2.1. Complex Formation with a 1:1 Host-Guest Stoichiometry: [(γ-CD)/C70] In Vacuo
At first two different interaction geometries between C
70 and the primary or secondary rim of γ-CD, are considered, as reported in the supplementary in
Figure S1. Using the same methodology proposed in previous work [
3], after an initial energy minimization, an MD run in vacuo (i.e., an apolar solvent) lasting for 10 ns, and the minimization of numerous geometries saved during the MD run, the most favorable interaction geometries are reported in
Figure 2 for the 1:1 host-guest stoichiometry [(γ-CD)/C
70]. The same geometry was also obtained carrying out the simulations in explicit water.
In the most favorable complex, C
70 interacts with the primary CD rim. In the isolated γ-CD, the primary rim is narrower than the secondary one, but it becomes considerably widened in the present case upon interaction with the fullerene. For instance, in the isolated γ-CD the diameter of the primary and secondary rims, defined as the average distance among the diametrically opposed O
6 and O
3 oxygens, respectively, amounted to 12.4 ± 1.7 Å (here the ± symbol refers to the standard deviation) and to 13.6 ± 0.8 Å in the order. On the other hand, upon complexation of C
70 at the primary rim these two values became 13.2 ± 0.3 Å and 12.7 ± 1.4 Å (in the same order), so that the primary rim becomes the wider one with a narrower distribution of distances due to the geometrical constraint of the complexed fullerene. The most stable geometry exhibits a favorable interaction energy E
int = −258 kJ/mol, while the inclusion complex in which C
70 interacts with the secondary γ-CD rim is less stable by 14.5 kJ/mol. It may be noted that the most stable arrangement displays an interaction energy that is more favorable by 17 kJ/mol compared to what found for the 1:1 complex between γ-CD with C
60 [
2]. Furthermore, it must be pointed out that C
70 best interacts with the primary rim of γ-CD, as found for C
60 in the most stable geometry for 1:1 complex.
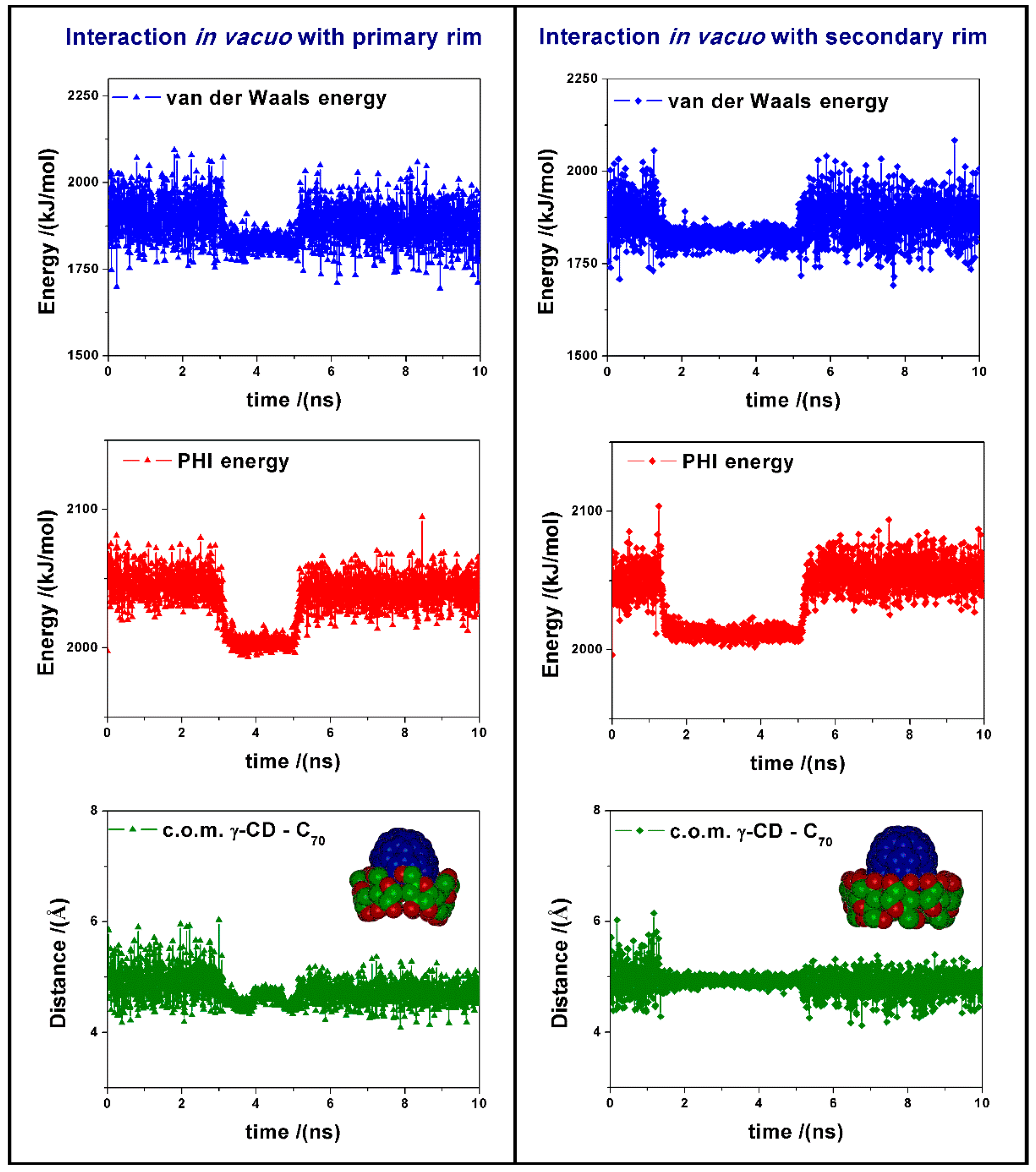
During the MD run we observe changes in the van der Waals energy and in the torsional energy (PHI energy), as reported in
Figure 3. These changes are due to the favorable interaction between the cyclodextrin and the fullerene, and to the variation in the dihedral angles of the γ-CD when the fullerene approaches the CD center. In vacuo the formation of the inclusion complexes is very fast, then we observe during the MD run some changes in the complex geometry and in parallel in the intermolecular van der Waals energy, together with final fluctuations around the average value from 5 to 10 ns. In this time range, the average potential energy of the 1:1 complex formed by interaction between C
70 and the primary rim of γ-CD is equal to 7310.2 kJ/mol, while the complex with the secondary rim is less stable by 25.2 kJ/mol. Moreover, in the most stable complex the average distance between the center of mass (c.o.m.) of the fullerene and of the γ-CD, reported in
Figure 3, is equal to 4.71 Å, whereas in the interaction with the secondary rim of γ-CD, this distance is slightly larger, being equal to 4.85 Å.
We can follow the inclusion process of the C
70 fullerene interacting with the primary rim of the γ-CD and the inclusion process of C
70 interacting with the secondary rim during the initial 5 ns of MD run in vacuo in the animation files reported in the links below
Figure S1.
2.2. Complex Formation with a 2:1 Host-Guest Stoichiometry: [(γ-CD)2/C70] In Vacuo
The possible complexes in a 2:1 stoichiometry [(γ-CD)
2/C
70] were modelled starting from four initial arrangements considering the two most stable geometries reported in
Figure 2 and facing them with another γ-CD having either the primary or the secondary rim close to the exposed surface of C
70 as reported in
Figure S2 for clarity. Using the simulation protocol proposed in previous work [
4,
5,
6,
7], after MD runs lasting for 10 ns and optimization of numerous conformations assumed by the system, the most stable geometries are reported in
Figure 4.
This most stable optimized geometry in vacuo for the PS complex (P refers to the first γ-CD in the most stable 1:1 stoichiometry and S to the second γ-CD) exhibits the lowest potential energy and also the most favorable interaction energy, equal to −593 kJ/mol. The SP complex, reported in
Figure S3, was obtained by starting from the less stable 1:1 complex, so that P refers to the second CD: this complex has a very similar in conformation, being less stable than 10.0 kJ/mol, whereas the SS complex is less stable than 20.0 kJ/mol and finally the PP complex than 64.0 kJ/mol. During the MD run at 300 K from 5 ns to 10 ns, the difference between the average potential energy for the two PS and SS complexes reported in
Figure 4 is even larger, amounting to 42.9 kJ/mol, thus confirming the significantly larger stability of the PS complex over the SS complex.
It should be pointed out that the C
60 fullerene and γ-CD do also form the most stable complex with a 2:1 stoichiometry, but adopting an unlike geometry. In fact, C
60 sits in a symmetrical cavity formed by two γ-CDs that interact through their wider secondary rims thanks to a large number of intermolecular H bonds, yielding a more favorable interaction energy of −652 kJ/mol. Conversely, C
70 is found in a non-symmetrical cavity formed by the two γ-CDs that interact through the secondary rim of one CD (the upper one in
Figure 4) and the primary rim of the other one (the lower one in
Figure 4) thanks to some local distortions of the macrocycle. In turn, the interaction in the PS complex allows for a large number of H-bonds (7 or 8) among the OH groups on either CD so that the fullerene is well screened from the outer environment. It should be pointed out in this context that in the less stable SS complex the number of intermolecular H-bonds amounts to 16, so that all the secondary OH groups are involved in vacuo. On the other hand, optimization of these interactions involves different penalties to the two CDs, in particular a penalty in the torsional energy and in the non-bonded repulsive energy (poor contacts) that eventually destabilize the SS complex compared to the SP complex, as mentioned above. It is also of interest to note that the principal axis of the fullerene is slightly tilted with respect to the axis passing through the mean CD planes in the PS, SP, and PP complexes, whereas in the SS complex the two axes are perfectly aligned in this symmetric cavity.
We can follow the process of the complex γ-CD-C
70 interacting with another γ-CD facing it with its primary or secondary rim, as reported in
Figure S2, during the initial 5 ns of MD run in vacuo in the animation files reported in the links below
Figure S2. From 5ns to 10 ns of the MD run, only fluctuations around the equilibrium positions are observed. The most stable complexes obtained in vacuo after minimization of fifty conformations periodically saved during MD run assumed by the system from 5 ns to 10 ns are reported in
Figure S3.
2.3. Complex Formation of [(γ-CD)/C70] and then of the Dimer [(γ-CD)2/C70] in Water
As anticipated, the next step is to consider the geometry and the stability of the 1:1 and of the 2:1 complexes in explicit water. For this purpose, we studied the interaction between C
70 and γ-CD adopting the same initial geometries as in vacuo (see
Figure S1) but now with explicit water molecules in a box with periodic boundary conditions.
We can follow the inclusion process of the C
70 fullerene interacting with the primary and with the secondary rim of the γ-CD during the MD run at 300 K in water lasting for 1 ns in the animation files (see links reported before
Figure S4 in Supplementary Materials).
The inclusion of hydrophobic C
70 in the hydrophobic cavity of γ-CD is very fast and the complex formed is stable during the MD run. The most stable geometry for the 1:1 complex [(γ-CD)/C
70] corresponds to the favorable interaction between the fullerene and the secondary rim of γ-CD, unlike to what was found in vacuo. Starting from this 1:1 complex and facing it in water with the second γ-CD in the same arrangements reported in
Figure S2 in SI, as well as starting from the less stable 1:1 complex with the interaction of the fullerene at the primary CD rim, we studied the formation of 2:1 host-guest stoichiometry inclusion complexes. After initial geometry optimizations using MM methods and then an MD run at 300 K lasting for 2 ns in explicit water, the final results show that the most stable host-guest stoichiometry in water corresponds again to the 2:1 complex. However, in water the most stable arrangement, reported at right in
Figure 5, shows that C
70 is hosted in the cavity formed by the two γ-CDs that interact through their primary rims without any intermolecular H bonds, as reported in
Figure S4. In fact, a large number of H bonds are present, but they only involve the terminal OH groups and the water molecules of the first hydration shell of the complex.
It is important to note that the most stable inclusion complex with a 2:1 stoichiometry [(γ-CD)
2/C
70] in water is the PP complex shown in
Figure 5, followed by the SS complex, which is less stable by 10.8 kJ/mol, and then by the SP complex, less stable by 18.6 kJ/mol. In water the stability of the three possible inclusion complexes is different from that obtained by the simulation in vacuo described before. The complexes formed in water are very peculiar and the arrangements of the two γ-CDs that include C
70 is very different with respect to that achieved with C
60. This difference in the inclusion complex geometry potentially has important consequences for the release of the fullerenes in amphiphilic liposomes or in cell membranes. In fact, in the case of C
70 the two γ-CDs rims approach only in part at the equatorial plane of the complex and do not form intermolecular H-bonds due to presence of the prolate fullerene in the cavity (see
Figure S4), unlike what was found for C
60. As a result, part of the hydrophobic C
70 surface is still exposed to the water molecules. All 2:1 host-guest complexes [(γ-CD)
2/C
70] formed in water are then amphiphilic inclusion complexes.
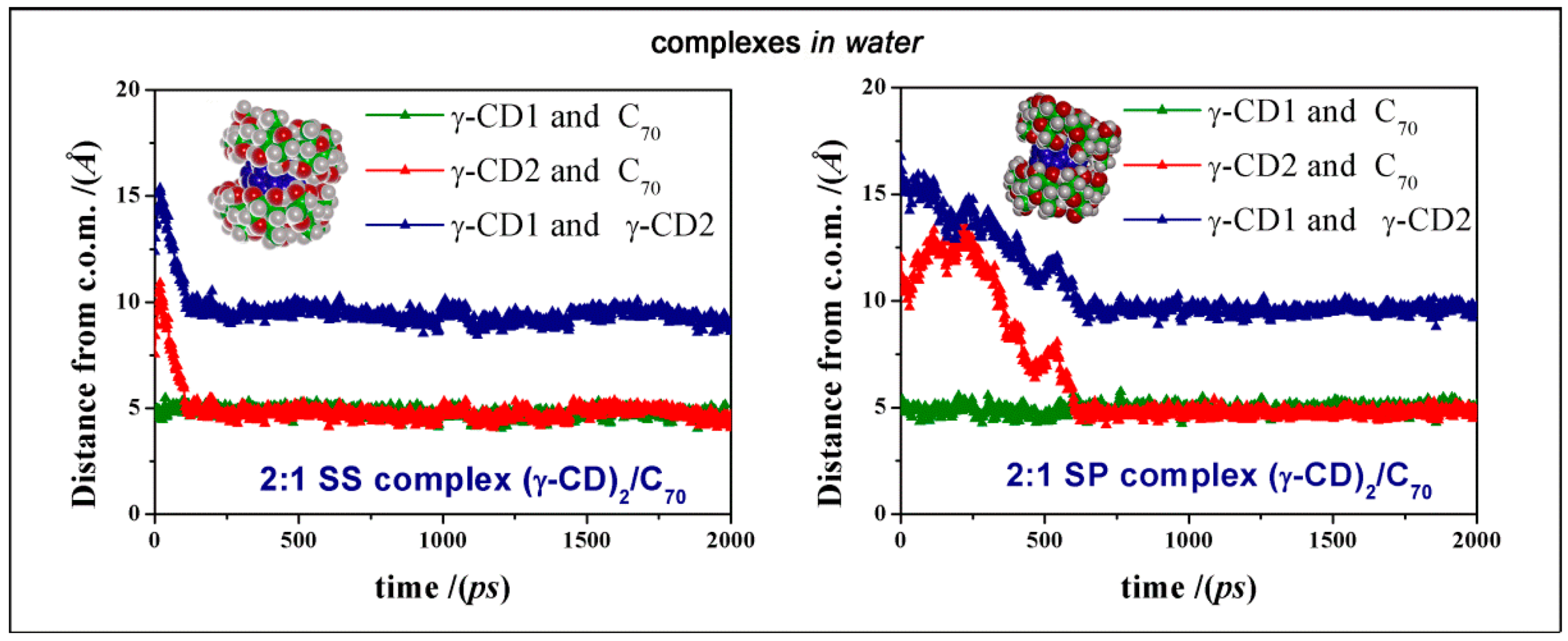
We also add that the most favorable geometry PP complex is stable during the MD run, as shown by the graphic reported in
Figure 6, showing that the second γ-CD approaches the initial complex formed between one γ-CD and C
70 and both remain at the same distance from the C
70 c.o.m. during the rest of MD simulation. A similar behavior was also found for the less stable SS and SP complexes, as reported in
Figure 7. In
Figure 6, at right, we also show the radial distribution function (RDF) of all the atoms of the two γ-CDs in the PP complex and of the oxygen atoms of water molecules measured as a function of the distance r from the c.o.m of C
70, calculated during the MD run from 1 to 2 ns. The distribution of the two γ-CDs is very similar, while the water molecules have a weak maximum at 7.3 Å from the C
70 c.o.m near the exposed surface of the fullerene. Moreover, the peak at 11.9 Å is the first hydration shell of the two cyclodextrins. Similar RDFs were found for the other amphiphilic complexes formed in water.
The stability of the most favorable geometry, where the two CD’s interact through their primary rims, is related to the favorable van der Waals interaction of the fullerene with the hydrophobic CD cavities but also by a number of the H-bonds among water molecules and the primary rims of two cyclodextrins, in addition to those formed within each γ-CD. For the PP [(γ-CD)
2/C
70] complex we note favorable H-bonds involving water molecules and the primary rims. The water molecules do form a bridge between the OH of the two CD’s when their distance is not too large. As a consequence, the most stable geometry is less constrained than what was observed in the case of C
60, where the number of intermolecular H-bonds is maximized. Therefore, in the amphiphilic complex thanks to these non-covalent interactions with C
70 the overall arrangement has the important possibility to open up in an appropriate environment. In this context, we additionally note that the other, less stable complexes show very few intermolecular H-bonds, involving spatially close OH groups of two cyclodextrins (see
Figure S4). Finally, we point also out that these geometries are not static, because the average planes through the CDs change their tilt angle with respect to the main axis of the fullerene, so that the groups that come into contact through H-bonds change continuously moving around the two rims.
Moreover, we can observe that all the complexes formed in water, as reported in
Figure S5a–c, have similar radii of gyration and solvent accessible surfaces. The PP and SS [(γ-CD)
2/C
70] complexes have similar dipole moment vectors, a bit inclined or roughly parallel to the equatorial plane separating the two CDs, while the less stable SP complex has a dipole moment vector roughly perpendicular to it with a larger moment. This difference in the dipole moment vector can be important when these amphiphilic complexes will be in contact with the polar surfaces of liposomes or cell membranes, because it affects the complex orientation, favoring the interaction with the exposed fullerene surface in the PP (and SS) complex.
We can follow the formation of the 2:1 complex when the 1:1 complex where C
70 interacts with the secondary rim of the first γ-CD is approached by the secondary rim or by the primary of the second γ-CD (respectively, the left and at right of
Figure 7 and in
Figure S4), during the initial MD run in water in the animations files in
Supplementary Materials (see the links reported below
Figure S4 in the Supplementary Materials). The same information about the most stable PP [(γ-CD)
2/C
70] complex in water is also reported in the
Supplementary Materials.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







