Differential Role of TGF-β in Extracellular Matrix Regulation During Trypanosoma cruzi-Host Cell Interaction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
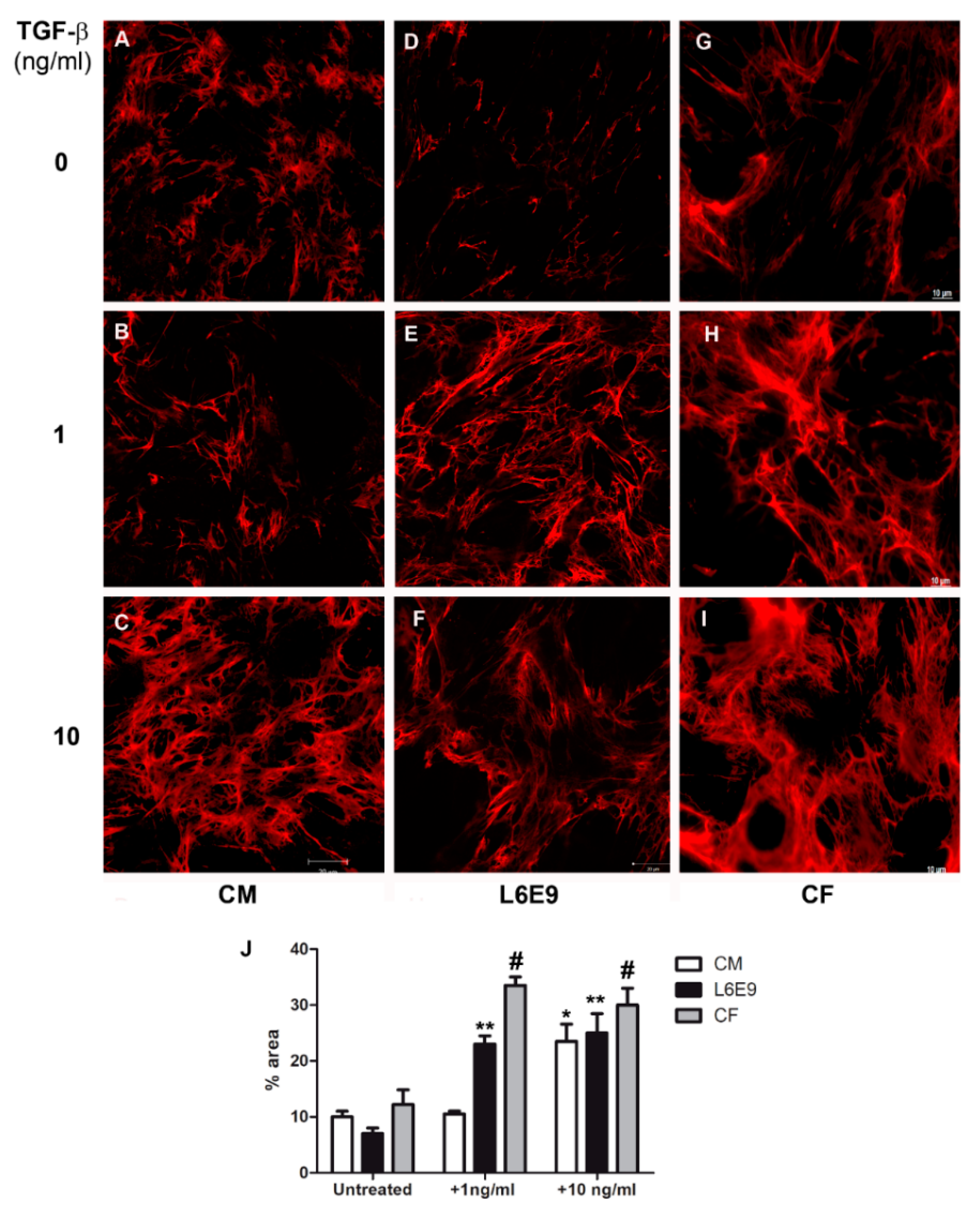
2.1. Modulation of Fibronectin Spatial Distribution in Different Cell Types after TGF-β Stimulation
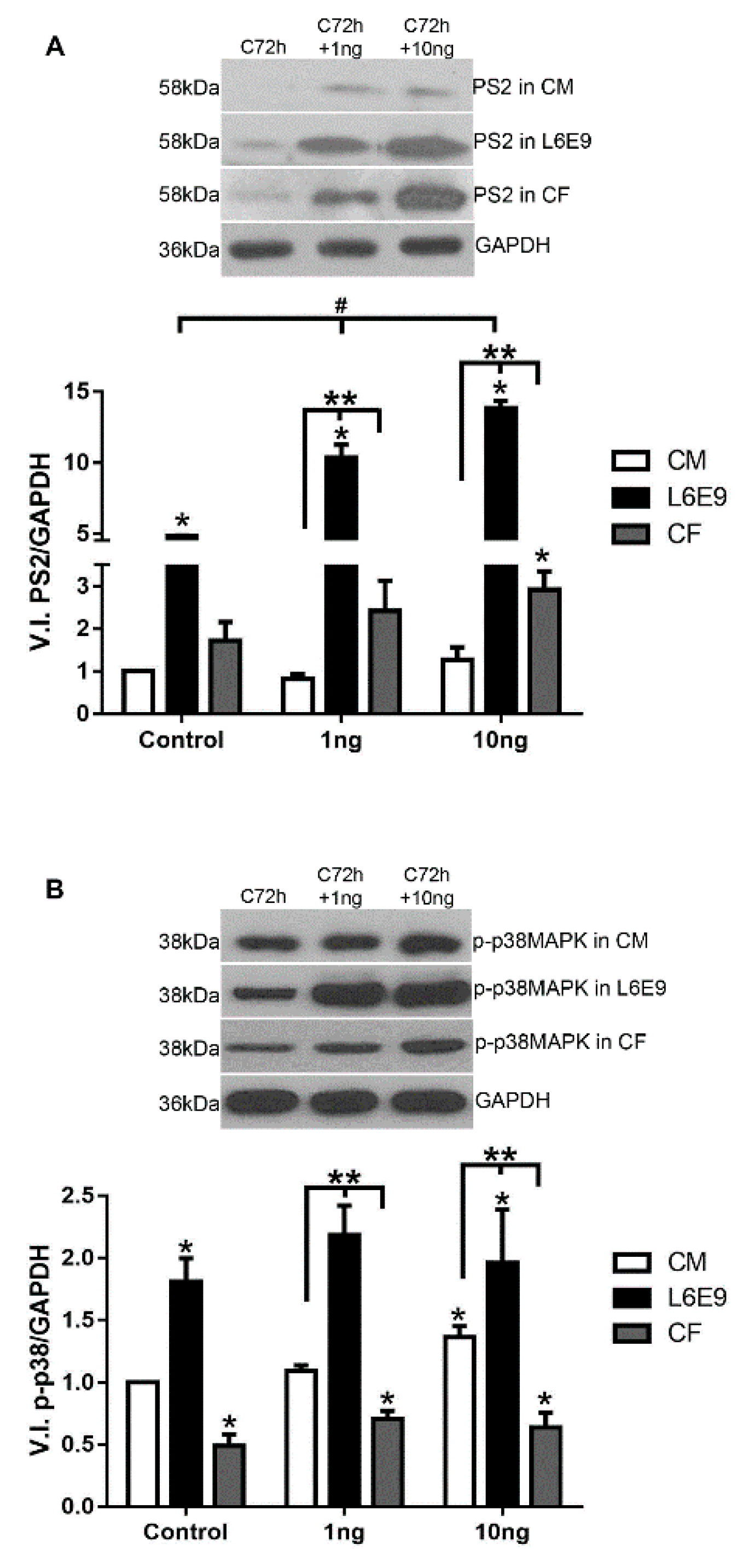
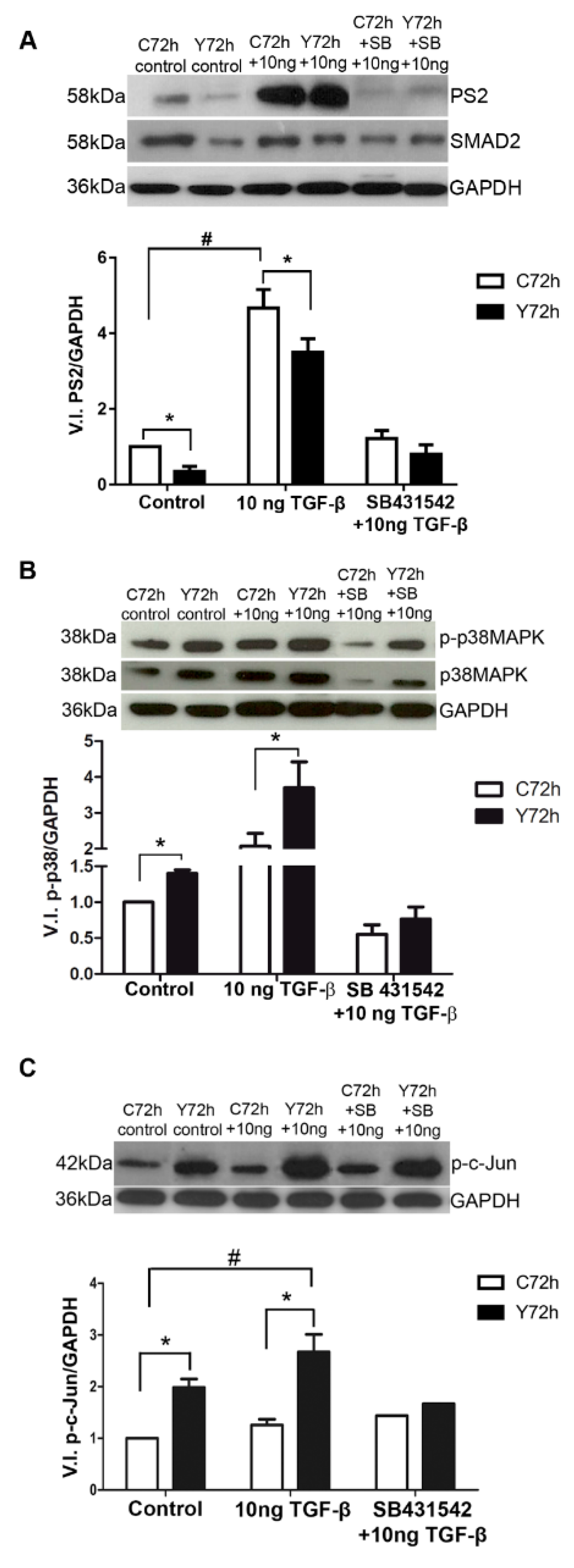
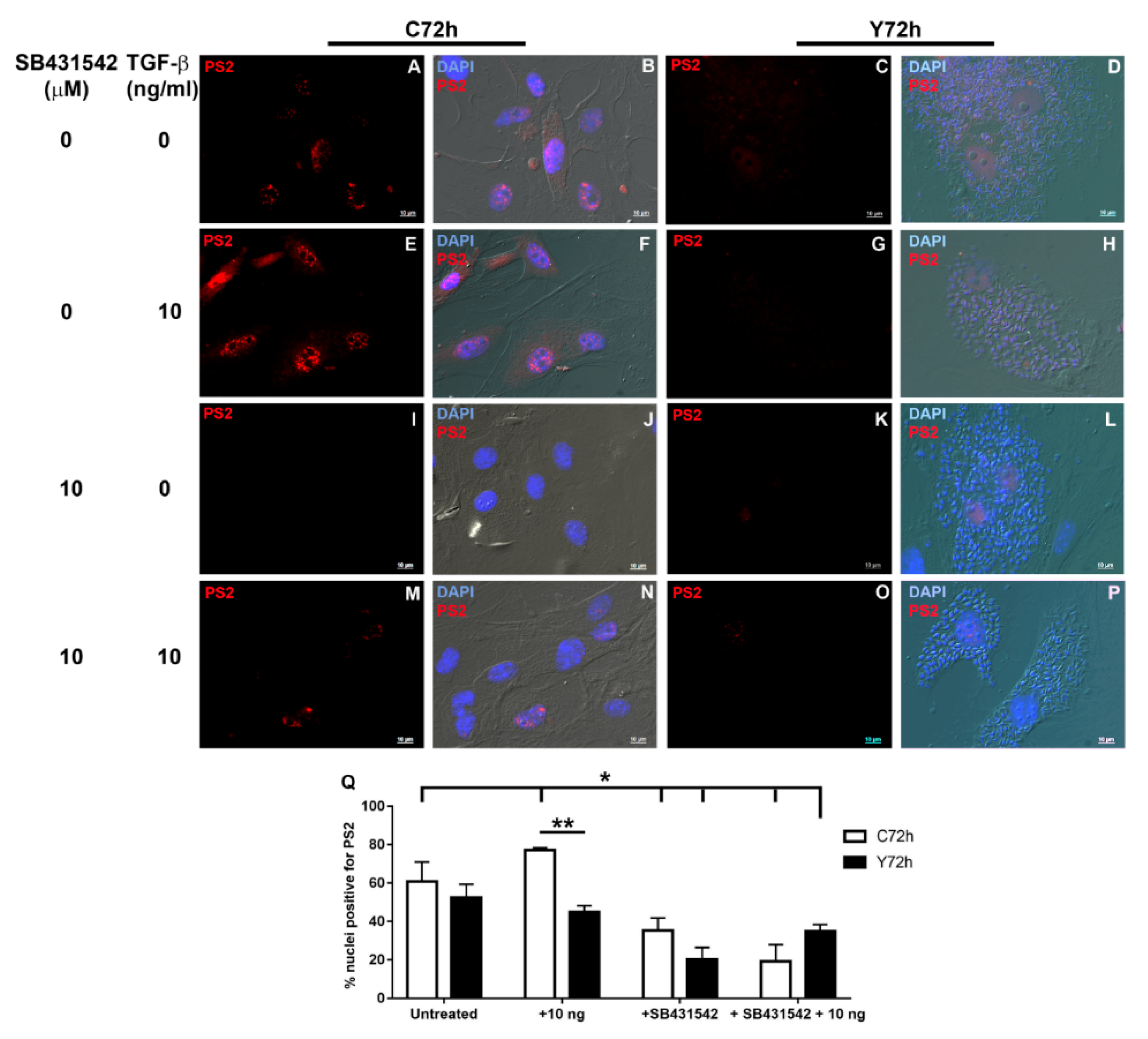
2.2. Signaling Pathways Triggered by TGF-β Stimulation
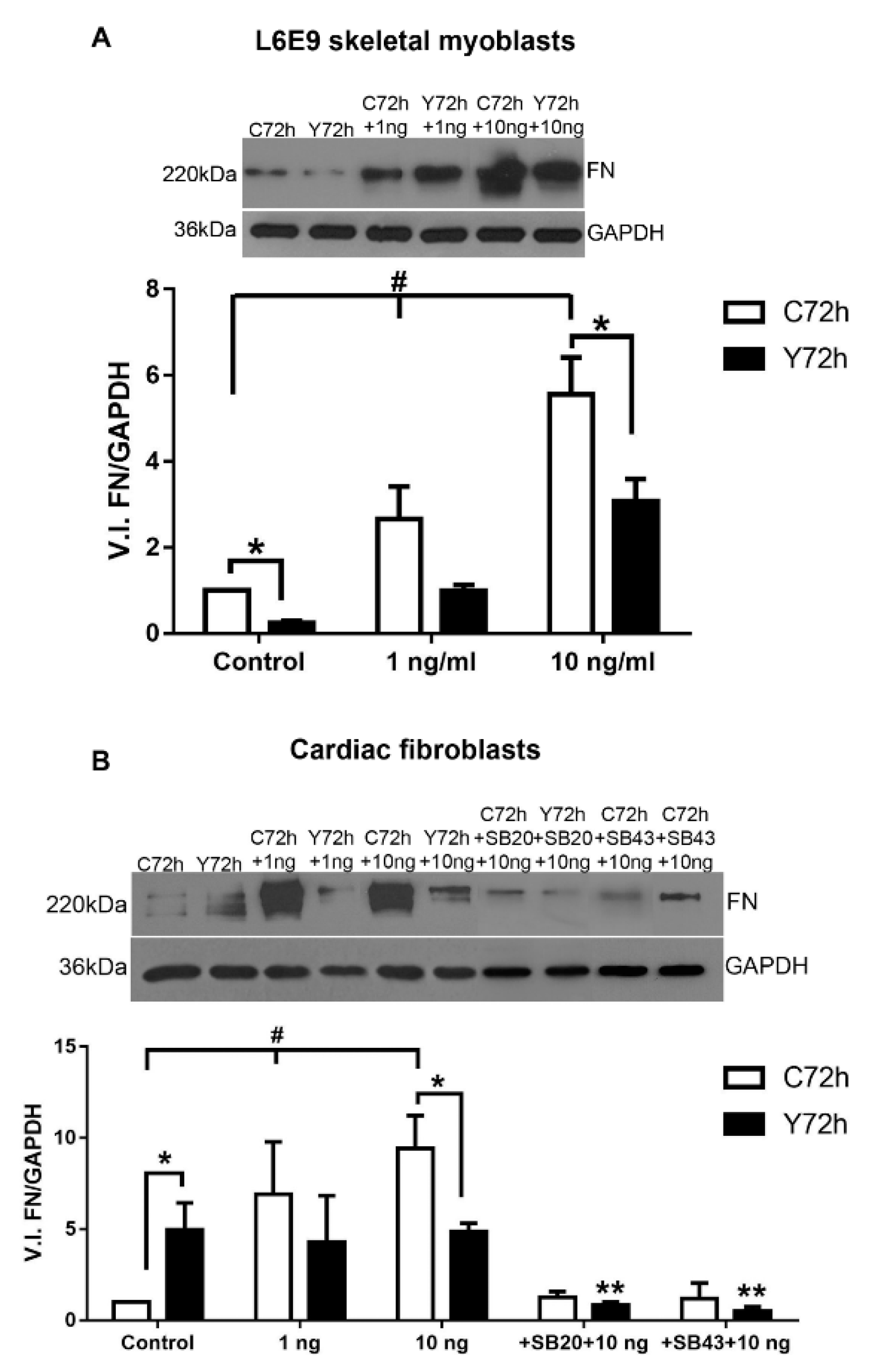
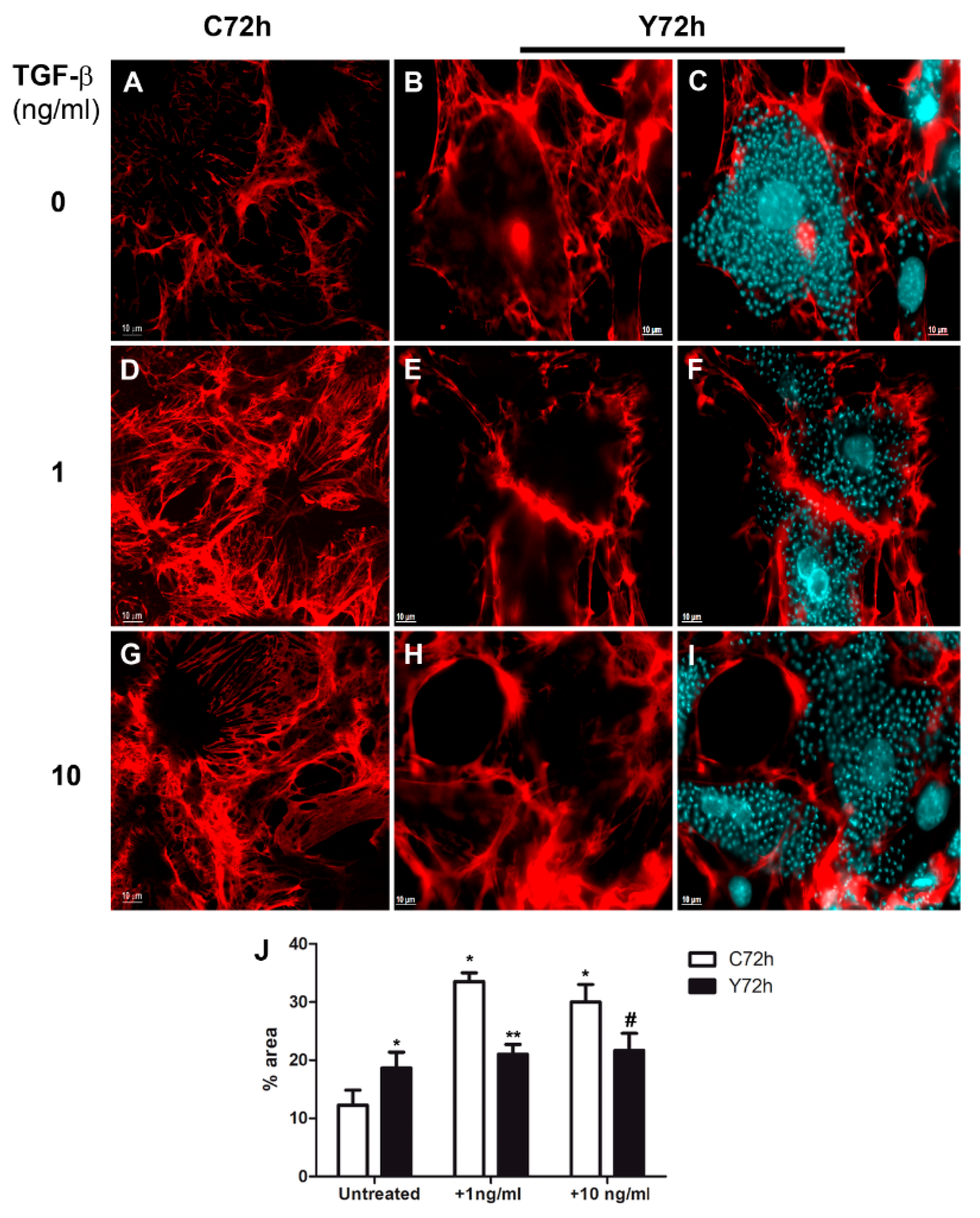
2.3. Differential Fibronectin Expression Induced by T. cruzi Infection and TGF-β Stimulation
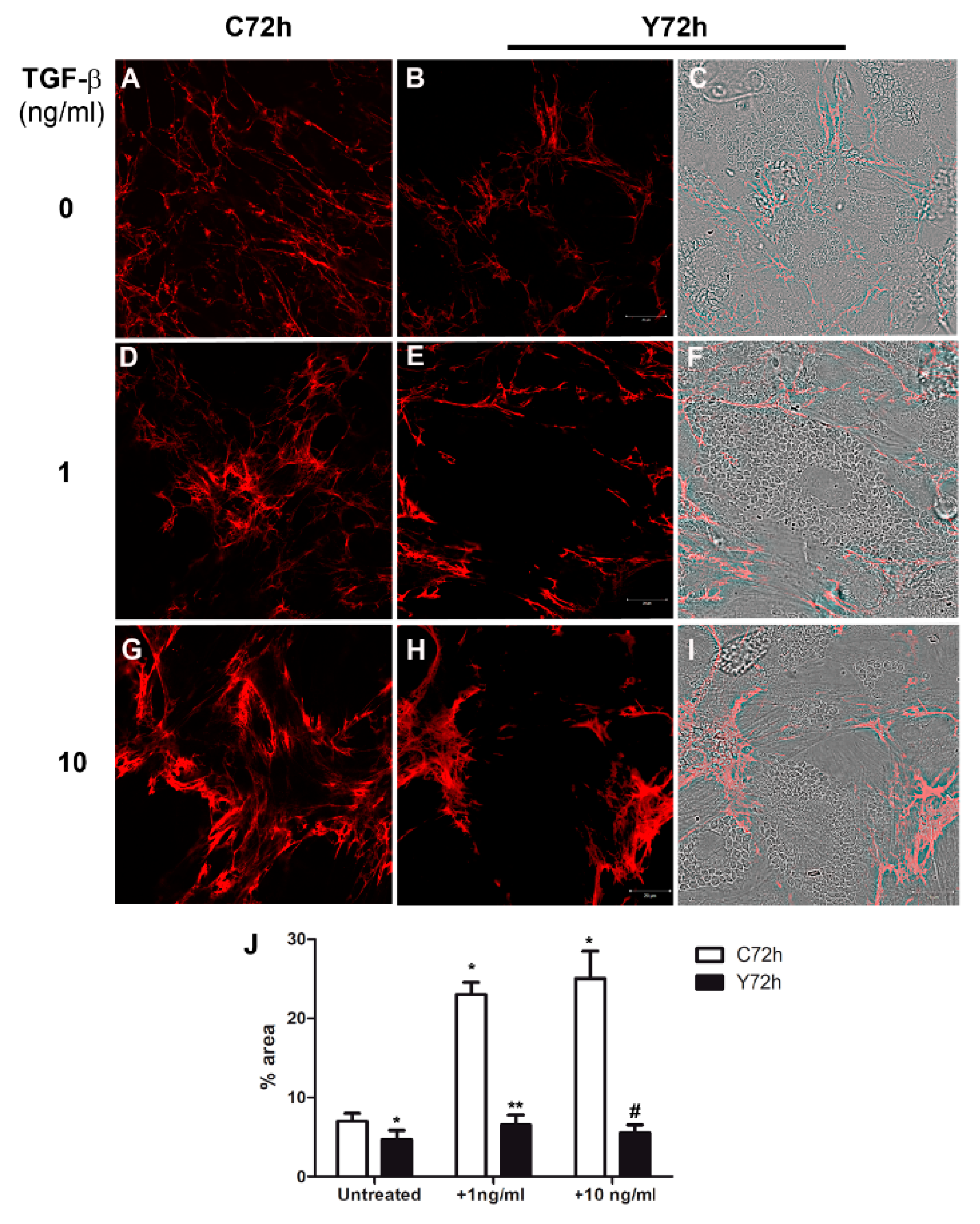
2.4. Regulation of Fibronectin Fibrillar Network Assembly by T. cruzi Infection
2.5. Signaling Pathways Involved in ECM Modulation Triggered by T. cruzi Cardiac Fibroblast Infection
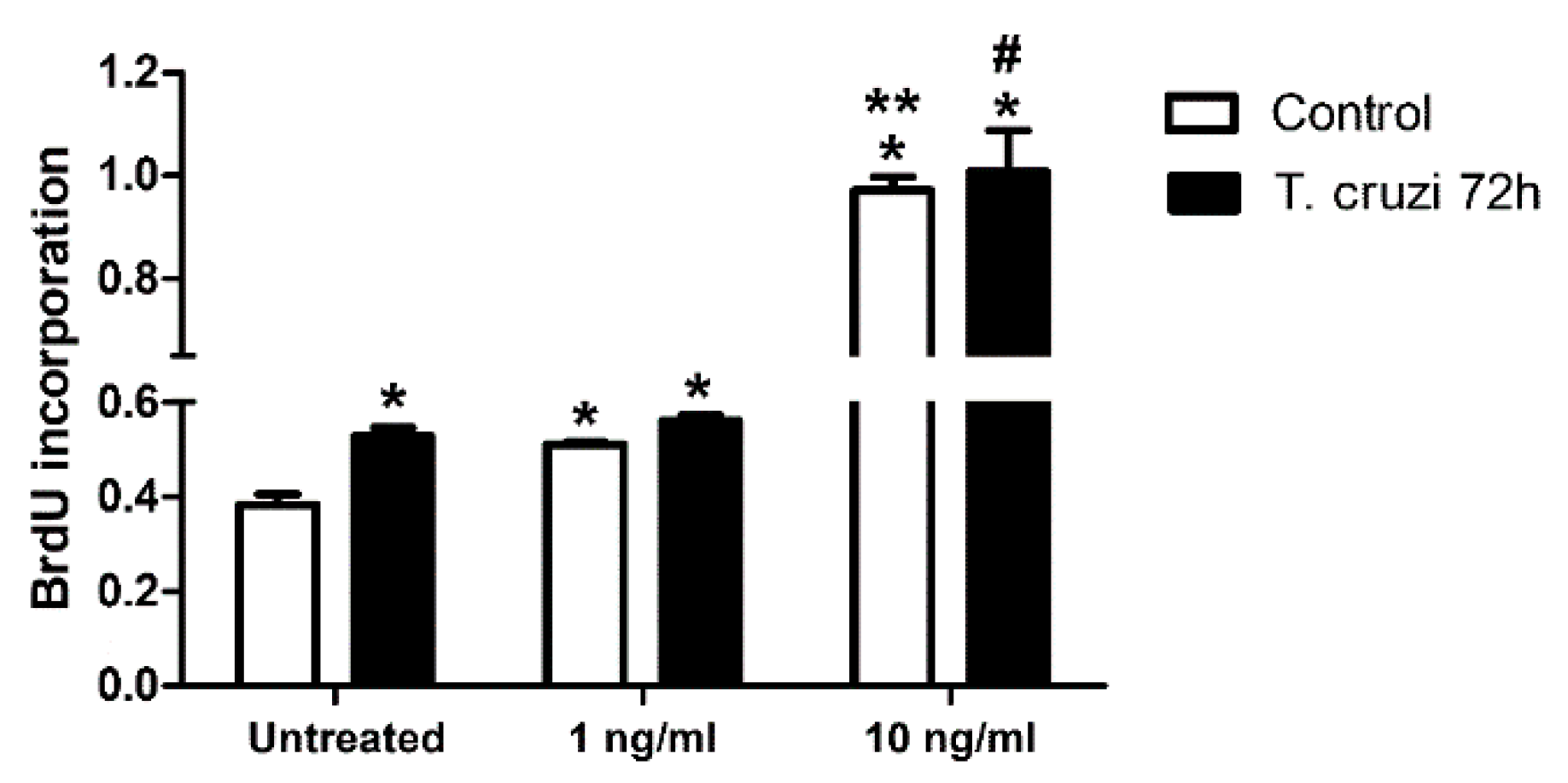
2.6. Induction of Cardiac Fibroblast Proliferation by T. cruzi Infection and TGF-β Stimulation
3. Discussion
4. Materials and Methods
4.1. Primary Cardiomyocyte Culture
4.2. Cardiac Fibroblasts and Skeletal Muscle Myoblasts Culture
4.3. Parasites and Cell Culture Infection
4.4. Treatment of Cardiomyocytes, Cardiac Fibroblasts, and Skeletal Myoblasts L6E9 with TGF-β
4.5. Indirect Immunofluorescence
4.6. Protein Extraction
4.7. Western Blot
4.8. Cardiac Fibroblasts Proliferation
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chagas, C. Nova tripanozomiaze humana. Mem. Inst. Oswaldo Cruz 1909, 1, 0074–0276. [Google Scholar] [CrossRef]
- Moncayo, Á.; Silveira, A.C. Current epidemiological trends of Chagas disease in Latin America and future challenges: Epidemiology, surveillance, and health policies. Am. Trypanos. Chagas Dis. 2017, 59–88. [Google Scholar] [CrossRef] [PubMed]
- WHO Chagas Disease- factsheet. Wkly. Epidemiol. Rec. 2012, 87, 519–522.
- Nunes, M.C.P.; Dones, W.; Morillo, C.A.; Encina, J.J.; Ribeiro, A.L. Chagas disease: An overview of clinical and epidemiological aspects. J. Am. Coll. Cardiol. 2013, 62, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, I.R.S.C.; Ferreira, M.L.; Camargos, E.R.S.; Chiari, E.; Machado, C.R.S. Skeletal muscle regeneration and Trypanosoma cruzi-induced myositis in rats. Histol. Histopathol. 2004, 19, 85–93. [Google Scholar]
- Rassi, A.; Marcondes de Rezende, J. American trypanosomiasis (Chagas disease). Infect. Dis. Clin. North Am. 2012, 26, 275–291. [Google Scholar] [CrossRef]
- Menezes Junior, A.D.S.; Lopes, C.C.; Cavalcante, P.F.; Martins, E. Chronic Chagas Cardiomyopathy Patients and Resynchronization Therapy: A Survival Analysis. Brazilian J. Cardiovasc. Surg. 2018, 33, 82–88. [Google Scholar]
- Benziger, C.P.; do Carmo, G.A.L.; Ribeiro, A.L.P. Chagas Cardiomyopathy: Clinical Presentation and Management in the Americas. Cardiol. Clin. 2017, 35, 31–47. [Google Scholar] [CrossRef]
- Rassi Jr, A.; Marin-Neto, J.A.; Rassi, A. Chronic Chagas cardiomyopathy: A review of the main pathogenic mechanisms and the efficacy of aetiological treatment following the BENznidazole Evaluation for Interrupting Trypanosomiasis (BENEFIT) trial. Mem. Inst. Oswaldo Cruz Rio Janeiro 2017, 112, 224–235. [Google Scholar] [CrossRef]
- Monteón, V.M.; Furuzawa-Carballeda, J.; Alejandre-Aguilar, R.; Aranda-Fraustro, A.; Rosales-Encina, J.L.; Reyes, P.A. American Trypanosomosis: In Situ and Generalized Features of Parasitism and Inflammation Kinetics in a Murine Model. Exp. Parasitol. 1996, 83, 267–274. [Google Scholar] [CrossRef]
- Fujiu, K.; Nagai, R. Fibroblast-mediated pathways in cardiac hypertrophy. J. Mol. Cell. Cardiol. 2014, 70, 64–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deb, A.; Ubil, E. Cardiac fibroblast in development and wound healing. J. Mol. Cell. Cardiol. 2014, 70, 47–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morikawa, M.; Derynck, R.; Miyazono, K. TGF- β and the TGF-β family: Context-dependent roles in cell and tissue physiology. Cold Spring Harb. Perspect. Biol. 2016, 8, a021873. [Google Scholar] [CrossRef] [PubMed]
- Araújo-Jorge, T.C.; Waghabi, M.C.; Hasslocher-Moreno, A.M.; Xavier, S.S.; Higuchi, M.D.L.; Keramidas, M.; Bailly, S.; Feige, J.-J. Implication of transforming growth factor-beta1 in Chagas disease myocardiopathy. J. Infect. Dis. 2002, 186, 1823–1828. [Google Scholar] [CrossRef] [PubMed]
- Araújo-Jorge, T.C.; Waghabi, M.C.; Bailly, S.; Feige, J.-J. The TGF-β pathway as an emerging target for Chagas disease therapy. Clin. Pharmacol. Ther. 2012, 92, 613–621. [Google Scholar] [PubMed]
- Zhao, B.; Chen, Y.-G. Regulation of TGF-β Signal Transduction. Scientifica (Cairo) 2014, 2014, 874065. [Google Scholar] [PubMed]
- Akhurst, R.J.; Hata, A. Targeting the TGFβ signalling pathway in disease. Nat. Rev. Drug Discov. 2012, 11, 790–811. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J. TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Mu, Y.; Gudey, S.K.; Landström, M. Non-Smad signaling pathways. Cell Tissue Res. 2012, 347, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Hall, B.S.; Pereira, M.A. Dual Role for Transforming Growth Factor β-Dependent Signaling in Trypanosoma cruzi Infection of Mammalian Cells. Infect. Immun. 2000, 68, 2077–2081. [Google Scholar] [CrossRef] [PubMed]
- Ming, M.; Ewen, M.E.; Pereira, M.E.A.; Street, W. Trypanosome Invasion of Mammalian Cells Requires Activation of the TGFp Signaling Pathway. Cell 1995, 82, 287–296. [Google Scholar] [CrossRef]
- Waghabi, M.C.; Keramidas, M.; Calvet, C.M.; Meuser, M.; Soeiro, M.D.N.C.; Mendonça-Lima, L.; Araújo-Jorge, T.C.; Feige, J.-J.; Bailly, S. SB-431542, a transforming growth factor β inhibitor, impairs Trypanosoma cruzi infection in cardiomyocytes and parasite cycle completion. Antimicrob. Agents Chemother. 2007, 51. [Google Scholar] [CrossRef] [PubMed]
- Waghabi, M.C.; Keramidas, M.; Feige, J.-J.; Araujo-Jorge, T.C.; Bailly, S. Activation of transforming growth factor beta by Trypanosoma cruzi. Cell. Microbiol. 2005, 7, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Waghabi, M.C.; Coutinho-Silva, R.; Feige, J.-J.; Higuchi, M.D.L.; Becker, D.; Burnstock, G.; Araújo-Jorge, T.C. Gap junction reduction in cardiomyocytes following transforming growth factor-beta treatment and Trypanosoma cruzi infection. Mem. Inst. Oswaldo Cruz 2009, 104, 1083–1090. [Google Scholar] [CrossRef] [PubMed]
- Waghabi, M.C.; Keramidas, M.; Bailly, S.; Degrave, W.; Mendonça-Lima, L.; Soeiro, M.D.N.C.; Meirelles, M.D.N.L.; Paciornik, S.; Araújo-Jorge, T.C.; Feige, J.J. Uptake of host cell transforming growth factor-beta by Trypanosoma cruzi amastigotes in cardiomyocytes: Potential role in parasite cycle completion. Am. J. Pathol. 2005, 167, 993–1003. [Google Scholar] [CrossRef]
- Ferrão, P.M.; d’Avila-Levy, C.M.; Araujo-Jorge, T.C.; Degrave, W.M.; Gonçalves, A.D.S.; Garzoni, L.R.; Lima, A.P.; Feige, J.J.; Bailly, S.; Mendonça-Lima, L.; et al. Cruzipain Activates Latent TGF-β from Host Cells during T. cruzi Invasion. PLoS One 2015, 10, e0124832. [Google Scholar] [CrossRef]
- Martin, D.L.; Postan, M.; Lucas, P.; Gress, R.; Tarleton, R.L. TGF-beta regulates pathology but not tissue CD8+ T cell dysfunction during experimental Trypanosoma cruzi infection. Eur. J. Immunol. 2007, 37, 2764–2771. [Google Scholar] [CrossRef]
- Waghabi, M.C.; Coutinho, C.M.L.M.; Soeiro, M.N.C.; Pereira, M.C.S.; Feige, J.; Keramidas, M.; Cosson, A.; Minoprio, P.; Van Leuven, F.; Arau, T.C.; et al. Increased Trypanosoma cruzi Invasion and Heart Fibrosis Associated with High Transforming Growth Factor β Levels in Mice Deficient in α2 -Macroglobulin. Infect. Immun. 2002, 70, 5115–5123. [Google Scholar] [CrossRef]
- Rodrigues, D.B.R.; dos Reis, M.A.; Romano, A.; Pereira, S.A.D.L.; Teixeira, V.D.P.A.; Tostes, S.; Rodrigues, V. In situ expression of regulatory cytokines by heart inflammatory cells in Chagas’ disease patients with heart failure. Clin. Dev. Immunol. 2012, 2012, 361730. [Google Scholar] [CrossRef]
- Ferreira, R.R.; de Souza, E.M.; de Oliveira, F.L.; Ferrão, P.M.; Gomes, L.H.F.; Mendonça-Lima, L.; Meuser-Batista, M.; Bailly, S.; Feige, J.J.; de Araujo-Jorge, T.C.; et al. Proteins involved on TGF-β pathway are up-regulated during the acute phase of experimental Chagas disease. Immunobiology 2016, 221, 587–594. [Google Scholar] [CrossRef]
- De Oliveira, F.L.; Araújo-Jorge, T.C.; de Souza, E.M.; de Oliveira, G.M.; Degrave, W.M.; Feige, J.-J.; Bailly, S.; Waghabi, M.C. Oral administration of GW788388, an inhibitor of transforming growth factor beta signaling, prevents heart fibrosis in Chagas disease. PLoS Negl. Trop. Dis. 2012, 6, e1696. [Google Scholar] [CrossRef]
- Bogoyevitch, M.A.; Boehm, I.; Oakley, A.; Ketterman, A.J.; Barr, R.K. Targeting the JNK MAPK cascade for inhibition: Basic science and therapeutic potential. Biochim. Biophys. Acta Proteins Proteomics 2004, 1697, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Calvet, C.M.; Oliveira, F.O.R.; Araújo-Jorge, T.C.; Pereira, M.C.S. Regulation of extracellular matrix expression and distribution in Trypanosoma cruzi-infected cardiomyocytes. Int. J. Med. Microbiol. 2009, 299, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Waghabi, M.C.; de Souza, E.M.; de Oliveira, G.M.; Keramidas, M.; Feige, J.-J.; Araújo-Jorge, T.C.; Bailly, S. Pharmacological inhibition of transforming growth factor beta signaling decreases infection and prevents heart damage in acute Chagas’ disease. Antimicrob. Agents Chemother. 2009, 53, 4694–4701. [Google Scholar] [CrossRef] [PubMed]
- Ferrão, P.; Nisimura, L.; Moreira, O.; Land, M.; Pereira, M.C.; de Mendonça-Lima, L.; Araujo-Jorge, T.; Waghabi, M.; Garzoni, L. Inhibition of TGF-β pathway reverts extracellular matrix remodeling in T. cruzi -infected cardiac spheroids. Exp. Cell Res. 2018, 362, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Calvet, C.M.; Meuser, M.; Almeida, D.; Meirelles, M.N.L.; Pereira, M.C.S. Trypanosoma cruzi-cardiomyocyte interaction: Role of fibronectin in the recognition process and extracellular matrix expression in vitro and in vivo. Exp. Parasitol. 2004, 107, 20–30. [Google Scholar] [CrossRef]
- Leask, A. TGFbeta, cardiac fibroblasts, and the fibrotic response. Cardiovasc. Res. 2007, 74, 207–212. [Google Scholar] [CrossRef]
- Ignotz, R.A.; Massaguù, J. Cell adhesion protein receptors as targets for transforming growth factor-beta action. Cell 1987, 51, 189–197. [Google Scholar] [CrossRef]
- García-Pozo, L.; Miquilena-Colina, M.E.; Lozano-Rodríguez, T.; García-Monzón, C. Endoglin: Structure, biological functions, and role in fibrogenesis. Rev. Española Enfermedades Dig. 2008, 100, 355–360. [Google Scholar]
- Rodríguez-Barbero, A.; Obreo, J.; Álvarez-Muñoz, P.; Pandiella, A.; Bernabeu, C.; Lopes-Novoa, J.M. Endoglin Modulation of TGF-β induced collagen synthesis is dependent on ERK1/2 MAPK Activation. Cell. Physiol. Biochem. 2006, 18, 135–142. [Google Scholar] [CrossRef]
- Kapur, N.K.; Wilson, S.; Yunis, A.A.; Qiao, X.; Mackey, E.; Paruchuri, V.; Baker, C.; Aronovitz, M.J.; Karumanchi, S.A.; Letarte, M.; et al. Reduced endoglin activity limits cardiac fibrosis and improves survival in heart failure. Circulation 2012, 125, 2728–2738. [Google Scholar] [CrossRef] [PubMed]
- Nomura-Kitabayashi, A.; Anderson, G.A.; Sleep, G.; Mena, J.; Karabegovic, A.; Karamath, S.; Letarte, M.; Puri, M.C. Endoglin is dispensable for angiogenesis, but required for endocardial cushion formation in the midgestation mouse embryo. Dev. Biol. 2009, 335, 66–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mott, G.A.; Costales, J.A.; Burleigh, B.A. A soluble factor from Trypanosoma cruzi inhibits transforming growth factor-ß-induced MAP kinase activation and gene expression in dermal fibroblasts. PLoS ONE 2011, 6, e23482. [Google Scholar] [CrossRef]
- Melo, T.G.; Tucci, A.R.; Nogueira, A.R.; Meirelles, M.D.N.S.L.; Pereira, M.C.S. The involvement of FAK and Src in the invasion of cardiomyocytes by Trypanosoma cruzi. Exp. Parasitol. 2014, 139, 49–57. [Google Scholar] [CrossRef] [Green Version]
- Pereira, M.C.; Costa, M.; Chagas Filho, C.; de Meirelles, M.N. Myofibrillar breakdown and cytoskeletal alterations in heart muscle cells during invasion by Trypanosoma cruzi: Immunological and ultrastructural study. J. Submicrosc. Cytol. Pathol. 1993, 25, 559–569. [Google Scholar] [PubMed]
- Calvet, C.M.; Silva, T.A.; De Melo, T.G.; De Araújo-Jorge, T.C.; De Souza Pereira, M.C. TGF-β receptor type II costameric localization in cardiomyocytes and host cell TGF-β response is disrupted by Trypanosoma cruzi infection. Parasitology 2016, 143. [Google Scholar] [CrossRef] [PubMed]
- Pereira, M.C.; Singer, R.H.; de Meirelles, M.N. Ultrastructural Distribution of Poly (A)+ RNA During Trypanosoma cruzi- Cardiomyocyte Interaction in vitro: A Quantitative Analysis of the Total mRNA Content by in situ Hybridization. J. Eukaryot. Microbiol. 2000, 47, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Machado, F.S.; Martins, G.A.; Aliberti, J.C.; Mestriner, F.L.; Cunha, F.Q.; Silva, J.S. Trypanosoma cruzi-infected cardiomyocytes produce chemokines and cytokines that trigger potent nitric oxide-dependent trypanocidal activity. Circulation 2000, 102, 3003–3008. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Chen, M.; Dawood, F.; Zurawska, U.; Li, J.Y.; Parker, T.; Kassiri, Z.; Kirshenbaum, L.A.; Arnold, M.; Khokha, R.; et al. Tumor necrosis factor-alpha mediates cardiac remodeling and ventricular dysfunction after pressure overload state. Circulation 2007, 115, 1398–1407. [Google Scholar] [CrossRef] [PubMed]
- Cuenda, A.; Rousseau, S. p38 MAP-kinases pathway regulation, function and role in human diseases. Biochim. Biophys. Acta 2007, 1773, 1358–1375. [Google Scholar] [CrossRef] [PubMed]
- Stempin, C.C.; Garrido, V.V.; Dulgerian, L.R.; Cerbán, F.M. Cruzipain and SP600125 induce p38 activation, alter NO/arginase balance and favor the survival of Trypanosoma cruzi in macrophages. Acta Trop. 2008, 106, 119–127. [Google Scholar] [PubMed]
- Molkentin, J.D.; Bugg, D.; Ghearing, N.; Dorn, L.E.; Kim, P.; Sargent, M.A.; Gunaje, J.; Otsu, K.; Davis, J. Fibroblast-specific genetic manipulation of p38 MAPK in vivo reveals its central regulatory role in fibrosis. Circulation 2017, 136, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Engel, M.E.; McDonnell, M.A.; Law, B.K.; Moses, H.L. Interdependent smad and jnk signaling in transforming growth factor- beta-mediated transcription. J. Biol. Chem. 1999, 274, 37413–37420. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.E. Non-Smad pathways in TGF-β signaling. Cell Res. 2010, 19, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Suman, S.; Rachakonda, G.; Mandape, S.N.; Sakhare, S.S.; Villalta, F.; Pratapid, S.; Lima, M.F.; Ndeid, P.N. Phospho-proteomic analysis of primary human colon epithelial cells during the early Trypanosoma cruzi infection phase. PLoS Negl. Trop. Dis. 2018, 12, e0006792. [Google Scholar] [CrossRef]
- Udoko, A.N.; Johnson, C.A.; Dykan, A.; Rachakonda, G.; Villalta, F.; Mandape, S.N.; Lima, M.F.; Pratap, S.; Nde, P.N. Early Regulation of Profibrotic Genes in Primary Human Cardiac Myocytes by Trypanosoma cruzi. PLoS Negl. Trop. Dis. 2016, 10, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Wyler, D.J.; Libby, P.; Prakash, S.; Prioli, R.P.; Pereira, M.E. Elaboration by mammalian mesenchymal cells infected with Trypanosoma cruzi of a fibroblast-stimulating factor that may contribute to chagasic cardiomyopathy. Infect. Immun. 1987, 55, 3188–3191. [Google Scholar] [Green Version]
- Arias, J.I.; Sepulveda, C.; Bravo, P.; Hamilton-West, C.; Maldonado, I.; Ferreira, A. Comparative effect of human and Trypanosoma cruzi calreticulin in wound healing. J. Tissue Eng. Regen. Med. 2015, 9, 41–54. [Google Scholar] [CrossRef]
- Reis, M.M.; Higuchi, M.L.; Benvenuti, L.A.; Aiello, V.D.; Gutierrez, P.S.; Bellotti, G.; Pileggi, F. An in situ quantitative immunohistochemical study of cytokines and IL-2R+ in chronic human chagasic myocarditis: Correlation with the presence of myocardial Trypanosoma cruzi antigens. Clin. Immunol. Immunopathol. 1997, 83, 165–172. [Google Scholar]
- Li, Q.; Chang, L.; Su, D.M.; Ma, X. Effects of tetrandrine on proliferation and activation of cardiac fibroblasts. Beijing Da Xue Xue Bao Yi Xue Ban 2018, 50, 331–334. [Google Scholar]
- Meirelles, M.N.S.L.; de Araújo-Jorge, T.C.; Miranda, C.F.; de Souza, W.; Barbosa, H.S. Interaction of Trypanosoma cruzi with heart muscle cells: Ultrastructural and cytochemical analysis of endocytic vacuole formation and effect upon myogenesis in vitro. Eur. J. Cell Biol. 1986, 41, 198–206. [Google Scholar]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva, T.A.; Ferreira, L.F.d.C.; Pereira, M.C.d.S.; Calvet, C.M. Differential Role of TGF-β in Extracellular Matrix Regulation During Trypanosoma cruzi-Host Cell Interaction. Int. J. Mol. Sci. 2019, 20, 4836. https://doi.org/10.3390/ijms20194836
Silva TA, Ferreira LFdC, Pereira MCdS, Calvet CM. Differential Role of TGF-β in Extracellular Matrix Regulation During Trypanosoma cruzi-Host Cell Interaction. International Journal of Molecular Sciences. 2019; 20(19):4836. https://doi.org/10.3390/ijms20194836
Chicago/Turabian StyleSilva, Tatiana Araújo, Luis Felipe de Carvalho Ferreira, Mirian Claudia de Souza Pereira, and Claudia Magalhães Calvet. 2019. "Differential Role of TGF-β in Extracellular Matrix Regulation During Trypanosoma cruzi-Host Cell Interaction" International Journal of Molecular Sciences 20, no. 19: 4836. https://doi.org/10.3390/ijms20194836