Towards Clinical Translation of CD8+ Regulatory T Cells Restricted by Non-Classical Major Histocompatibility Complex Ib Molecules

 ,
, 
Abstract
1. Introduction
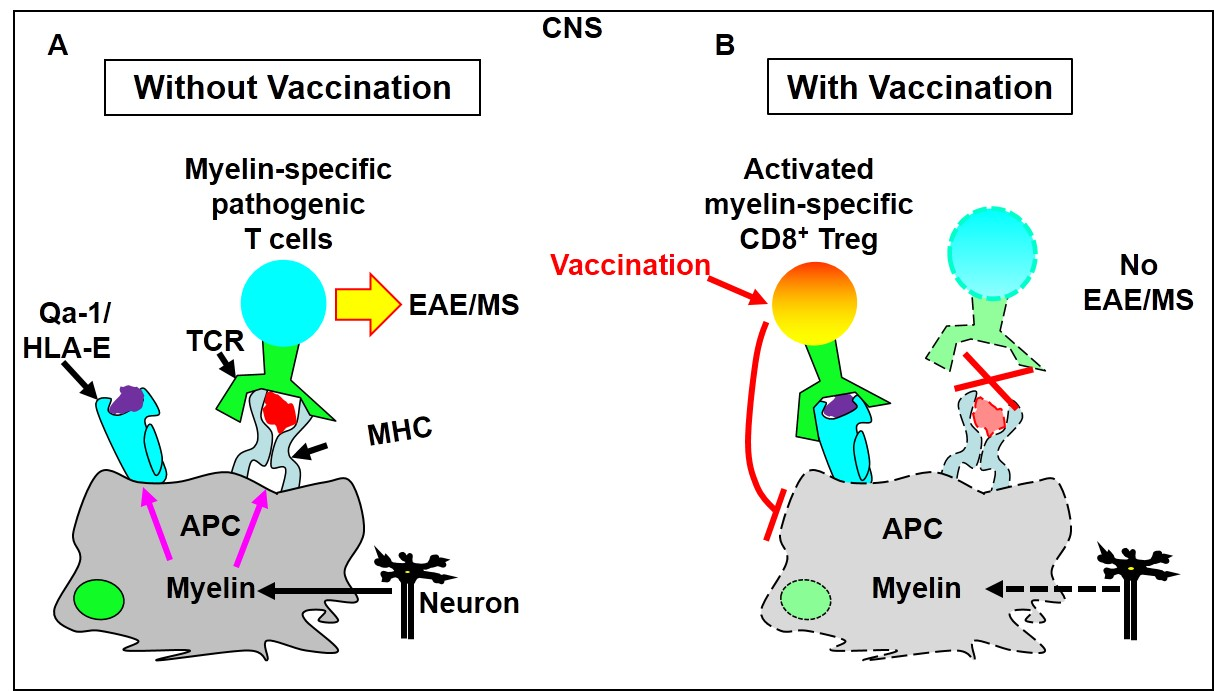
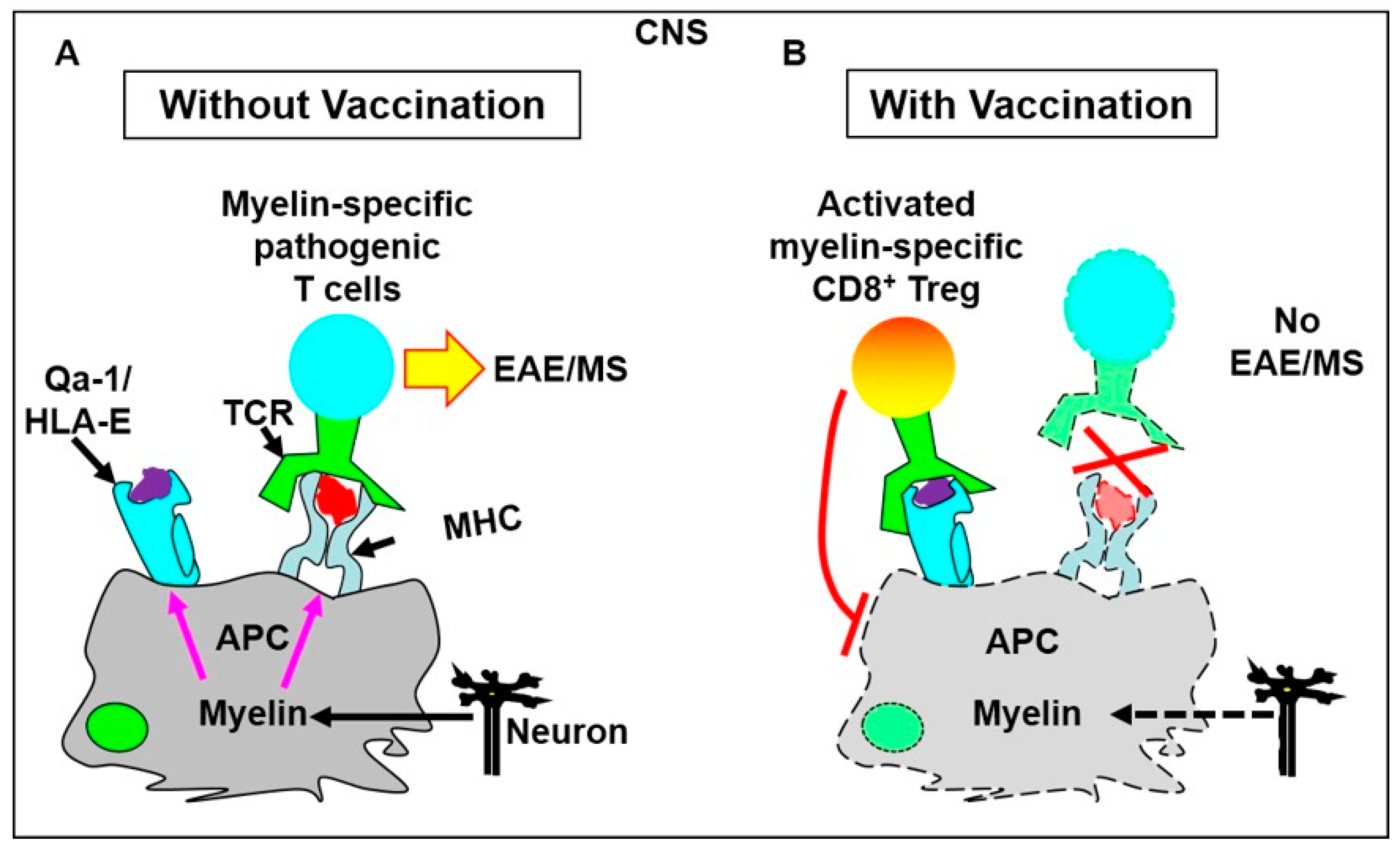
2. Evidence That CD8+ T Cells Use TCRS to Interact with Qa-1/Peptide Complexes on the Cell Surface of Target Cells to Execute Immune Regulation
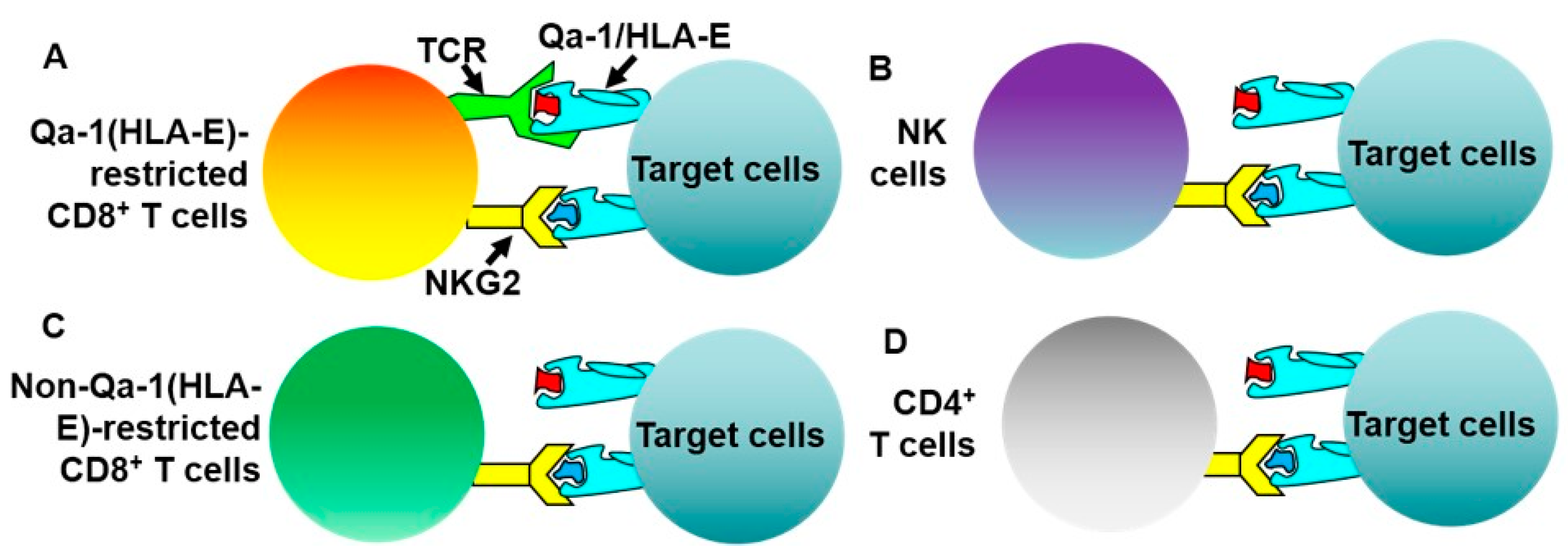
3. Potential Regulatory Mechanisms of Qa-1-Restricted CD8+ Treg Cells
4. Harnessing Qa-1-Restricted CD8+ Treg Cells for the Treatment of Immune-Mediated Diseases
4.1. Studies on the Strategies that Can Enhance the Ability of Qa-1-Restricted CD8+ Treg Cells to Suppress Pathogenic Autoreactive CD4+ T Cells
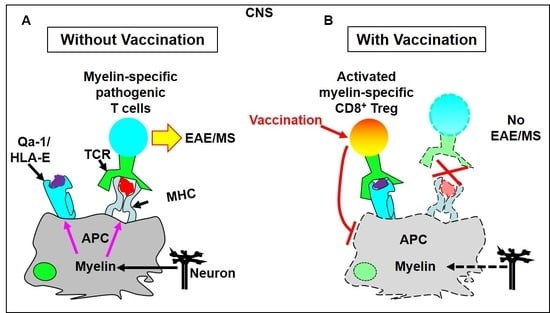
4.1.1. T Cell Vaccination Enhances the Feedback Regulatory Function of TCRVβ-Specific Qa-1-Restricted CD8+ Treg Cells in Vivo
4.1.2. Vaccination with a TCRVβ8.2-Derived, Qa-1-Binding Peptide Prevents the Induction of EAE in Which Pathogenic CD4+ T Cells Predominantly Utilize TCRVβ8.2
4.1.3. Immunization with Qa-1-Binding Peptides Derived from 60 KDa Heat Shock Protein (HSP60) Suppresses Pathogenic Autoreactive CD4+ T Cells in Several Autoimmune Disease Models
4.1.4. A Potential Novel Approach to Promote the Clinical Application of Qa-1-Restricted CD8+ Treg Cells for the Treatment of Autoimmune Diseases
4.2. Studies on the Therapeutic Potentials of Qa-1-Restricted CD8+ T Cells for Cancers and Infections
5. Current Status of Human Studies
6. Future Directions
Acknowledgments
Conflicts of Interest
Abbreviations
| APCs | Antigen-presenting cells. |
| CD4 | Cluster of differentiation 4. |
| CD8 | Cluster of differentiation 8. |
| CNS | Central nervous system. |
| DCs | Dendritic cells. |
| EAE | Experimental allergic encephalomyelitis. |
| ERAAP | Endoplasmic reticulum aminopeptidase associated with antigen processing. |
| Foxp3 | Forkhead box P3. |
| GVAX | Tumor cells engineered to overexpress granulocyte-macrophage colony-stimulating factor. |
| hHSP60sp | Human HSP60 signal peptide (QMRPVSRVL). |
| HLA-E | Human leukocyte antigen-E. Human homologue of murine Qa-1. |
| HSP60 | 60 KDa heat shock protein. |
| IFN-γ | Interferon-γ. |
| IL-15 | Interlecukin-15. |
| MHC | Major histocompatibility complex. |
| mHSP60sp | Murine HSP60 signal peptide (QMRPVSRAL). |
| mHSP60p216 | Murine HSP60 peptide amino acid216-224 (GMKFDRGYI). |
| MOG | Myelin oligodendrocyte glycoprotein |
| MOG196-204 | A Qa-1-binding peptide from murine myelin oligodendrocyte glycoprotein. |
| MS | Multiple sclerosis. |
| Qa-1 | A group of murine non-classical major histocompatibility complex Ib molecules. |
| SEB | Staphylococcus enterotoxin B. |
| TAP | Transporter associated with antigen processing. |
| TCRs | T cell receptors. |
| TCRVβ8 | T cell receptor β chain variable region 8. |
| Tfh cells | T follicular helper cells. |
| Th cells | T helper cells. |
| Treg cells | Regulatory T cells. |
References
- Enouz, S.; Carrie, L.; Merkler, D.; Bevan, M.J.; Zehn, D. Autoreactive T cells bypass negative selection and respond to self-antigen stimulation during infection. J. Exp. Med. 2012, 209, 1769–1779. [Google Scholar] [CrossRef] [PubMed]
- Bouneaud, C.; Kourilsky, P.; Bousso, P. Impact of negative selection on the T cell repertoire reactive to a self-peptide: A large fraction of T cell clones escapes clonal deletion. Immunity 2000, 13, 829–840. [Google Scholar] [CrossRef]
- Christoffersson, G.; von Herrath, M. Regulatory Immune Mechanisms beyond Regulatory T Cells. Trends Immunol. 2019, 40, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Sakaguchi, N.; Asano, M.; Itoh, M.; Toda, M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 1995, 155, 1151–1164. [Google Scholar]
- Hori, S.; Nomura, T.; Sakaguchi, S. Control of regulatory T cell development by the transcription factor Foxp3. Science 2003, 299, 1057–1061. [Google Scholar] [CrossRef]
- Fontenot, J.D.; Gavin, M.A.; Rudensky, A.Y. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 2003, 4, 330–336. [Google Scholar] [CrossRef]
- Atarashi, K.; Tanoue, T.; Shima, T.; Imaoka, A.; Kuwahara, T.; Momose, Y.; Cheng, G.; Yamasaki, S.; Saito, T.; Ohba, Y.; et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science 2011, 331, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Sujino, T.; London, M.; Hoytema van Konijnenburg, D.P.; Rendon, T.; Buch, T.; Silva, H.M.; Lafaille, J.J.; Reis, B.S.; Mucida, D. Tissue adaptation of regulatory and intraepithelial CD4(+) T cells controls gut inflammation. Science 2016, 352, 1581–1586. [Google Scholar] [CrossRef]
- Yang, S.; Fujikado, N.; Kolodin, D.; Benoist, C.; Mathis, D. Immune tolerance. Regulatory T cells generated early in life play a distinct role in maintaining self-tolerance. Science 2015, 348, 589–594. [Google Scholar] [CrossRef]
- Ito, M.; Komai, K.; Mise-Omata, S.; Iizuka-Koga, M.; Noguchi, Y.; Kondo, T.; Sakai, R.; Matsuo, K.; Nakayama, T.; Yoshie, O.; et al. Brain regulatory T cells suppress astrogliosis and potentiate neurological recovery. Nature 2019, 565, 246–250. [Google Scholar] [CrossRef]
- Delgoffe, G.M.; Woo, S.R.; Turnis, M.E.; Gravano, D.M.; Guy, C.; Overacre, A.E.; Bettini, M.L.; Vogel, P.; Finkelstein, D.; Bonnevier, J.; et al. Stability and function of regulatory T cells is maintained by a neuropilin-1-semaphorin-4a axis. Nature 2013, 501, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Bluestone, J.A.; Buckner, J.H.; Fitch, M.; Gitelman, S.E.; Gupta, S.; Hellerstein, M.K.; Herold, K.C.; Lares, A.; Lee, M.R.; Li, K.; et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci. Transl. Med. 2015, 7, 315. [Google Scholar] [CrossRef] [PubMed]
- Marek-Trzonkowska, N.; Mysliwiec, M.; Dobyszuk, A.; Grabowska, M.; Derkowska, I.; Juscinska, J.; Owczuk, R.; Szadkowska, A.; Witkowski, P.; Mlynarski, W.; et al. Therapy of type 1 diabetes with CD4(+)CD25(high)CD127-regulatory T cells prolongs survival of pancreatic islets - results of one year follow-up. Clin. Immunol. 2014, 153, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Desreumaux, P.; Foussat, A.; Allez, M.; Beaugerie, L.; Hebuterne, X.; Bouhnik, Y.; Nachury, M.; Brun, V.; Bastian, H.; Belmonte, N.; et al. Safety and efficacy of antigen-specific regulatory T-cell therapy for patients with refractory Crohn’s disease. Gastroenterology 2012, 143, 1207–1217. [Google Scholar] [CrossRef] [PubMed]
- Di Ianni, M.; Falzetti, F.; Carotti, A.; Terenzi, A.; Castellino, F.; Bonifacio, E.; Del Papa, B.; Zei, T.; Ostini, R.I.; Cecchini, D.; et al. Tregs prevent GVHD and promote immune reconstitution in HLA-haploidentical transplantation. Blood 2011, 117, 3921–3928. [Google Scholar] [CrossRef] [PubMed]
- Brunstein, C.G.; Miller, J.S.; Cao, Q.; McKenna, D.H.; Hippen, K.L.; Curtsinger, J.; Defor, T.; Levine, B.L.; June, C.H.; Rubinstein, P.; et al. Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: Safety profile and detection kinetics. Blood 2011, 117, 1061–1070. [Google Scholar] [CrossRef] [PubMed]
- Martelli, M.F.; Di Ianni, M.; Ruggeri, L.; Falzetti, F.; Carotti, A.; Terenzi, A.; Pierini, A.; Massei, M.S.; Amico, L.; Urbani, E.; et al. HLA-haploidentical transplantation with regulatory and conventional T-cell adoptive immunotherapy prevents acute leukemia relapse. Blood 2014, 124, 638–644. [Google Scholar] [CrossRef] [PubMed]
- Trzonkowski, P.; Bieniaszewska, M.; Juscinska, J.; Dobyszuk, A.; Krzystyniak, A.; Marek, N.; Mysliwska, J.; Hellmann, A. First-in-man clinical results of the treatment of patients with graft versus host disease with human ex vivo expanded CD4+ CD25+ CD127− T regulatory cells. Clin. Immunol. 2009, 133, 22–26. [Google Scholar] [CrossRef]
- Brunstein, C.G.; Blazar, B.R.; Miller, J.S.; Cao, Q.; Hippen, K.L.; McKenna, D.H.; Curtsinger, J.; McGlave, P.B.; Wagner, J.E. Adoptive transfer of umbilical cord blood-derived regulatory T cells and early viral reactivation. Biol. Blood Marrow Transpl.. 2013, 19, 1271–1273. [Google Scholar] [CrossRef]
- Theil, A.; Tuve, S.; Oelschlagel, U.; Maiwald, A.; Dohler, D.; Ossmann, D.; Zenkel, A.; Wilhelm, C.; Middeke, J.M.; Shayegi, N.; et al. Adoptive transfer of allogeneic regulatory T cells into patients with chronic graft-versus-host disease. Cytotherapy 2015, 17, 473–486. [Google Scholar] [CrossRef]
- Bacchetta, R.; Lucarelli, B.; Sartirana, C.; Gregori, S.; Lupo Stanghellini, M.T.; Miqueu, P.; Tomiuk, S.; Hernandez-Fuentes, M.; Gianolini, M.E.; Greco, R.; et al. Immunological Outcome in Haploidentical-HSC Transplanted Patients Treated with IL-10-Anergized Donor T Cells. Front. Immunol. 2014, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Todo, S.; Yamashita, K.; Goto, R.; Zaitsu, M.; Nagatsu, A.; Oura, T.; Watanabe, M.; Aoyagi, T.; Suzuki, T.; Shimamura, T.; et al. A pilot study of operational tolerance with a regulatory T-cell-based cell therapy in living donor liver transplantation. Hepatology 2016, 64, 632–643. [Google Scholar] [CrossRef] [PubMed]
- Marek-Trzonkowska, N.; Mysliwiec, M.; Dobyszuk, A.; Grabowska, M.; Techmanska, I.; Juscinska, J.; Wujtewicz, M.A.; Witkowski, P.; Mlynarski, W.; Balcerska, A.; et al. Administration of CD4+CD25highCD127- regulatory T cells preserves beta-cell function in type 1 diabetes in children. Diabetes Care 2012, 35, 1817–1820. [Google Scholar] [CrossRef] [PubMed]
- Marek-Trzonkowska, N.; Mysliwec, M.; Siebert, J.; Trzonkowski, P. Clinical application of regulatory T cells in type 1 diabetes. Pediatr Diabetes 2013, 14, 322–332. [Google Scholar] [CrossRef] [PubMed]
- Marek-Trzonkowska, N.; Mysliwiec, M.; Iwaszkiewicz-Grzes, D.; Gliwinski, M.; Derkowska, I.; Zalinska, M.; Zielinski, M.; Grabowska, M.; Zielinska, H.; Piekarska, K.; et al. Factors affecting long-term efficacy of T regulatory cell-based therapy in type 1 diabetes. J. Transl Med. 2016, 14, 332. [Google Scholar] [CrossRef]
- Gitelman, S.E.; Bluestone, J.A. Regulatory T cell therapy for type 1 diabetes: May the force be with you. J. Autoimmun 2016, 71, 78–87. [Google Scholar] [CrossRef]
- Christoffersson, G.; Chodaczek, G.; Ratliff, S.S.; Coppieters, K.; von Herrath, M.G. Suppression of diabetes by accumulation of non-islet-specific CD8(+) effector T cells in pancreatic islets. Sci. Immunol. 2018, 3. [Google Scholar] [CrossRef]
- Gershon, R.K.; Kondo, K. Cell interactions in the induction of tolerance: The role of thymic lymphocytes. Immunology 1970, 18, 723–737. [Google Scholar]
- Kronenberg, M.; Steinmetz, M.; Kobori, J.; Kraig, E.; Kapp, J.A.; Pierce, C.W.; Sorensen, C.M.; Suzuki, G.; Tada, T.; Hood, L. RNA transcripts for I-J polypeptides are apparently not encoded between the I-A and I-E subregions of the murine major histocompatibility complex. Proc. Natl.Acad. Sci. USA 1983, 80, 5704–5708. [Google Scholar] [CrossRef]
- Rifa’i, M.; Kawamoto, Y.; Nakashima, I.; Suzuki, H. Essential roles of CD8+ CD122+ regulatory T cells in the maintenance of T cell homeostasis. J. Exp. Med. 2004, 200, 1123–1134. [Google Scholar] [CrossRef]
- Akane, K.; Kojima, S.; Mak, T.W.; Shiku, H.; Suzuki, H. CD8+ CD122+ CD49 dlow regulatory T cells maintain T-cell homeostasis by killing activated T cells via Fas/FasL-mediated cytotoxicity. Proc. Natl. Acad. Sci. USA 2016, 113, 2460–2465. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Maricic, I.; Purohit, N.; Bakamjian, B.; Reed-Loisel, L.M.; Beeston, T.; Jensen, P.; Kumar, V. Regulation of immunlity by a novel population of Qa-1-restricted CD8 alpha alpha+TCR alpha beta(+) T cells. J. Immunol. 2006, 177, 7645–7655. [Google Scholar] [CrossRef]
- Hu, D.; Ikizawa, K.; Lu, L.; Sanchirico, M.E.; Shinohara, M.L.; Cantor, H. Analysis of regulatory CD8 T cells in Qa-1-deficient mice. Nat. Immunol. 2004, 5, 516–523. [Google Scholar] [CrossRef]
- Jiang, H.; Ware, R.; Stall, A.; Flaherty, L.; Chess, L.; Pernis, B. Murine CD8+ T cells that specifically delete autologous CD4+ T cells expressing V beta 8 TCR: A role of the Qa-1 molecule. Immunity 1995, 2, 185–194. [Google Scholar] [CrossRef]
- Boyden, A.W.; Brate, A.A.; Karandikar, N.J. Early IFNgamma-Mediated and Late Perforin-Mediated Suppression of Pathogenic CD4 T Cell Responses Are Both Required for Inhibition of Demyelinating Disease by CNS-Specific Autoregulatory CD8 T Cells. Front. Immunol. 2018, 9, 2336. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Park, N.; Park, J.Y.; Kaplan, B.L.F.; Pruett, S.B.; Park, J.W.; Park, Y.H.; Seo, K.S. Induction of Immunosuppressive CD8(+) CD25(+) FOXP3(+) Regulatory T Cells by Suboptimal Stimulation with Staphylococcal Enterotoxin C1. J. Immunol. 2018, 200, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.L.; Smith, T.R.; Kumar, V. Specific control of immunity by regulatory CD8 T cells. Cell Mol. Immunol. 2005, 2, 11–19. [Google Scholar]
- Kim, H.J.; Cantor, H. Regulation of self-tolerance by Qa-1-restricted CD8(+) regulatory T cells. Semin. Immunol. 2011, 23, 446–452. [Google Scholar] [CrossRef]
- Smith, T.R.; Kumar, V. Revival of CD8+ Treg-mediated suppression. Trends. Immunol. 2008, 29, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Chess, L. The specific regulation of immune responses by CD8+ T cells restricted by the MHC class Ib molecule, Qa-1. Annu. Rev. Immunol. 2000, 18, 185–216. [Google Scholar] [CrossRef]
- Strong, R.K.; Holmes, M.A.; Li, P.; Braun, L.; Lee, N.; Geraghty, D.E. HLA-E allelic variants. Correlating differential expression, peptide affinities, crystal structures, and thermal stabilities. J. Biol. Chem. 2003, 278, 5082–5090. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Kanaseki, T.; Shionoya, Y.; Tokita, S.; Miyamoto, S.; Saka, E.; Kochin, V.; Takasawa, A.; Hirohashi, Y.; Tamura, Y.; et al. Microenvironmental stresses induce HLA-E/Qa-1 surface expression and thereby reduce CD8(+) T-cell recognition of stressed cells. Eur. J. Immunol. 2016, 46, 929–940. [Google Scholar] [CrossRef] [PubMed]
- Stanton, T.H.; Carbon, S. Gene(s) affecting the expression of Qa-1. Immunogenetics 1982, 16, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Romero, P.; Ortega, C.; Palma, A.; Molina, I.J.; Pena, J.; Santamaria, M. Expression of CD94 and NKG2 molecules on human CD4(+) T cells in response to CD3-mediated stimulation. J. Leukoc. Biol. 2001, 70, 219–224. [Google Scholar] [PubMed]
- Bellon, T.; Heredia, A.B.; Llano, M.; Minguela, A.; Rodriguez, A.; Lopez-Botet, M.; Aparicio, P. Triggering of effector functions on a CD8+ T cell clone upon the aggregation of an activatory CD94/kp39 heterodimer. J. Immunol. 1999, 162, 3996–4002. [Google Scholar] [PubMed]
- Vance, R.E.; Kraft, J.R.; Altman, J.D.; Jensen, P.E.; Raulet, D.H. Mouse CD94/NKG2A is a natural killer cell receptor for the nonclassical major histocompatibility complex (MHC) class I molecule Qa-1(b). J. Exp. Med. 1998, 188, 1841–1848. [Google Scholar] [CrossRef] [PubMed]
- Vance, R.E.; Jamieson, A.M.; Raulet, D.H. Recognition of the class Ib molecule Qa-1(b) by putative activating receptors CD94/NKG2C and CD94/NKG2E on mouse natural killer cells. J. Exp. Med. 1999, 190, 1801–1812. [Google Scholar] [CrossRef]
- Braud, V.M.; Allan, D.S.; O’Callaghan, C.A.; Soderstrom, K.; D’Andrea, A.; Ogg, G.S.; Lazetic, S.; Young, N.T.; Bell, J.I.; Phillips, J.H.; et al. HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature 1998, 391, 795–799. [Google Scholar] [CrossRef]
- Eardley, D.D.; Hugenberger, J.; McVay-Boudreau, L.; Shen, F.W.; Gershon, R.K.; Cantor, H. Immunoregulatory circuits among T-cell sets. I. T-helper cells induce other T-cell sets to exert feedback inhibition. J. Exp. Med. 1978, 147, 1106–1115. [Google Scholar] [CrossRef]
- Cantor, H.; McVay-Boudreau, L.; Hugenberger, J.; Naidorf, K.; Shen, F.W.; Gershon, R.K. Immunoregulatory circuits among T-cell sets. II. Physiologic role of feedback inhibition in vivo: Absence in NZB mice. J. Exp. Med. 1978, 147, 1116–1125. [Google Scholar] [CrossRef]
- Cantor, H.; Hugenberger, J.; McVay-Boudreau, L.; Eardley, D.D.; Kemp, J.; Shen, F.W.; Gershon, R.K. Immunoregulatory circuits among T-cell sets. Identification of a subpopulation of T-helper cells that induces feedback inhibition. J. Exp. Med. 1978, 148, 871–877. [Google Scholar] [CrossRef] [PubMed]
- Gonzalo, J.A.; Baixeras, E.; Gonzalez-Garcia, A.; George-Chandy, A.; Van Rooijen, N.; Martinez, C.; Kroemer, G. Differential in vivo effects of a superantigen and an antibody targeted to the same T cell receptor. Activation-induced cell death vs passive macrophage-dependent deletion. J. Immunol. 1994, 152, 1597–1608. [Google Scholar] [PubMed]
- Kawabe, Y.; Ochi, A. Programmed cell death and extrathymic reduction of Vbeta8+ CD4+ T cells in mice tolerant to Staphylococcus aureus enterotoxin B. Nature 1991, 349, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Kawabe, Y.; Ochi, A. Selective anergy of V beta 8+,CD4+ T cells in Staphylococcus enterotoxin B-primed mice. J. Exp. Med. 1990, 172, 1065–1070. [Google Scholar] [CrossRef] [PubMed]
- Rellahan, B.L.; Jones, L.A.; Kruisbeek, A.M.; Fry, A.M.; Matis, L.A. In vivo induction of anergy in peripheral V beta 8+ T cells by staphylococcal enterotoxin B. J. Exp. Med. 1990, 172, 1091–1100. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Verbinnen, B.; Tang, X.; Lu, L.; Cantor, H. Inhibition of follicular T-helper cells by CD8(+) regulatory T cells is essential for self tolerance. Nature 2010, 467, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Kim, H.J.; Werneck, M.B.; Cantor, H. Regulation of CD8+ regulatory T cells: Interruption of the NKG2A-Qa-1 interaction allows robust suppressive activity and resolution of autoimmune disease. Proc. Natl. Acad. Sci. USA 2008, 105, 19420–19425. [Google Scholar] [CrossRef] [PubMed]
- Alvarez Arias, D.A.; Kim, H.J.; Zhou, P.; Holderried, T.A.; Wang, X.; Dranoff, G.; Cantor, H. Disruption of CD8+ Treg activity results in expansion of T follicular helper cells and enhanced antitumor immunity. Cancer Immunol. Res. 2014, 2, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Holderried, T.A.; Lang, P.A.; Kim, H.J.; Cantor, H. Genetic disruption of CD8+ Treg activity enhances the immune response to viral infection. Proc. Natl. Acad. Sci. USA 2013, 110, 21089–21094. [Google Scholar] [CrossRef]
- Tang, X.L.; Maricic, I.; Kumar, V. Anti-TCR antibody treatment activates a novel population of nonintestinal CD8 alpha alpha+TCR alpha beta(+) regulatory T cells and prevents experimental autoimmune encephalomyelitis. J. Immunol. 2007, 178, 6043–6050. [Google Scholar] [CrossRef]
- Beeston, T.; Smith, T.R.; Maricic, I.; Tang, X.; Kumar, V. Involvement of IFN-gamma and perforin, but not Fas/FasL interactions in regulatory T cell-mediated suppression of experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2010, 229, 91–97. [Google Scholar] [CrossRef]
- Leclerc, M.; Voilin, E.; Gros, G.; Corgnac, S.; de Montpreville, V.; Validire, P.; Bismuth, G.; Mami-Chouaib, F. Regulation of antitumour CD8 T-cell immunity and checkpoint blockade immunotherapy by Neuropilin-1. Nat. Commun. 2019, 10, 3345. [Google Scholar] [CrossRef]
- Shevach, E.M. Biological functions of regulatory T cells. Adv. Immunol 2011, 112, 137–176. [Google Scholar]
- Xu, Y.; Cheng, Y.; Baylink, D.J.; Wasnik, S.; Goel, G.; Huang, M.; Cao, H.; Qin, X.; Lau, K.W.; Chan, C.; et al. In Vivo Generation of Gut-Homing Regulatory T Cells for the Suppression of Colitis. J. Immunol. 2019, 202, 3447–3457. [Google Scholar] [CrossRef]
- Nedeva, C.; Menassa, J.; Puthalakath, H. Sepsis: Inflammation Is a Necessary Evil. Front. Cell Dev. Biol. 2019, 7, 108. [Google Scholar] [CrossRef]
- Li, C.H.; Zhang, J.; Baylink, D.J.; Wang, X.; Goparaju, N.B.; Xu, Y.; Wasnik, S.; Cheng, Y.; Berumen, E.C.; Qin, X.; et al. Dendritic cells, engineered to overexpress 25-hydroxyvitamin D 1alpha-hydroxylase and pulsed with a myelin antigen, provide myelin-specific suppression of ongoing experimental allergic encephalomyelitis. FASEB J. 2017, 31, 2996–3006. [Google Scholar] [CrossRef]
- Wagner, D.H. Of the multiple mechanisms leading to type 1 diabetes, T cell receptor revision may play a prominent role (is type 1 diabetes more than a single disease?). Clin. Exp. Immunol. 2016, 185, 271–280. [Google Scholar] [CrossRef]
- Varthaman, A.; Clement, M.; Khallou-Laschet, J.; Fornasa, G.; Gaston, A.T.; Dussiot, M.; Caligiuri, G.; Cantor, H.; Kaveri, S.; Nicoletti, A. Physiological induction of regulatory Qa-1-restricted CD8+ T cells triggered by endogenous CD4+ T cell responses. PLoS ONE 2011, 6, e21628. [Google Scholar] [CrossRef]
- Jiang, H.; Kashleva, H.; Xu, L.X.; Forman, J.; Flaherty, L.; Pernis, B.; Braunstein, N.S.; Chess, L. T cell vaccination induces T cell receptor Vbeta-specific Qa-1-restricted regulatory CD8(+) T cells. Proc. Natl. Acad. Sci. USA 1998, 95, 4533–4537. [Google Scholar] [CrossRef]
- Panoutsakopoulou, V.; Huster, K.M.; McCarty, N.; Feinberg, E.; Wang, R.; Wucherpfennig, K.W.; Cantor, H. Suppression of autoimmune disease after vaccination with autoreactive T cells that express Qa-1 peptide complexes. J. Clin. Invest. 2004, 113, 1218–1224. [Google Scholar] [CrossRef]
- Acha-Orbea, H.; Mitchell, D.J.; Timmermann, L.; Wraith, D.C.; Tausch, G.S.; Waldor, M.K.; Zamvil, S.S.; McDevitt, H.O.; Steinman, L. Limited heterogeneity of T cell receptors from lymphocytes mediating autoimmune encephalomyelitis allows specific immune intervention. Cell 1988, 54, 263–273. [Google Scholar] [CrossRef]
- Urban, J.L.; Kumar, V.; Kono, D.H.; Gomez, C.; Horvath, S.J.; Clayton, J.; Ando, D.G.; Sercarz, E.E.; Hood, L. Restricted use of T cell receptor V genes in murine autoimmune encephalomyelitis raises possibilities for antibody therapy. Cell 1988, 54, 577–592. [Google Scholar] [CrossRef]
- Smith, T.R.F.; Tang, X.L.; Maricic, I.; Garcia, Z.; Fanchiang, S.; Kumar, V. Dendritic Cells Use Endocytic Pathway for Cross-Priming Class Ib MHC-Restricted CD8 alpha alpha+TCR alpha beta(+) T Cells with Regulatory Properties. J. Immunol. 2009, 182, 6959–6968. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, P.V.; Forsthuber, T.; Miller, A.; Sercarz, E.E. Spreading of T-cell autoimmunity to cryptic determinants of an autoantigen. Nature 1992, 358, 155–157. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zhang, L.; Liang, B.; Saenger, Y.; Li, J.; Chess, L.; Jiang, H. Perceiving the avidity of T cell activation can be translated into peripheral T cell regulation. Proc. Natl. Acad. Sci. USA 2007, 104, 20472–20477. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zheng, Z.; Jiang, Y.; Chess, L.; Jiang, H. The specificity of T cell regulation that enables self-nonself discrimination in the periphery. Proc. Natl. Acad. Sci. USA 2009, 106, 534–539. [Google Scholar] [CrossRef] [PubMed]
- Leavenworth, J.W.; Tang, X.L.; Kim, H.J.; Wang, X.Y.; Cantor, H. Amelioration of arthritis through mobilization of peptide-specific CD8(+) regulatory T cells. J. Clin. Invest. 2013, 123, 1382–1389. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, J.; Baylink, D.J.; Li, C.H.; Watts, D.M.; Xu, Y.; Qin, X.; Walter, M.H.; Tang, X. Targeting Non-classical Myelin Epitopes to Treat Experimental Autoimmune Encephalomyelitis. Sci. Rep. 2016, 6, 36064. [Google Scholar] [CrossRef]
- Xu, Y.; Wasnik, S.; Baylink, D.J.; Berumen, E.C.; Tang, X. Overlapping Peptide Library to Map Qa-1 Epitopes in a Protein. J. Vis. Exp. Jove. 2017. [Google Scholar] [CrossRef]
- Oliveira, C.C.; van Veelen, P.A.; Querido, B.; de Ru, A.; Sluijter, M.; Laban, S.; Drijfhout, J.W.; van der Burg, S.H.; Offringa, R.; van Hall, T. The nonpolymorphic MHC Qa-1b mediates CD8+ T cell surveillance of antigen-processing defects. J. Exp. Med. 2010, 207, 207–221. [Google Scholar] [CrossRef]
- Nagarajan, N.A.; Gonzalez, F.; Shastri, N. Nonclassical MHC class Ib-restricted cytotoxic T cells monitor antigen processing in the endoplasmic reticulum. Nat. Immunol. 2012, 13, 579–586. [Google Scholar] [CrossRef]
- Anderson, C.K.; Reilly, E.C.; Lee, A.Y.; Brossay, L. Qa-1-Restricted CD8(+) T Cells Can Compensate for the Absence of Conventional T Cells during Viral Infection. Cell Rep. 2019. [Google Scholar] [CrossRef]
- Paladini, F.; Belfiore, F.; Cocco, E.; Carcassi, C.; Cauli, A.; Vacca, A.; Fiorillo, M.T.; Mathieu, A.; Cascino, I.; Sorrentino, R. HLA-E gene polymorphism associates with ankylosing spondylitis in Sardinia. Arthritis Res. 2009, 11, R171. [Google Scholar] [CrossRef]
- Lin, Y.J.; Wan, L.; Wu, J.Y.; Sheu, J.J.; Lin, C.W.; Lan, Y.C.; Lai, C.H.; Hung, C.H.; Tsai, Y.; Tsai, C.H.; et al. HLA-E gene polymorphism associated with susceptibility to Kawasaki disease and formation of coronary artery aneurysms. Arthritis Rheum. 2009, 60, 604–610. [Google Scholar] [CrossRef]
- Iwaszko, M.; Swierkot, J.; Kolossa, K.; Jeka, S.; Wiland, P.; Bogunia-Kubik, K. Polymorphisms within the human leucocyte antigen-E gene and their associations with susceptibility to rheumatoid arthritis as well as clinical outcome of anti-tumour necrosis factor therapy. Clin. Exp. Immunol. 2015, 182, 270–277. [Google Scholar] [CrossRef]
- Hodgkinson, A.D.; Millward, B.A.; Demaine, A.G. The HLA-E locus is associated with age at onset and susceptibility to type 1 diabetes mellitus. Hum. Immunol. 2000, 61, 290–295. [Google Scholar] [CrossRef]
- Bhanusali, D.G.; Sachdev, A.; Rahmanian, A.; Gerlach, J.A.; Tong, J.C.; Seiffert-Sinha, K.; Sinha, A.A. HLA-E*0103X is associated with susceptibility to Pemphigus vulgaris. Exp. Derm. 2013, 22, 108–112. [Google Scholar] [CrossRef]
- Sokolik, R.; Gebura, K.; Iwaszko, M.; Swierkot, J.; Korman, L.; Wiland, P.; Bogunia-Kubik, K. Significance of association of HLA-C and HLA-E with psoriatic arthritis. Hum. Immunol. 2014, 75, 1188–1191. [Google Scholar] [CrossRef]
- Schulte, D.; Vogel, M.; Langhans, B.; Kramer, B.; Korner, C.; Nischalke, H.D.; Steinberg, V.; Michalk, M.; Berg, T.; Rockstroh, J.K.; et al. The HLA-E(R)/HLA-E(R) genotype affects the natural course of hepatitis C virus (HCV) infection and is associated with HLA-E-restricted recognition of an HCV-derived peptide by interferon-gamma-secreting human CD8(+) T cells. J. Infect. Dis. 2009, 200, 1397–1401. [Google Scholar] [CrossRef][Green Version]
- Araujo, R.C.; Dias, F.C.; Bertol, B.C.; Silva, D.M.; Almeida, P.H.; Teixeira, A.C.; Souza, F.F.; Villanova, M.G.; Ramalho, L.N.Z.; Donadi, E.A.; et al. Liver HLA-E Expression Is Associated with Severity of Liver Disease in Chronic Hepatitis, C. J. Immunol. Res. 2018. [Google Scholar] [CrossRef]
- Boujelbene, N.; Ben Yahia, H.; Babay, W.; Gadria, S.; Zemni, I.; Azaiez, H.; Dhouioui, S.; Zidi, N.; McHiri, R.; Mrad, K.; et al. HLA-G, HLA-E, and IDO overexpression predicts a worse survival of Tunisian patients with vulvar squamous cell carcinoma. HLA 2019, 94, 11–24. [Google Scholar] [CrossRef]
- Jorgensen, P.B.; Livbjerg, A.H.; Hansen, H.J.; Petersen, T.; Hollsberg, P. Epstein-Barr virus peptide presented by HLA-E is predominantly recognized by CD8(bright) cells in multiple sclerosis patients. PLoS ONE 2012, 7, e46120. [Google Scholar] [CrossRef]
- Correale, J.; Villa, A. Isolation and characterization of CD8+ regulatory T cells in multiple sclerosis. J. Neuroimmunol. 2008, 195, 121–134. [Google Scholar] [CrossRef]
- Jiang, H.; Canfield, S.M.; Gallagher, M.P.; Jiang, H.H.; Jiang, Y.; Zheng, Z.; Chess, L. HLA-E-restricted regulatory CD8(+) T cells are involved in development and control of human autoimmune type 1 diabetes. J. Clin. Invest. 2010, 120, 3641–3650. [Google Scholar] [CrossRef]
- Michaelsson, J.; Teixeira de Matos, C.; Achour, A.; Lanier, L.L.; Karre, K.; Soderstrom, K. A signal peptide derived from hsp60 binds HLA-E and interferes with CD94/NKG2A recognition. J. Exp. Med. 2002, 196, 1403–1414. [Google Scholar] [CrossRef]
- Salerno-Goncalves, R.; Fernandez-Vina, M.; Lewinsohn, D.M.; Sztein, M.B. Identification of a human HLA-E-restricted CD8+ T cell subset in volunteers immunized with Salmonella enterica serovar Typhi strain Ty21a typhoid vaccine. J. Immunol. 2004, 173, 5852–5862. [Google Scholar] [CrossRef]
- Pietra, G.; Romagnani, C.; Mazzarino, P.; Falco, M.; Millo, E.; Moretta, A.; Moretta, L.; Mingari, M.C. HLA-E-restricted recognition of cytomegalovirus-derived peptides by human CD8+ cytolytic T lymphocytes. Proc. Natl. Acad. Sci. USA 2003, 100, 10896–10901. [Google Scholar] [CrossRef]
- Jouand, N.; Bressollette-Bodin, C.; Gerard, N.; Giral, M.; Guerif, P.; Rodallec, A.; Oger, R.; Parrot, T.; Allard, M.; Cesbron-Gautier, A.; et al. HCMV triggers frequent and persistent UL40-specific unconventional HLA-E-restricted CD8 T-cell responses with potential autologous and allogeneic peptide recognition. PLoS Pathog. 2018, 14, e1007041. [Google Scholar] [CrossRef]
- van Meijgaarden, K.E.; Haks, M.C.; Caccamo, N.; Dieli, F.; Ottenhoff, T.H.; Joosten, S.A. Human CD8+ T-cells recognizing peptides from Mycobacterium tuberculosis (Mtb) presented by HLA-E have an unorthodox Th2-like, multifunctional, Mtb inhibitory phenotype and represent a novel human T-cell subset. PLoS Pathog. 2015, 11, e1004671. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Diseases | Phase | # of Patients | Types of Treg Cells | Results | References |
|---|---|---|---|---|---|
| GvHD | I | 2 | In vitro expanded CD4+ CD25+CD127− Treg cells | Safe. Chronic GvHD: significant symptom alleviation and reduced immune suppression for the longest time within all immunosuppressants used. Acute GvHD: transient improvement. | [18] |
| I | 23 | In vitro expanded CD4+ CD25+ Treg cells | Safe but increased early opportunistic infections when Treg cells were present. Acute GvHD: Reduced incidence of grade II-IV. | [16,19] | |
| I | 28 | Freshly isolated CD4+ CD25+ Treg cells. | Safe. Reduced GvHD incidence. Enhanced immune reconstitution.Unaltered graft-versus-leukemia effect. | [15] | |
| II | 43 | Freshly isolated CD4+ CD25+ Treg cells. | Safe. Reduced GvHD incidence. Enhanced immune reconstitution. Reduced leukemia relapse. | [17] | |
| I | 5 | In vitro expanded CD4+ CD25+ Treg cells | Cancers found in 2 out of 5 patients. Improved chronic GvHD in 2 out of 5 patients. Stable chronic GvHD for 21 months in 3 out of 5 patients. | [20] | |
| I | 12 | IL-10-tolerized donor T cells | Safe. Four patients were disease- and immunosuppressant-free for at least 7.2 years after haplo-HSCT. | [21] | |
| Solid Organ Transplantation | I | 10 | Donor-specifically tolerized lymphocytes. | Safe. Seven patients reached immunosuppressants-free for 16-33 months. Three patients developed mild rejection during weaning of immunosuppressants and resumed conventional immunosuppressants. | [22] |
| Type 1 Diabetes | I | 10 | In vitro expanded CD4+ CD25+CD127− Treg cells. | Safe. 4–5 months after Treg cell infusion, eight patients still required <0.5 UI/Kg body wt of insulin daily. Two patients were completely insulin-free. 2 years after Treg cell infusion, the disease progressed and all patients were insulin-dependent. | [13,23,24,25] |
| I | 14 | In vitro expanded CD4+ CD25+CD127− Treg cells. | Safe. | [12,26] | |
| Refractory Crohn′s Disease | I/II | 20 | In vitro cloned OVA-specific Tr1 | Safe. 40% response rate based on a reduction in Crohn′s Disease Activity Index (CDAI). | [14] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wasnik, S.; Baylink, D.J.; Leavenworth, J.; Liu, C.; Bi, H.; Tang, X. Towards Clinical Translation of CD8+ Regulatory T Cells Restricted by Non-Classical Major Histocompatibility Complex Ib Molecules. Int. J. Mol. Sci. 2019, 20, 4829. https://doi.org/10.3390/ijms20194829
Wasnik S, Baylink DJ, Leavenworth J, Liu C, Bi H, Tang X. Towards Clinical Translation of CD8+ Regulatory T Cells Restricted by Non-Classical Major Histocompatibility Complex Ib Molecules. International Journal of Molecular Sciences. 2019; 20(19):4829. https://doi.org/10.3390/ijms20194829
Chicago/Turabian StyleWasnik, Samiksha, David J. Baylink, Jianmei Leavenworth, Chenfan Liu, Hongzheng Bi, and Xiaolei Tang. 2019. "Towards Clinical Translation of CD8+ Regulatory T Cells Restricted by Non-Classical Major Histocompatibility Complex Ib Molecules" International Journal of Molecular Sciences 20, no. 19: 4829. https://doi.org/10.3390/ijms20194829
APA StyleWasnik, S., Baylink, D. J., Leavenworth, J., Liu, C., Bi, H., & Tang, X. (2019). Towards Clinical Translation of CD8+ Regulatory T Cells Restricted by Non-Classical Major Histocompatibility Complex Ib Molecules. International Journal of Molecular Sciences, 20(19), 4829. https://doi.org/10.3390/ijms20194829