530. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2017. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | (D9)-PC | (D4)-PE | (D4)-PE P | (D9)-SM | (D9)-LPC |
| (% labeled) | (% labeled) | (% labeled) | (% labeled) | (% labeled) | |
| (nmol/mg) | (nmol/mg) | (nmol/mg) | (pmol/mg) | (pmol/mg) | |
| Control | 13.6 ± 4.9 | 25.8 ± 2.7 | 9.4 ± 1.3 | 1.0 ± 0.3 | 4.5 ± 1.6 |
| 12.8 ± 4.7 | 8.2 ± 1.6 | 2.2 ± 0.2 | 245 ± 74 | 65 ± 23 | |
| Dex | 11.4 ± 3.5** | 21.3 ± 2.3 ** | 8.1 ± 1.2 * | 0.6 ± 0.2 ** | 4.3 ± 1.1 |
| 10.7 ± 3.1 | 6.6 ± 2.4 | 2.1 ± 0.4 | 162 ± 56 | 80 ± 26 | |
| Control | 6.0 ± 1.0 | 14.3 ± 1.5 | 7.0 ± 0.4 | 0.5 ± 0.0 | 2.0 ± 0.1 |
| 4.6 ± 0.8 | 2.9 ± 0.6 | 2.8 ± 0.3 | 115 ± 28 | 26 ± 3 | |
| Dex | 5.4 ± 1.8 | 11.3 ± 1.3 ** | 6.8 ± 0.6 | 0.3 ± 0.0 *** | 2.1 ± 0.4 |
| 4.0 ± 1.4 | 2.1 ± 0.3 | 2.7 ± 0.4 | 87 ± 22 | 27 ± 7 | |
| Dex + RU 486 | 5.9 ± 1.6 | 12.4 ± 1.5 | 7.3 ± 0.7* | 0.4 ± 0.0 ** | 2.0 ± 0.4 |
| 4.5 ± 1.4 | 2.3 ± 0.4 | 3.0 ± 0.4 | 105 ± 32 | 27 ± 8 | |
| Control | 11.8 ± 3.2 | 22.6 ± 2.9 | 8.0 ± 1.9 | 0.9 ± 0.2 | 3.9 ± 1.2 |
| 11.9 ± 3.8 | 7.6 ± 2.6 | 2.1 ± 0.4 | 222 ± 61 | 59 ± 20 | |
| Terbutaline | 12.5 ± 4.1 | 23.8 ± 1.8 | 8.4 ± 1.1 | 0.8 ± 0.2 | 5.0 ± 1.3 * |
| 11.5 ± 4.7 | 7.1 ± 1.2 | 2.0 ± 0.3 | 202 ± 69 | 74 ± 23 | |
| Epinephrine | 12.8 ± 4.9 | 24.2 ± 2.3 | 8.8 ± 1.3 | 0.9 ± 0.2 | 5.4 ± 1.6 * |
| 11.3 ± 4.3 | 7.2 ± 1.4 | 2.0 ± 0.1 | 197 ± 63 | 75 ± 26 | |
| Carbachol | 12.5 ± 4.8 | 24.1 ± 2.8 | 8.5 ± 1.9 | 0.9 ± 0.3 | 4.7 ± 1.7 |
| 12.1 ± 5.7 | 7.7 ± 2.8 | 2.1 ± 0.6 | 217 ± 92 | 81 ± 38 | |
| Pilocarpine | 12.8 ± 5.4 | 24.5 ± 2.7 * | 8.6 ± 1.7 | 0.9 ± 0.3 | 5.3 ± 2.0** |
| 12.5 ± 4.9 | 8.4 ± 3.7 | 2.2 ± 0.7 | 237 ± 88 | 90 ± 36 |
| Treatment | [3H]-Choline-Labeled PLs (% Released PLs) | [14C]-Ethanolamine-Labeled PLs (% Released PLs) |
|---|---|---|
| Control | 4.97 ± 1.04 | 2.19 ± 0.65 |
| Dexamethasone | 4.80 ± 1.44 | 2.09 ± 0.69 |
| Control | 5.03 ± 0.78 | 1.79 ± 0.33 |
| Terbutaline | 4.97 ± 1.20 | 2.06 ± 0.57 |
| Epinephrine | 5.08 ± 1.18 | ND |
| Carbachol | 4.67 ± 1.22 | 2.14 ± 0.68 |
| Pilocarpine | 5.19 ± 1.55 | ND |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sluzalska, K.D.; Liebisch, G.; Ishaque, B.; Schmitz, G.; Rickert, M.; Steinmeyer, J. The Effect of Dexamethasone, Adrenergic and Cholinergic Receptor Agonists on Phospholipid Metabolism in Human Osteoarthritic Synoviocytes. Int. J. Mol. Sci. 2019, 20, 342. https://doi.org/10.3390/ijms20020342
Sluzalska KD, Liebisch G, Ishaque B, Schmitz G, Rickert M, Steinmeyer J. The Effect of Dexamethasone, Adrenergic and Cholinergic Receptor Agonists on Phospholipid Metabolism in Human Osteoarthritic Synoviocytes. International Journal of Molecular Sciences. 2019; 20(2):342. https://doi.org/10.3390/ijms20020342
Chicago/Turabian StyleSluzalska, Katarzyna D., Gerhard Liebisch, Bernd Ishaque, Gerd Schmitz, Markus Rickert, and Juergen Steinmeyer. 2019. "The Effect of Dexamethasone, Adrenergic and Cholinergic Receptor Agonists on Phospholipid Metabolism in Human Osteoarthritic Synoviocytes" International Journal of Molecular Sciences 20, no. 2: 342. https://doi.org/10.3390/ijms20020342