Microbiome Influence in the Pathogenesis of Prion and Alzheimer’s Diseases



{kind=link}
{kind=link}
Abstract
1. Introduction
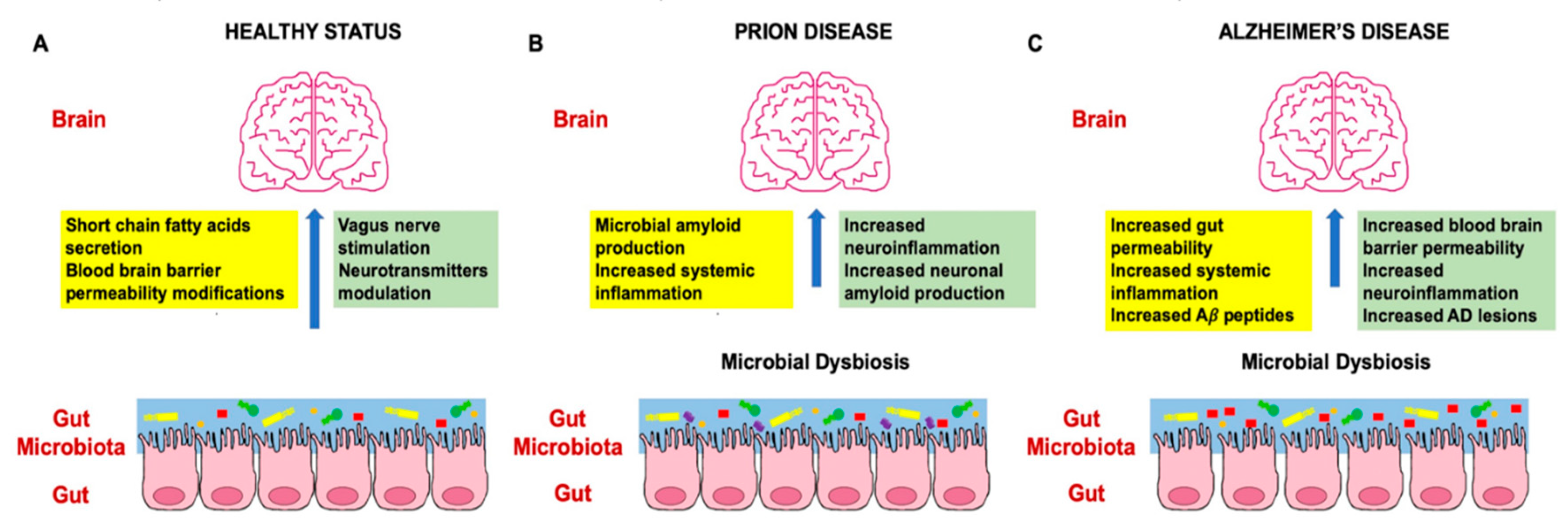
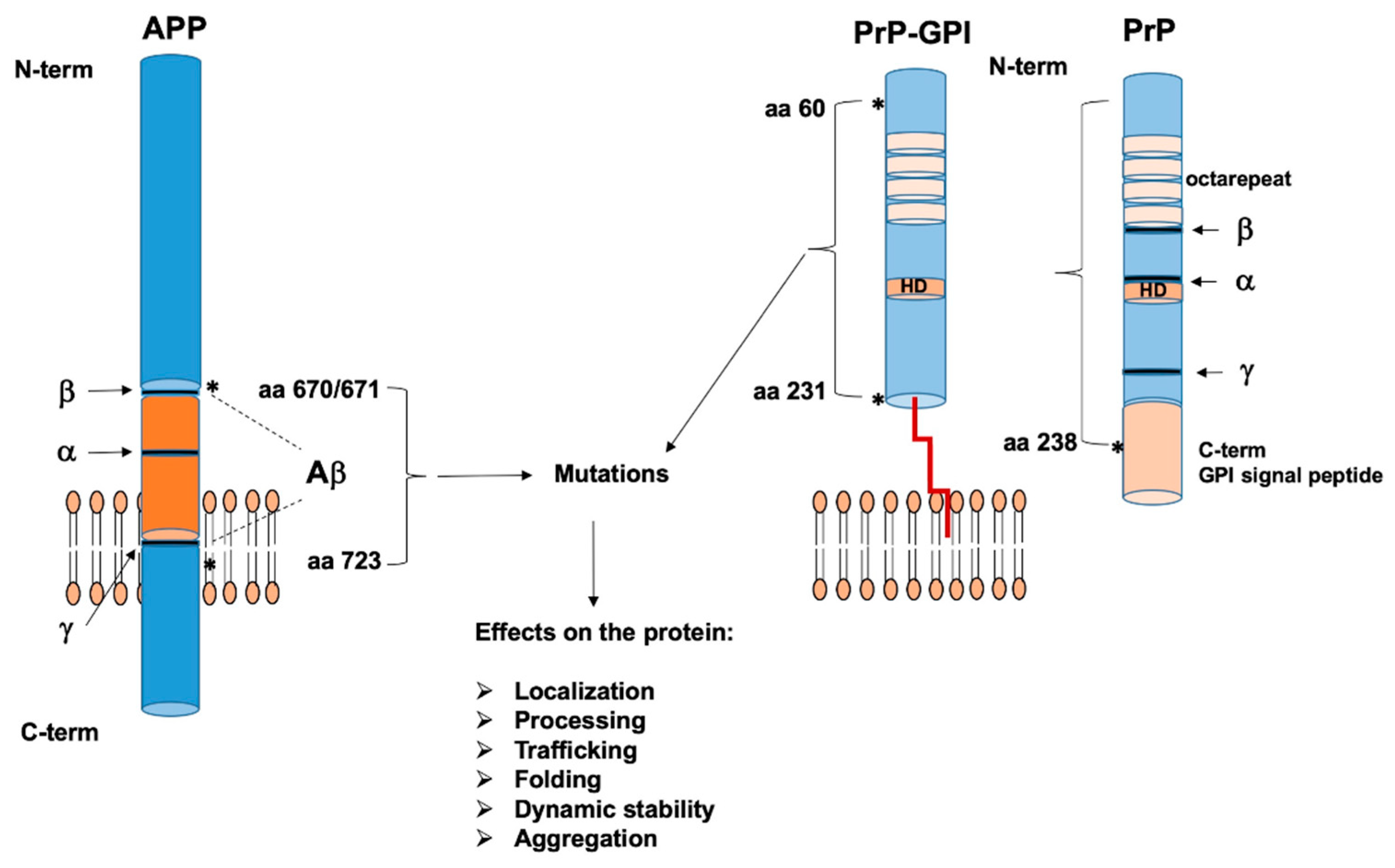
2. The Role of Microbiome in Prion and Alzheimer’s Disease
2.1. Human Microbiota and Prion Disease Development
2.2. Human Microbiota and Alzheimer’s Disease Development
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| PrPC | Cellular prion protein |
| Aβ | Amyloid beta |
| AD | Alzheimer’s disease |
| APP | Aβ precursor protein |
| PRNP | PrP gene |
| PrPSc | Prion protein infectious isoform |
| BSE | Bovine encephalopathies |
| CJD | Creutzfeldt–Jacob diseases |
| GSS | Gerstman–Sträussler–Scheinker |
| FFI | Fatal familial insomnia |
| GPI | Glycosylphosphatidylinositol |
| HD | Hydrophobic domain |
| FTD | Frontotemporal dementia |
| Hpl | Human platelet lysate |
| ctm | C-terminus transmembrane |
| ER | Endoplasmic reticulum |
| HRdup | Hydrophobic region duplication |
| PS1 | Presenilin 1 |
| PS2 | Presenilin 2 |
| BACE1 | Beta secretase 1 |
| MAMs | Mitochondrial-ER associated membranes |
| NMR | Nuclear magnetic resonance |
| TM | Transmembrane |
| CLU | Clusterin |
| CR1 | Complement component 3b/4b receptor 1 |
| TREM2 | Triggering receptor myeloid 2 cells |
| APOE | Apolipoprotein E |
| SORL-1 | Sortilin related receptor 1 |
| TGF-β | Transforming growth factor-beta |
| PGE2 | Prostaglandin E2 |
| SiglecF | Sialic acid binding Ig-like lectin F |
| CD200R | Cell surface transmembrane glycoprotein CD200 receptor 1 |
| Fcg | Fragment crystallizable gamma receptor |
| MFGE-8 | Milk fat globule—epidermal growth factor—factor VIII |
| CD14 | Cluster of differentiation 14 |
| SCFAs | Short-chain fatty acids |
| LPS | Lipopolysaccharide |
| SPF | Specific pathogen free |
| WHO | World Health Organization |
| NFTs | Neurofibrillary tangles |
| TMAO | Trimethylamine N-oxide |
References
- Puig, B.; Altmeppen, H.; Glatzel, M. The GPI-anchoring of PrP: Implications in sorting and pathogenesis. Prion 2014, 8, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Swietnicki, W.; Petersen, R.B.; Gambetti, P.; Surewicz, W.K. Familial mutations and the thermodynamic stability of the recombinant human prion protein. J. Biol. Chem. 1998, 273, 31048–31052. [Google Scholar] [CrossRef] [PubMed]
- Sarnataro, D. Attempt to untangle the prion-like misfolding mechanism for neurodegenerative diseases. Int. J. Mol. Sci. 2018, 19, 3081. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Clavaguera, F.; Tolnay, M. The propagation of prion-like protein inclusions in neurodegenerative diseases. Trends Neurosc. 2010, 33, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Jucker, M.; Walker, L.C. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 2013, 501, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Sarnataro, D.; Pepe, A.; Zurzolo, C. Cell biology of prion protein. Prog. Mol. Biol. Transl. Sci. 2017, 150, 57–82. [Google Scholar] [PubMed]
- Kujala, P.; Raymond, C.R.; Romeijn, M.; Godsave, S.F.; van Kasteren, S.I.; Wille, H.; Prusiner, S.B.; Mabbott, N.A.; Peters, P.J. Prion uptake in the gut: Identification of the first uptake and replication sites. PLoS Pathog. 2011, 7, e1002449. [Google Scholar] [CrossRef] [PubMed]
- Friedland, R.P.; Chapman, M.R. The role of microbial amyloid in neurodegeneration. PLos Pathog. 2017, 13, e1006654. [Google Scholar] [CrossRef]
- Theriault, P.; ElAli, A.; Rivest, S. High fat diet exacerbates Alzheimer’s disease related pathology in APPswe/PS1 mice. Oncotarget 2016, 7, 67808–67827. [Google Scholar] [CrossRef]
- D’Argenio, V.; Salvatore, F. The role of the gut microbiome in the healthy adult status. Clin. Chim. Acta 2015, 451, 97–102. [Google Scholar] [CrossRef]
- D’Argenio, V. Human microbiome acquisition and bioinformatic challenges in metagenomic studies. Int. J. Mol. Sci. 2018, 19, 383. [Google Scholar] [CrossRef] [PubMed]
- D’Argenio, V. The Prenatal Microbiome: A New Player for Human Health. High Throughput 2018, 7, 38. [Google Scholar] [CrossRef]
- Imhann, F.; Vich Vila, A.; Bonder, M.J.; Fu, J.; Gevers, D.; Visschedijk, M.C.; Spekhorst, L.M.; Alberts, R.; Franke, L.; van Dullemen, H.M.; et al. Interplay of host genetics and gut microbiota underlying the onset and clinical presentation of inflammatory bowel disease. Gut 2018, 67, 108–119. [Google Scholar] [CrossRef] [PubMed]
- D’Argenio, V.; Casaburi, G.; Precone, V.; Pagliuca, C.; Colicchio, R.; Sarnataro, D.; Discepolo, V.; Kim, S.M.; Russo, I.; Del Vecchio Blanco, G.; et al. Metagenomics reveals dysbiosis and a potentially pathogenic n. flavescens strain in duodenum of adult celiac patients. Am. J. Gastroenterol. 2016, 111, 879–890. [Google Scholar] [PubMed]
- D’Argenio, V.; Casaburi, G.; Precone, V.; Pagliuca, C.; Colicchio, R.; Sarnataro, D.; Discepolo, V.; Kim, S.M.; Russo, I.; Del Vecchio Blanco, G.; et al. No change in the mucosal gut microbiome is associated with celiac disease-specific microbiome alteration in adult patients. Am. J. Gastroenterol. 2016, 111, 1659–1661. [Google Scholar] [PubMed]
- D’Argenio, V.; Torino, M.; Precone, V.; Casaburi, G.; Esposito, M.V.; Iaffaldano, L.; Malapelle, U.; Troncone, G.; Coto, I.; Cavalcanti, P.; et al. The cause of death of a child in the 18th century solved by bone microbiome typing using laser microdissection and next generation sequencing. Int. J. Mol. Sci. 2017, 18, 109. [Google Scholar] [CrossRef] [PubMed]
- Iaffaldano, L.; Granata, I.; Pagliuca, C.; Esposito, M.V.; Casaburi, G.; Salerno, G.; Colicchio, R.; Piccirillo, M.; Ciacci, C.; Del Vecchio Blanco, G.; et al. Oropharyngeal microbiome evaluation highlights Neisseria abundance in active celiac patients. Sci. Rep. 2018, 8, 11047. [Google Scholar] [CrossRef] [PubMed]
- D’Argenio, V.; Casaburi, G.; Precone, V.; Moccia, L.G.; Postiglione, I.; Bocchino, M.; Sanduzzi, A. A common microbial signature is present in the lower airways of interstitial lung diseases including sarcoidosis. Sarcoidosis Vasc. Diffuse Lung Dis. 2018, 35, 354–362. [Google Scholar]
- Labruna, G.; Nanayakkara, M.; Pagliuca, C.; Nunziato, M.; Iaffaldano, L.; D’Argenio, V.; Colicchio, R.; Budelli, A.L.; Nigro, R.; Salvatore, P.; et al. Celiac disease-associated Neisseria flavescens decreases mitochondrial respiration in CaCo-2 epithelial cells: Impact of Lactobacillus paracasei CBA L74 on bacterial-induced cellular imbalance. Cell Microbiol. 2019, 21, e13035. [Google Scholar] [CrossRef]
- Strzępa, A.; Lobo, F.M.; Majewska-Szczepani, K.M.; Szczepanik, M. Antibiotics and autoimmune and allergy diseases: Causative factor or treatment? Int. Immunopharmacol. 2018, 65, 328–341. [Google Scholar] [CrossRef]
- D’Argenio, V.; Precone, V.; Casaburi, G.; Miele, E.; Martinelli, M.; Staiano, A.; Salvatore, F.; Sacchetti, L. An altered gut microbiome profile in a child affected by Crohn’s disease normalized after nutritional therapy. Am. J. Gastroenterol. 2013, 108, 851–852. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.; Li, J.; Panagiotou, G. A molecular-level landscape of diet-gut microbiome interactions: Toward dietary interventions targeting bacterial genes. MBio 2015, 6, e01263-15. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.; Yacyshyn, B.R.; Yacyshyn, M.B. Gut microbiota and obesity: An opportunity to alter obesity through Fecal Microbiota Transplant (FMT). Diabetes Obes. Metab. 2018, 21, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Sender, R.; Fuchs, S.; Milo, R. Are we really vastly outnumbered? Revisiting the ratio of bacterial to host cells in humans. Cell 2016, 164, 337–340. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.M.; Clement, C.; Pogue, A.I.; Bhattacharjee, S.; Zhao, Y.; Lukiw, W.J. Pathogenic microbes, the microbiome, and Alzheimer’s disease (AD). Front. Aging Neurosci. 2014, 6, 127. [Google Scholar] [PubMed]
- Heijtz, R.D.; Wang, S.; Anuar, F.; Qian, Y.; Björkholm, B.; Samuelsson, A.; Hibberd, M.L.; Forssberg, H.; Pettersson, S. Normal gut microbiota modulates brain development and behavior. Proc. Natl. Acad. Sci. USA 2011, 108, 3047–3052. [Google Scholar] [CrossRef] [PubMed]
- Ogbonnaya, E.S.; Clarke, G.; Shanahan, F.; Dinan, T.G.; Cryan, J.F.; O’Leary, O.F. Adult hippocampal neurogenesis is regulated by the microbiome. Biol. Psychiatry 2015, 78, e7–e9. [Google Scholar] [CrossRef]
- Fung, T.C.; Olson, C.A.; Hsiao, E.Y. Interactions between the microbiota, immune and nervous systems in health and disease. Nat. Neurosci. 2017, 20, 145–155. [Google Scholar] [CrossRef]
- Erny, D.; Hrabě de Angelis, A.L.; Jaitin, D.; Wieghofer, P.; Staszewski, O.; David, E.; Keren-Shaul, H.; Mahlakoiv, T.; Jakobshagen, K.; Buch, T.; et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat. Neurosci. 2015, 18, 965–977. [Google Scholar] [CrossRef]
- Braniste, V.; Al-Asmakh, M.; Kowal, C.; Anuar, F.; Abbaspour, A.; Tóth, M.; Korecka, A.; Bakocevic, N.; Ng, L.G.; Kundu, P.; et al. The gut microbiota influences blood-brain barrier permeability in mice. Sci. Transl. Med. 2014, 6, 263ra158. [Google Scholar] [CrossRef]
- Strandwitz, P. Neurotransmitter modulation by the gut microbiota. Brain Res. 2018, 1693 Pt B, 128–133. [Google Scholar] [CrossRef]
- Ge, X.; Zhao, W.; Ding, C.; Tian, H.; Xu, L.; Wang, H.; Ni, L.; Jiang, J.; Gong, J.; Zhu, W.; et al. Potential role of fecal microbiota from patients with slow transit constipation in the regulation of gastrointestinal motility. Sci. Rep. 2017, 7, 441. [Google Scholar] [CrossRef] [PubMed]
- Luczynski, P.; Tramullas, M.; Viola, M.; Shanahan, F.; Clarke, G.; O’Mahony, S.; Dinan, T.G.; Cryan, J.F. Microbiota regulates visceral pain in the mouse. Elife 2017, 6, e25887. [Google Scholar] [CrossRef]
- Kelly, J.R.; Borre, Y.; O’ Brien, C.; Patterson, E.; El Aidy, S.; Deane, J.; Kennedy, P.J.; Beers, S.; Scott, K.; Moloney, G.; et al. Transferring the blues: Depression-associated gut microbiota induces neurobehavioural changes in the rat. J. Psychiatr. Res. 2016, 82, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Zheng, P.; Zeng, B.; Zhou, C.; Liu, M.; Fang, Z.; Xu, X.; Zeng, L.; Chen, J.; Fan, S.; Du, X.; et al. Gut microbiome remodeling induces depressive-like behaviors through a pathway mediated by the host’s metabolism. Mol. Psychiatry 2016, 21, 786–796. [Google Scholar] [CrossRef] [PubMed]
- De Palma, G.; Lynch, M.D.; Lu, J.; Dang, V.T.; Deng, Y.; Jury, J.; Umeh, G.; Miranda, P.M.; Pigrau Pastor, M.; Sidani, S.; et al. Transplantation of fecal microbiota from patients with irritable bowel syndrome alters gut function and behavior in recipient mice. Sci. Transl. Med. 2017, 9, eaaf6397. [Google Scholar] [CrossRef] [PubMed]
- Sampson, T.R.; Debelius, J.W.; Thron, T.; Janssen, S.; Shastri, G.G.; Ilhan, Z.E.; Challis, C.; Schretter, C.E.; Rocha, S.; Gradinaru, V.; et al. Gut microbiota regulate motor deficits and neuroinflammation in a model of parkinson’s disease. Cell 2016, 167, 1469–1480. [Google Scholar] [CrossRef] [PubMed]
- Minter, M.R.; Zhang, C.; Leone, V.; Ringus, D.L.; Zhang, X.; Oyler-Castrillo, P.; Musch, M.W.; Liao, F.; Ward, J.F.; Holtzman, D.M.; et al. Antibiotic-induced perturbations in gut microbial diversity influences neuro-inflammation and amyloidosis in a murine model of Alzheimer’s disease. Sci. Rep. 2016, 6, 30028. [Google Scholar] [CrossRef]
- Berer, K.; Gerdes, L.A.; Cekanaviciute, E.; Jia, X.; Xiao, L.; Xia, Z.; Liu, C.; Klotz, L.; Stauffer, U.; Baranzini, S.E.; et al. Gut microbiota from multiple sclerosis patients enables spontaneous autoimmune encephalomyelitis in mice. Proc. Natl. Acad. Sci. USA 2017, 114, 10719–10724. [Google Scholar] [CrossRef]
- Cekanaviciute, E.; Yoo, B.B.; Runia, T.F.; Debelius, J.W.; Singh, S.; Nelson, C.A.; Kanner, R.; Bencosme, Y.; Lee, Y.K.; Hauser, S.L.; et al. Gut bacteria from multiple sclerosis patients modulate human T cells and exacerbate symptoms in mouse models. Proc. Natl. Acad. Sci. USA 2017, 114, 10713–10718. [Google Scholar] [CrossRef]
- Benakis, C.; Brea, D.; Caballero, S.; Faraco, G.; Moore, J.; Murphy, M.; Sita, G.; Racchumi, G.; Ling, L.; Pamer, E.G.; et al. Commensal microbiota affects ischemic stroke outcome by regulating intestinal gammadelta T cells. Nat. Med. 2016, 22, 516–523. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, E.Y.; McBride, S.W.; Hsien, S.; Sharon, G.; Hyde, E.R.; McCue, T.; Codelli, J.A.; Chow, J.; Reisman, S.E.; Petrosino, J.F.; et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 2013, 155, 1451–1463. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, D.S.; Mabbott, N.A. The influence of the commensal and pathogenic gut microbiota on prion disease pathogenesis. J. Gen. Virol. 2016, 97, 1725–3178. [Google Scholar] [CrossRef] [PubMed]
- Collinge, J. Prion diseases of humans and animals: Their causes and molecular basis. Annu. Rev. Neurosci. 2001, 24, 519–550. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.O.; Takada, L.T.; Wong, K.; Forner, S.A.; Geschwind, M.D. Genetic PrP prion diseases. Cold Spring Harb. Perspect. Biol. 2018, 10, a033134. [Google Scholar] [CrossRef] [PubMed]
- Frost, B.; Diamond, M.I. Prion-like mechanism in neurodegenerative diseases. Nat. Rev. Neurosci. 2010, 11, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Campana, V.; Caputo, A.; Sarnataro, D.; Paladino, S.; Tivodar, S.; Zurzolo, C. Characterization of the properties and trafficking of an anchorless form of the prion protein. J. Biol. Chem. 2007, 282, 22747–22756. [Google Scholar] [CrossRef]
- Pepe, A.; Avolio, R.; Matassa, D.S.; Esposito, F.; Nitsch, L.; Zurzolo, C.; Paladino, S.; Sarnataro, D. Regulation of subcompartmental targeting and folding properties of the prion-like protein Shadoo. Sci. Rep. 2017, 7, 3731. [Google Scholar] [CrossRef]
- Caputo, A.; Sarnataro, D.; Campana, V.; Costanzo, M.; Negro, A.; Sorgato, M.C.; Zurzolo, C. Doppel and PrPC co-immunoprecipitate in detergent-resistant membrane domains of epithelial cells. Biochem. J. 2009, 425, 341–351. [Google Scholar] [CrossRef]
- Sarnataro, D.; Caputo, A.; Casanova, P.; Puri, C.; Paladino, S.; Tivodar, S.S.; Campana, V.; Tacchetti, C.; Zurzolo, C. Lipid rafts and clathrin cooperate in the internalization of PrP in epithelial FRT cells. PLoS ONE 2009, 4, e5829. [Google Scholar] [CrossRef]
- Sarnataro, D.; Campana, V.; Zurzolo, C. Lipid rafts in trafficking and processing of prion protein and amyloid precursor protein. In Lipid Rafts and Caveolae: From Membrane Biophysics to Cell Biology; Fielding, C.J., Ed.; Cardiovascular Research Institute, Department of Medicine, University of California: San Francisco, CA, USA, 2006; pp. 205–231. ISBN 978-352731261-0. [Google Scholar]
- Campana, V.; Sarnataro, D.; Fasano, C.; Casanova, P.; Paladino, S.; Zurzolo, C. Detergent-resistant membrane domains but not the proteasome are involved in the misfolding of a PrP mutant retained in the endoplasmic reticulum. J. Cell Sci. 2006, 119, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, D.S.; Bradford, B.M.; Artis, D.; Mabbott, N.A. Reciprocal regulation of lymphoid tissue development in the large intestine by IL-25 and IL-23. Mucosal Immunol. 2015, 8, 582–595. [Google Scholar] [CrossRef] [PubMed]
- Hamada, H.; Hiroi, T.; Nishiyama, Y.; Takahashi, H.; Masunaga, Y.; Hachimura, S.; Kaminogawa, S.; Takahashi-Iwanaga, H.; Iwanaga, T.; Kiyono, H.; et al. Identification of multiple isolated lymphoid follicles on the antimesenteric wall of the mouse small intestine. J. Immunol. 2002, 168, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, D.S.; Else, K.J.; Mabbott, N.A. The gut-associated lymphoid tissues in the small intestine, not the large intestine, play a major role in oral prion disease pathogenesis. J. Virol. 2015, 15, 9532–9547. [Google Scholar] [CrossRef] [PubMed]
- Glaysher, B.R.; Mabbott, N.A. Role of the GALT in scrapie agent neuroinvasion from the intestine. J. Immunol. 2007, 178, 3757–3766. [Google Scholar] [CrossRef] [PubMed]
- De Lucia, C.; Rinchon, A.; Olmos-Alonso, A.; Riecken, K.; Fehse, B.; Boche, D.; Perry, V.H.; Gomez-Nicola, D. Microglia regulate hippocampal neurogenesis during chronic neurodegeneration. Brain Behav. Immun. 2015, 55, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Kranich, J.; Krautler, N.J.; Falsig, J.; Ballmer, B.; Li, S.; Hutter, G.; Schwarz, P.; Moos, R.; Julius, C.; Miele, G.; et al. Engulfment of cerebral apoptotic bodies controls the course of prion disease in a mouse strain-dependent manner. J. Exp. Med. 2010, 207, 2271–2281. [Google Scholar] [CrossRef]
- Prinz, M.; Priller, J. Microglia and brain macrophages in the molecular age. Nat. Rev. Neurosci. 2014, 15, 300–312. [Google Scholar] [CrossRef]
- Zhan, Y.; Paolicelli, R.C.; Sforazzini, F.; Weinhard, L.; Bolasco, G.; Pagani, F.; Vyssotski, A.L.; Bifone, A.; Gozzi, A.; Ragozzino, D.; et al. Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behaviour. Nat. Neurosci. 2014, 17, 400–406. [Google Scholar] [CrossRef]
- Lunnon, K.; Teeling, J.L.; Tutt, A.L.; Cragg, M.S.; Glennie, M.J.; Perry, V.H. Systemic inflammation modulates Fc receptor expression on microglia during chronic neurodegeneration. J. Immunol. 2011, 186, 7215–7224. [Google Scholar] [CrossRef]
- Vincenti, J.E.; Murphy, L.; Grabert, K.; McColl, B.W.; Cancellotti, E.; Freeman, T.C.; Manson, J.C. Defining the microglial response during the time course of chronic neurodegeneration. J. Virol. 2016, 90, 3003–3017. [Google Scholar] [CrossRef] [PubMed]
- Sakai, K.; Hasebe, R.; Takahashi, Y.; Song, C.H.; Suzuki, A.; Yamasaki, T.; Horiuchi, M. Absence of CD14 delays progression of prion diseases accompanied by increased microglial activation. J. Virol. 2013, 87, 13433–13445. [Google Scholar] [CrossRef] [PubMed]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Spielman, L.J.; Gibson, D.L.; Klegeris, A. Unhealthy gut, unhealthy brain: The role of the intestinal microbiota in neurodegenerative diseases. Neurochem. Int. 2018, 120, 149–163. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, A.; Harty, S.; Lehto, S.M.; Moeller, A.H.; Dinan, T.G.; Dunbar, R.I.M.; Cryan, J.F.; Burnet, P.W.J. The Microbiome in Psychology and Cognitive Neuroscience. Trends Cogn. Sci. 2018, 22, 611–636. [Google Scholar] [CrossRef]
- Claesson, M.J.; Jeffery, I.B.; Conde, S.; Power, S.E.; O’Connor, E.M.; Cusack, S.; Harris, H.M.; Coakley, M.; Lakshminarayanan, B.; O’Sullivan, O.; et al. Gut microbiota composition correlates with diet and health in the elderly. Nature 2012, 488, 178–184. [Google Scholar] [CrossRef]
- Harach, T.; Marungruang, N.; Duthilleul, N.; Cheatham, V.; Mc Coy, K.D.; Frisoni, G.; Neher, J.J.; Fåk, F.; Jucker, M.; Lasser, T.; et al. Reduction of Abeta amyloid pathology in APPPS1 transgenic mice in the absence of gut microbiota. Sci. Rep. 2017, 7, 41802. [Google Scholar] [CrossRef]
- Kawano, M.; Miyoshi, M.; Ogawa, A.; Sakai, F.; Kadooka, Y. Lactobacillus gasseri SBT2055 inhibits adipose tissue inflammation and intestinal permeability in mice fed a high-fat diet. J. Nutr. Sci. 2016, 5, e23. [Google Scholar] [CrossRef]
- Cani, P.D.; Bibiloni, R.; Knauf, C.; Waget, A.; Neyrinck, A.M.; Delzenne, N.M.; Burcelin, R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 2008, 57, 1470–1481. [Google Scholar] [CrossRef]
- Zhang, R.; Miller, R.G.; Gascon, R.; Champion, S.; Katz, J.; Lancero, M.; Narvaez, A.; Honrada, R.; Ruvalcaba, D.; McGrath, M.S. Circulating endotoxin and systemic immune activation in sporadic amyotrophic lateral sclerosis (sALS). J. Neuroimmunol. 2009, 206, 121–124. [Google Scholar] [CrossRef]
- Calsolaro, V.; Edison, P. Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimers Dement. 2016, 12, 719–732. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Cong, L.; Jaber, V.; Lukiw, W.J. Microbiome-derived lipopolysaccharide enriched in the perinuclear region of Alzheimer’s disease brain. Front. Immunol. 2017, 8, 1064. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Lin, C.L.; Kao, C.H. Irritable bowel syndrome is associated with an increased risk of dementia: A nationwide population-based study. PLoS ONE 2016, 11, e0144589. [Google Scholar] [CrossRef] [PubMed]
- Caini, S.; Bagnoli, S.; Palli, D.; Saieva, C.; Ceroti, M.; Bendinelli, B.; Assedi, M.; Masala, G. Total and cancer mortality in a cohort of ulcerative colitis and Crohn’s disease patients: The Florence inflammatory bowel disease study, 1978-2010. Dig. Liver Dis. 2016, 48, 1162–1167. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, A.; Cattane, N.; Galluzzi, S.; Provasi, S.; Lopizzo, N.; Festari, C.; Ferrari, C.; Guerra, U.P.; Paghera, B.; Muscio, C.; et al. Association of brain amyloidosis with pro-inflammatory gut bacterial taxa and peripheral inflammation markers in cognitively impaired elderly. Neurobiol. Aging 2017, 49, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Haran, J.P.; Bhattarai, S.K.; Foley, S.E.; Dutta, P.; Ward, D.V.; Bucci, V.; McCormick, B.A. Alzheimer’s Disease Microbiome Is Associated with Dysregulation of the Anti-Inflammatory P-Glycoprotein Pathway. MBio 2019, 10, 3. [Google Scholar] [CrossRef] [PubMed]
- Block, J. Alzheimer’s disease might depend on enabling pathogens which do not necessarily cross the blood-brain barrier. Med. Hypotheses 2019, 125, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Wang, Q. Towards understanding brain-gut-microbiome connections in Alzheimer’s disease. BMC Syst. Biol. 2016, 10 (Suppl. 3), 63. [Google Scholar] [CrossRef] [PubMed]
- Larsen, P.; Nielsen, J.L.; Otzen, D.; Nielsen, P.H. Amyloid-like adhesins produced by floc-forming and filamentous bacteria in activated sludge. Appl. Environ. Microbiol. 2008, 74, 1517–1526. [Google Scholar] [CrossRef] [PubMed]
- Oli, M.W.; Otoo, H.N.; Crowley, P.J.; Heim, K.P.; Nascimento, M.M.; Ramsook, C.B.; Lipke, P.N.; Brady, L.J. Functional amyloid formation by Streptococcus mutans. Microbiology 2012, 158, 2903–2916. [Google Scholar] [CrossRef]
- Pistollato, F.; Sumalla Cano, S.; Elio, I.; Masias Vergara, M.; Giampieri, F.; Battino, M. Role of gut microbiota and nutrients in amyloid formation and pathogenesis of Alzheimer disease. Nutr. Rev. 2016, 74, 624–634. [Google Scholar] [CrossRef] [PubMed]
- Asti, A.; Gioglio, L. Can a bacterial endotoxin be a key factor in the kinetics of amyloid fibril formation? J. Alzheimers Dis. 2014, 39, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Galloway, S.; Takechi, R.; Pallebage-Gamarallage, M.M.; Dhaliwal, S.S.; Mamo, J.C. Amyloid-beta colocalizes with apolipoprotein B in absorptive cells of the small intestine. Lipids Health Dis. 2009, 8, 46. [Google Scholar] [CrossRef] [PubMed]
- Takechi, R.; Galloway, S.; Pallebage-Gamarallage, M.; Wellington, C.; Johnsen, R.; Mamo, J.C. Three-dimensional colocalization analysis of plasma-derived apolipoprotein B with amyloid plaques in APP/PS1 transgenic mice. Histochem. Cell Biol. 2009, 131, 661–666. [Google Scholar]
- Zlokovic, B.V. Cerebrovascular transport of Alzheimer’s amyloid beta and apolipoproteins J and E: Possible anti-amyloidogenic role of the blood-brain barrier. Life Sci. 1996, 59, 1483–1497. [Google Scholar] [CrossRef]
- Friedland, R.P. Mechanisms of molecular mimicry involving the microbiota in neurodegeneration. J. Alzheimers Dis. 2015, 45, 349–362. [Google Scholar] [CrossRef]
- Holmqvist, S.; Chutna, O.; Bousset, L.; Aldrin-Kirk, P.; Li, W.; Bjorklund, T.; Wang, Z.Y.; Roybon, L.; Melki, R.; Li, J.Y. Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol. 2014, 128, 805–820. [Google Scholar] [CrossRef]
- Singhrao, S.K.; Harding, A.; Poole, S.; Kesavalu, L.; Crean, S. Porphyromonas gingivalis periodontal infection and its putative links with Alzheimer’s disease. Mediators Inflamm. 2015, 2015. [Google Scholar] [CrossRef]
- Ishida, N.; Ishihara, Y.; Ishida, K.; Tada, H.; Funaki-Kato, Y.; Hagiwara, M.; Ferdous, T.; Abdullah, M.; Mitani, A.; Michikawa, M.; et al. Periodontitis induced by bacterial infection exacerbates features of Alzheimer’s disease in transgenic mice. NPJ Aging Mech. Dis. 2017, 3, 15. [Google Scholar] [CrossRef]
- Chu, J.; Lauretti, E.; Pratic, D. Caspase-3-dependent cleavage of Akt modulates tau phosphorylation via GSK3ß kinase: Implications for Alzheimer’s disease. Mol. Psychiatry 2017, 22, 1002–1008. [Google Scholar] [CrossRef]
- Dominy, S.S.; Lynch, C.; Ermini, F.; Benedyk, M.; Marczyk, A.; Konradi, A.; Nguyen, M.; Haditsch, U.; Raha, D.; Griffin, C.; et al. Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci. Adv. 2019, 5, eaau3333. [Google Scholar] [CrossRef] [PubMed]
- Rivest, S. TREM2 enables amyloid beta clearance by microglia. Cell Res. 2015, 25, 535–536. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Lukiw, W.J. Microbiome-generated amyloid and potential impact on amyloidogenesis in Alzheimer’s disease (AD). J. Nat. Sci. 2015, 1, e138. [Google Scholar] [PubMed]
- Melchior, B.; Garcia, A.E.; Hsiung, B.K.; Lo, K.M.; Doose, J.M.; Thrash, J.C.; Stalder, A.K.; Staufenbiel, M.; Neumann, H.; Carson, M.J. Dual induction of TREM2 and tolerance-related transcript, Tmem176b, in amyloid transgenic mice: Implications for vaccine-based therapies for Alzheimer’s disease. ASN Neuro 2010, 2, e00037. [Google Scholar] [CrossRef] [PubMed]
- Gawish, R.; Martins, R.; Bohm, B.; Wimberger, T.; Sharif, O.; Lakovits, K.; Schmidt, M.; Knapp, S. Triggering receptor expressed on myeloid cells-2 fine-tunes inflammatory responses in murine Gram-negative sepsis. Faseb J. 2015, 29, 1247–1257. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Jaber, V.; Lukiw, W.J. Over-expressed pathogenic miRNAs in Alzheimer’s disease (AD) and prion disease (PrD) drive deficits in TREM2-mediated Abeta42 peptide clearance. Front. Aging Neurosci. 2016, 8, 140. [Google Scholar] [CrossRef]
- Mahmoudian Dehkordi, S.; Arnold, M.; Nho, K.; Ahmad, S.; Jia, W.; Xie, G.; Louie, G.; Kueider-Paisley, A.; Moseley, M.A.; Thompson, J.W.; et al. Alzheimer’s Disease Neuroimaging Initiative and the Alzheimer Disease Metabolomics Consortium. Altered bile acid profile associates with cognitive impairment in Alzheimer’s disease-An emerging role for gut microbiome. Alzheimers Dement. 2019, 15, 76–92. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Ho, L.; Faith, J.; Ono, K.; Janle, E.M.; Lachcik, P.J.; Cooper, B.R.; Jannasch, A.H.; D’Arcy, B.R.; Williams, B.A.; et al. Role of intestinal microbiota in the generation of polyphenolderived phenolic acid mediated attenuation of Alzheimer’s disease beta-amyloid oligomerization. Mol. Nutr. Food Res. 2015, 59, 1025–1040. [Google Scholar] [CrossRef]
- Kawashima, K.; Misawa, H.; Moriwaki, Y.; Fujii, Y.X.; Fujii, T.; Horiuchi, Y.; Yamada, T.; Imanaka, T.; Kamekura, M. Ubiquitous expression of acetylcholine and its biological functions in life forms without nervous systems. Life Sci. 2007, 80, 2206–2209. [Google Scholar] [CrossRef]
- Wall, R.; Cryan, J.F.; Ross, R.P.; Fitzgerald, G.F.; Dinan, T.G.; Stanton, C. Bacterial neuroactive compounds produced by psychobiotics. Adv. Exp. Med. Biol. 2014, 817, 221–239. [Google Scholar]
- Hu, X.; Wang, T.; Jin, F. Alzheimer’s disease and gut microbiota. Sci. China Life Sci. 2016, 59, 1006–1023. [Google Scholar] [CrossRef] [PubMed]
- Pyndt Jorgensen, B.; Hansen, J.T.; Krych, L.; Larsen, C.; Klein, A.B.; Nielsen, D.S.; Josefsen, K.; Hansen, A.K.; Sorensen, D.B. A possible link between food and mood: Dietary impact on gut microbiota and behavior in BALB/c mice. PLoS ONE 2014, 9, e103398. [Google Scholar] [CrossRef]
- Minter, M.R.; Hinterleitner, R.; Meisel, M.; Zhang, C.; Leone, V.; Zhang, X.; Oyler-Castrillo, P.; Zhang, X.; Musch, M.W.; Shen, X.; et al. Antibiotic-induced perturbations in microbial diversity during post-natal development alters amyloid pathology in an aged APPSWE/PS1DeltaE9 murine model of Alzheimer’s disease. Sci. Rep. 2017, 7, 10411. [Google Scholar] [CrossRef] [PubMed]
- Abraham, D.; Feher, J.; Scuderi, G.L.; Szabo, D.; Dobolyi, A.; Cservenak, M.; Juhasz, J.; Ligeti, B.; Pongor, S.; Gomez-Cabrera, M.C.; et al. Exercise and probiotics attenuate the development of Alzheimer’s disease in transgenic mice: Role of microbiome. Exp. Gerontol. 2019, 115, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Akbari, E.; Asemi, Z.; Daneshvar Kakhaki, R.; Bahmani, F.; Kouchaki, E.; Tamtaji, O.R.; Hamidi, G.A.; Salami, M. Effect of probiotic supplementation on cognitive function and metabolic status in Alzheimer’s disease: A randomized, double-blind and controlled trial. Front. Aging Neurosci. 2016, 8, 256. [Google Scholar] [CrossRef] [PubMed]
- Leblhuber, F.; Steiner, K.; Schuetz, B.; Fuchs, D.; Gostner, J.M. Probiotic Supplementation in Patients with Alzheimer’s Dementia-An Explorative Intervention Study. Med. J. Islam Repub. Iran 2017, 31, 103. [Google Scholar] [CrossRef] [PubMed]
- Athari Nik Azm, S.; Djazayeri, A.; Safa, M.; Azami, K.; Djalali, M.; Sharifzadeh, M.; Vafa, M. Probiotics improve insulin resistance status in an experimental model of Alzheimer’s disease. Curr. Alzheimer Res. 2018, 15, 1106–1113. [Google Scholar] [CrossRef]
- Agahi, A.; Hamidi, G.A.; Daneshvar, R.; Hamdieh, M.; Soheili, M.; Alinaghipour, A.; Esmaeili Taba, S.M.; Salami, M. Does Severity of Alzheimer’s Disease Contribute to Its Responsiveness to Modifying Gut Microbiota? A Double Blind Clinical Trial. Front. Neurol. 2018, 9, 662. [Google Scholar] [CrossRef]
- Sanborn, V.; Azcarate-Peril, M.A.; Updegraff, J.; Manderino, L.M.; Gunstad, J. A randomized clinical trial examining the impact of LGG probiotic supplementation on psychological status in middle-aged and older adults. Contemp. Clin. Trials Commun. 2018, 12, 192–197. [Google Scholar] [CrossRef]
- Asl, Z.R.; Sepehri, G.; Salami, M. Probiotic treatment improves the impaired spatial cognitive performance and restores synaptic plasticity in an animal model of Alzheimer’s disease. Behav. Brain Res. 2019, 112183. [Google Scholar] [CrossRef]
- Fernández-Sanz, P.; Ruiz-Gabarre, D.; García-Escudero, V. Modulating Effect of Diet on Alzheimer’s Disease. Diseases 2019, 7, 12. [Google Scholar]
- Dehhaghi, M.; Kazemi Shariat Panahi, H.; Guillemin, G.J. Microorganisms, Tryptophan Metabolism, and Kynurenine Pathway: A Complex Interconnected Loop Influencing Human Health Status. Int. J. Tryptophan Res. 2019, 12, 1178646919852996. [Google Scholar] [CrossRef] [PubMed]
- Nagpal, R.; Neth, B.J.; Wang, S.; Craft, S.; Yadav, H. Modified Mediterranean-ketogenic diet modulates gut microbiome and short-chain fatty acids in association with Alzheimer’s disease markers in subjects with mild cognitive impairment. EBioMedicine 2019. [Google Scholar] [CrossRef] [PubMed]
- McCann, A.; Jeffery, I.B.; Ouliass, B.; Ferland, G.; Fu, X.; Booth, S.L.; Tran, T.T.; O’Toole, P.W.; O’Connor, E.M. Exploratory analysis of covariation of microbiota-derived vitamin K and cognition in older adults. Am. J. Clin. Nutr. 2019. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Argenio, V.; Sarnataro, D. Microbiome Influence in the Pathogenesis of Prion and Alzheimer’s Diseases. Int. J. Mol. Sci. 2019, 20, 4704. https://doi.org/10.3390/ijms20194704
D’Argenio V, Sarnataro D. Microbiome Influence in the Pathogenesis of Prion and Alzheimer’s Diseases. International Journal of Molecular Sciences. 2019; 20(19):4704. https://doi.org/10.3390/ijms20194704
Chicago/Turabian StyleD’Argenio, Valeria, and Daniela Sarnataro. 2019. "Microbiome Influence in the Pathogenesis of Prion and Alzheimer’s Diseases" International Journal of Molecular Sciences 20, no. 19: 4704. https://doi.org/10.3390/ijms20194704
APA StyleD’Argenio, V., & Sarnataro, D. (2019). Microbiome Influence in the Pathogenesis of Prion and Alzheimer’s Diseases. International Journal of Molecular Sciences, 20(19), 4704. https://doi.org/10.3390/ijms20194704