Comparison of Whole Plastome Sequences between Thermogenic Skunk Cabbage Symplocarpus renifolius and Nonthermogenic S. nipponicus (Orontioideae; Araceae) in East Asia




Abstract
1. Introduction
2. Results and Discussion
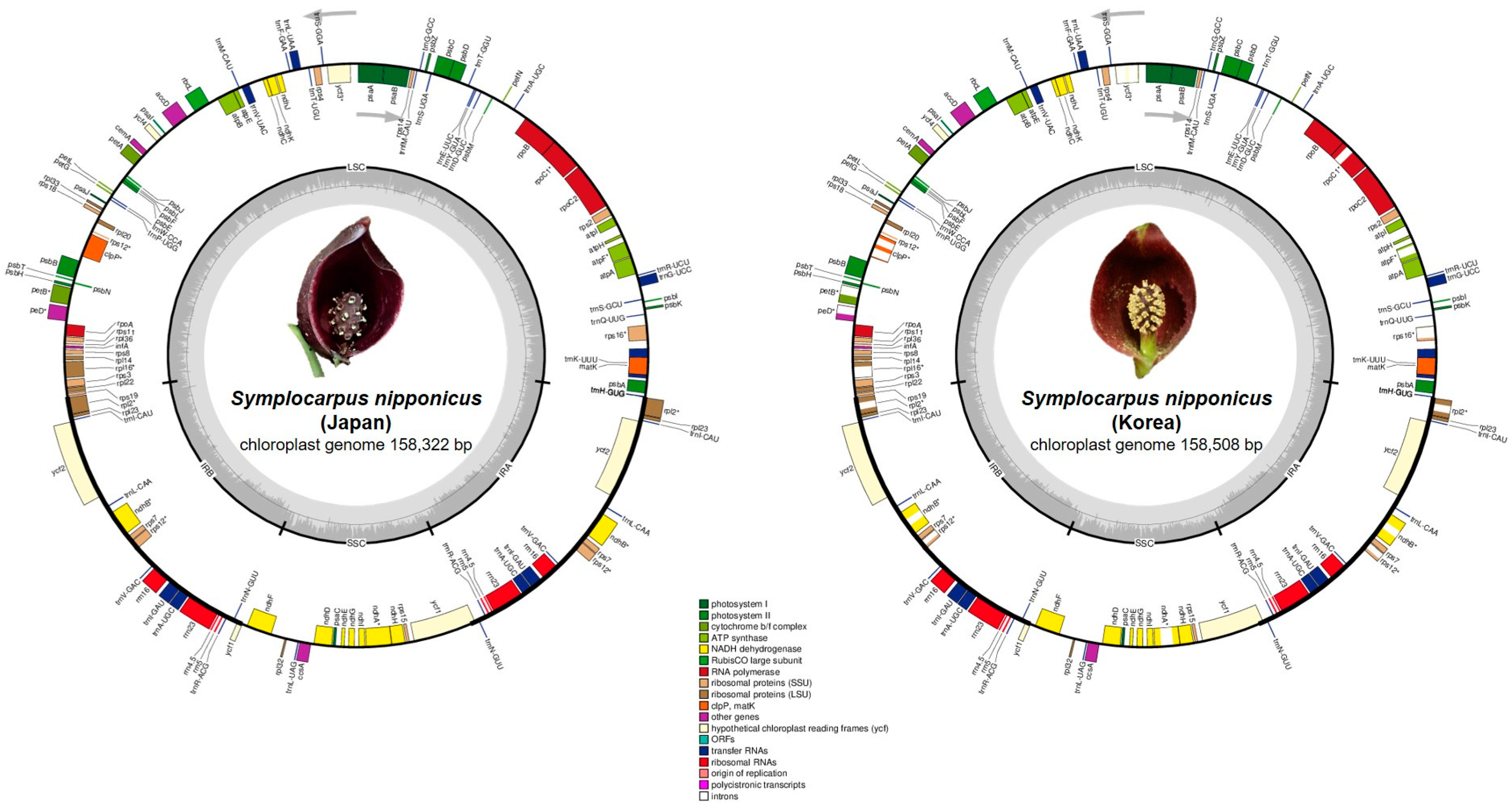
2.1. Genome Size and Features
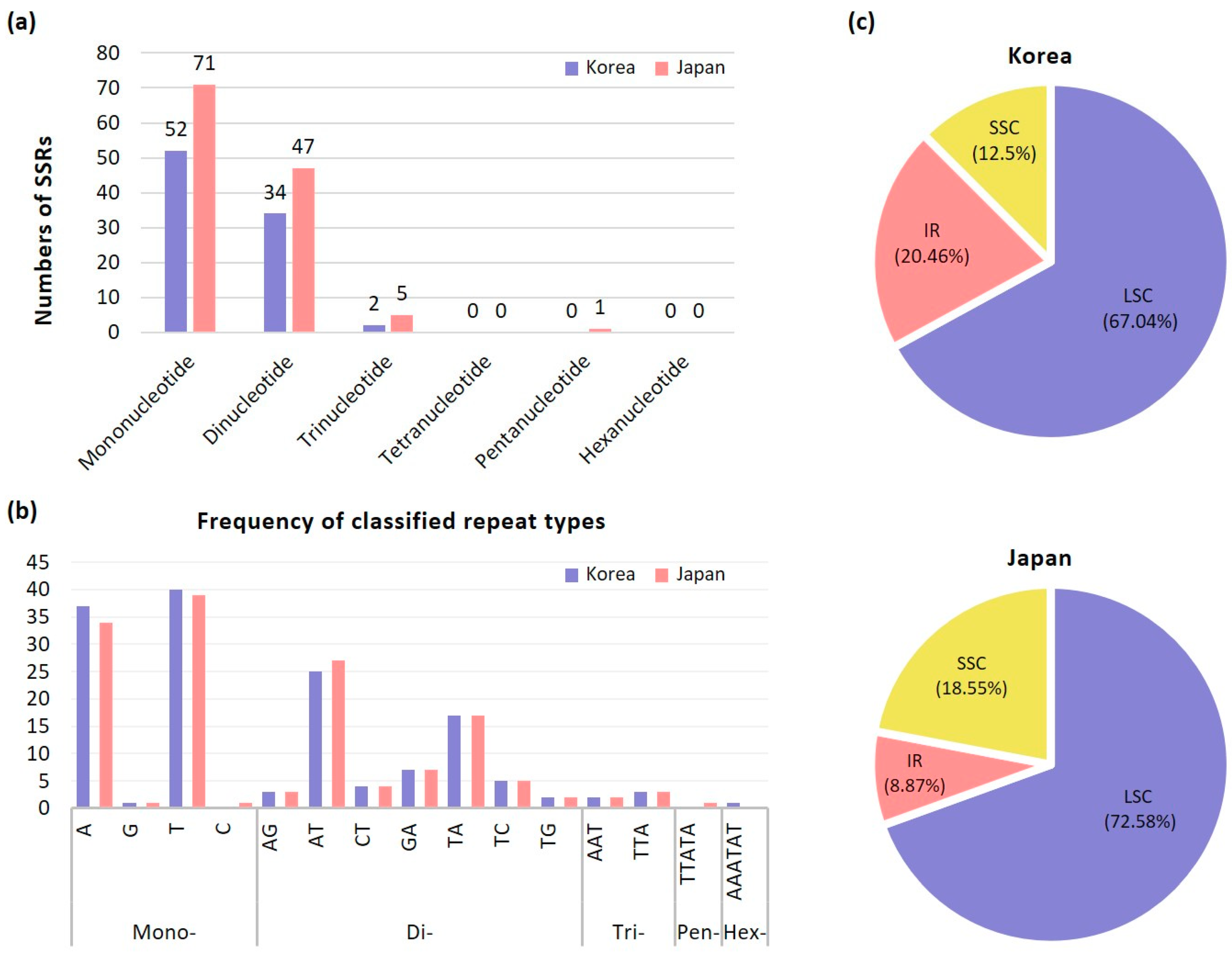
2.2. Microsatellites Analysis
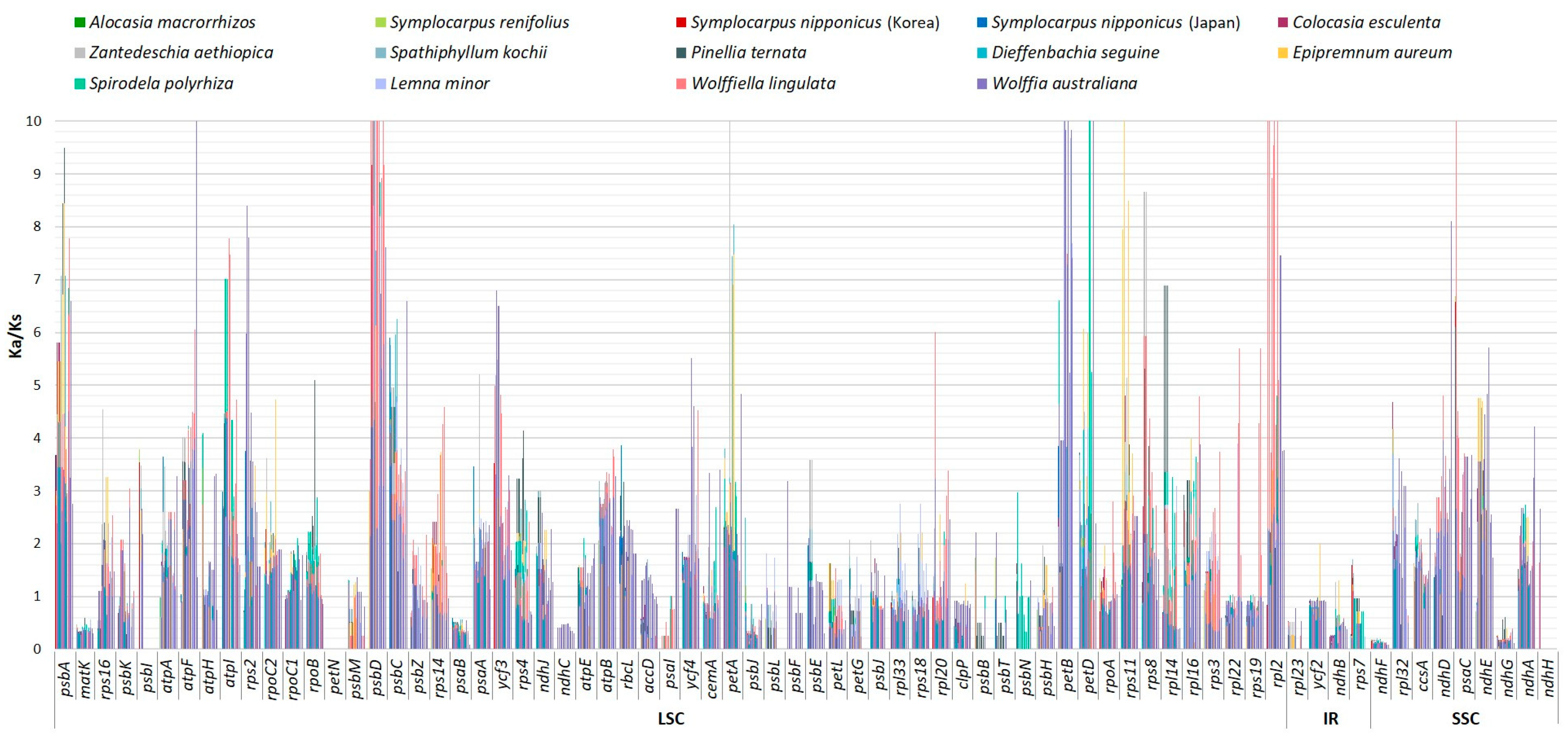
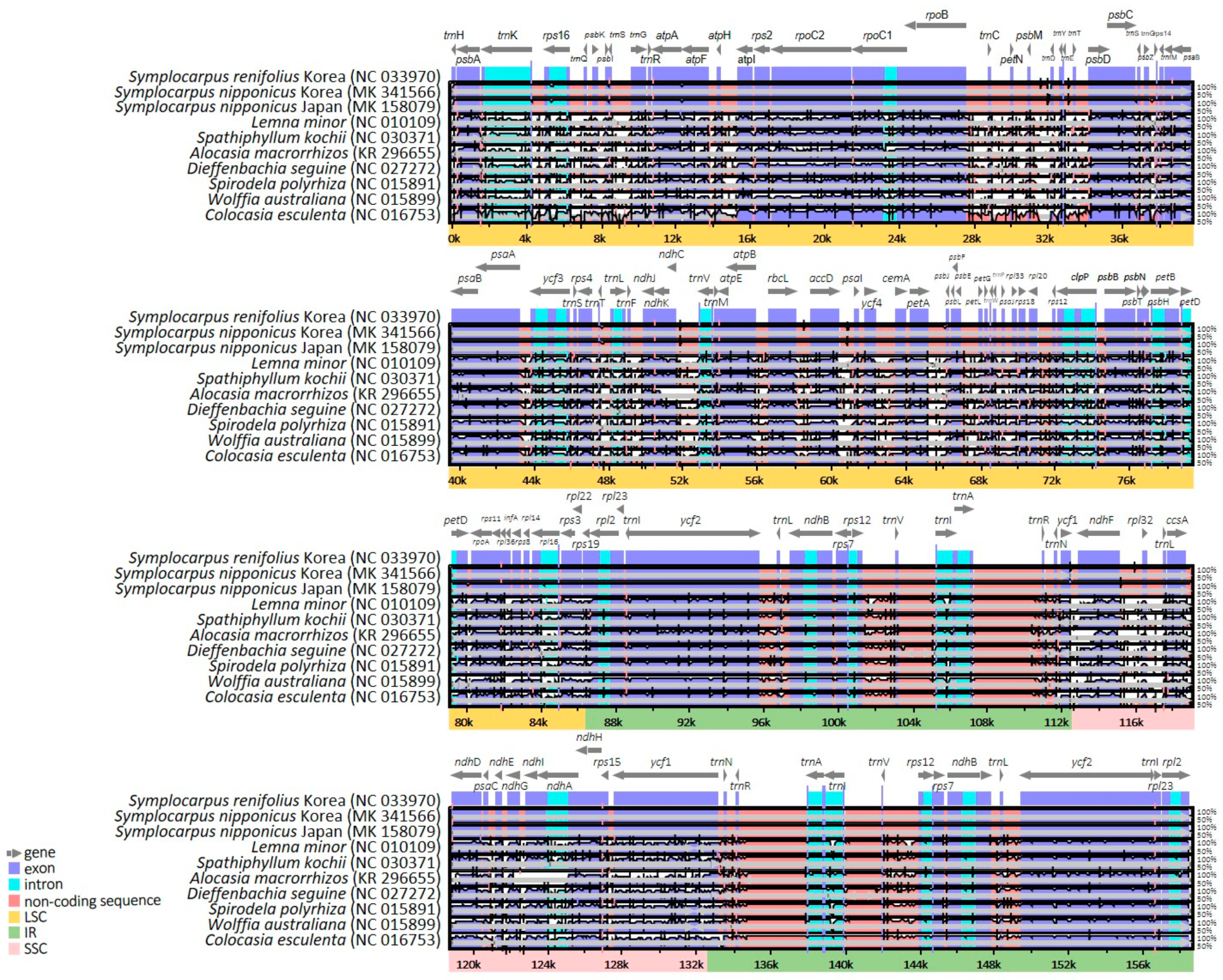
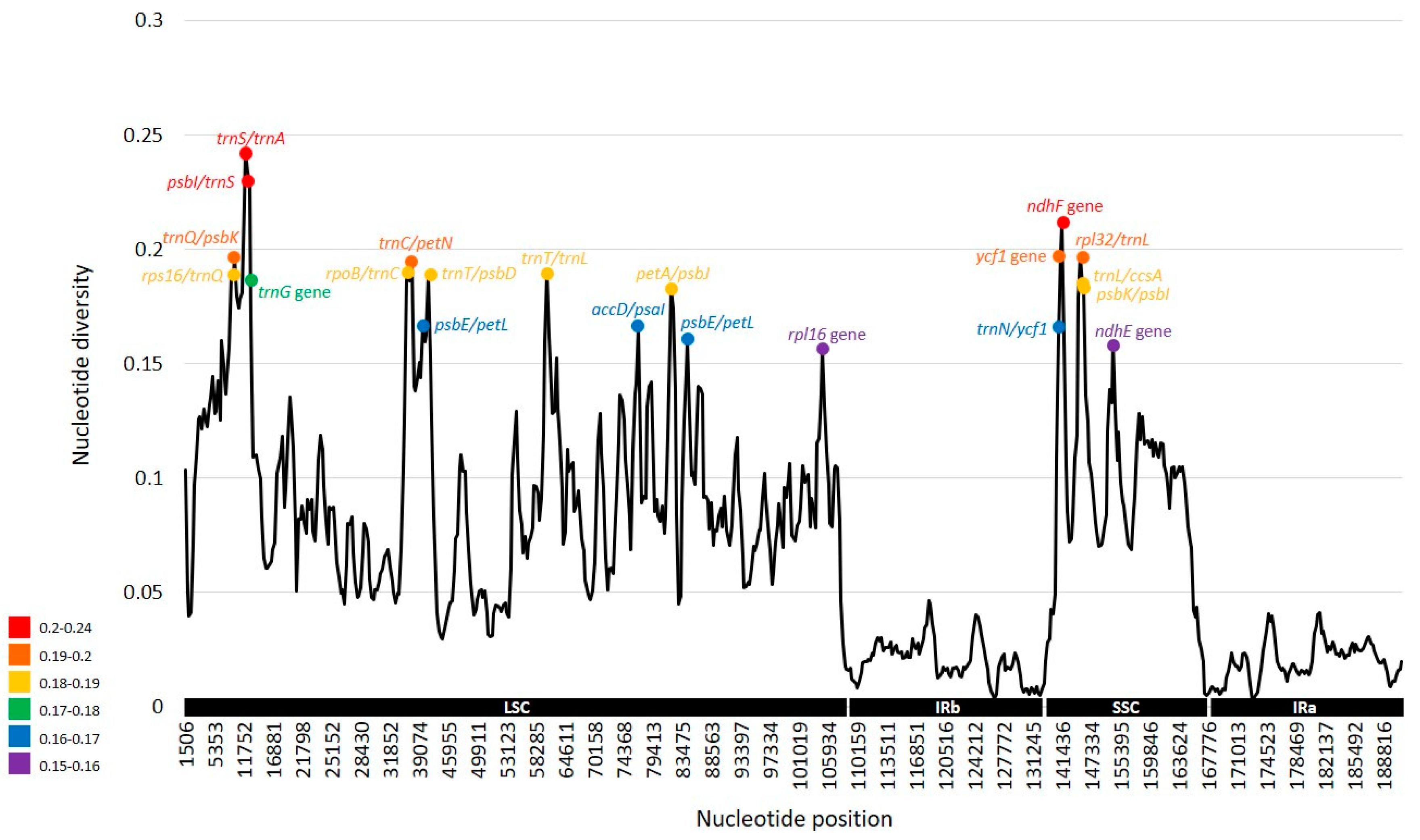
2.3. Comparative Analysis of Genome Structure
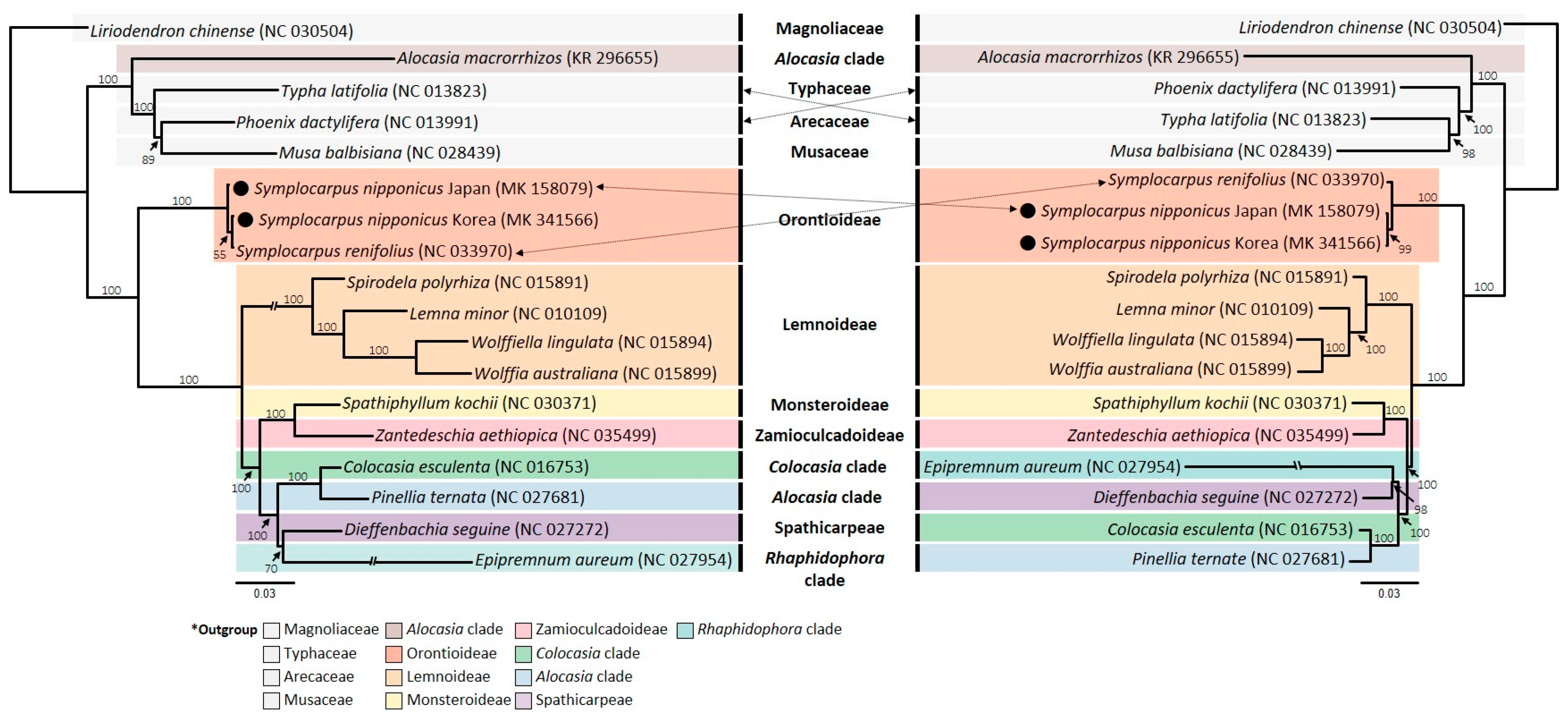
2.4. Phylogenetic Analysis
3. Materials and Methods
3.1. Plastome Sequencing, Assembly, and Annotation
3.2. Comparative Plastome Analysis
3.3. Tandem Repeat and Microsatellite Analysis
3.4. Phylogenetic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Nie, Z.-L.; Sun, H.; Li, H.; Wen, J. Intercontinental biogeography of subfamily Orontioideae (Symplocarpus, Lysichiton, and Orontium) of Araceae in eastern Asia and North America. Mol. Phylogenet. Evol. 2006, 40, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Mayo, S.J.; Bogner, J.; Boyce, P.C. The Genera Araceae; Royal Botanic Gardens, Kew: Richmond, UK, 1997. [Google Scholar]
- Otsuka, K.; Ryuichi, W.; Inoue, K. A new species of Symplocarpus (Araceae) from Nagano Prefecture, Central Japan. J. Jpn. Bot. 2002, 77, 96–100. [Google Scholar]
- Pavlova, N.S.; Nechaev, V.A. A new species of the genus Symplocarpus (Araceae) from the southern Russian Far East. Bot. Zhurn. (Mosc. Leningr.) 2005, 90, 753–758. [Google Scholar]
- Knutson, R.M. Heat production and temperature regulation in eastern skunk cabbage. Science 1974, 186, 746–747. [Google Scholar] [CrossRef] [PubMed]
- Knutson, R.M. Plants in heat. Nat. Hist. 1979, 88, 42–47. [Google Scholar]
- Wada, N.; Uemura, S. Seed dispersal and predation by small rodents on the herbaceous understory plant Symplocarpus renifolius. Am. Midl. Nat. 1994, 132, 320–327. [Google Scholar] [CrossRef]
- Wada, N.; Uemura, S. Size-dependent flowering behavior and heat production of a sequential hermaphrodite, Symplocarpus renifolius (Araceae). Am. J. Bot. 2000, 87, 1489–1494. [Google Scholar] [CrossRef]
- Ito, K.; Ito, T.; Onda, Y.; Uemura, S. Temperature-triggered periodical thermogenic oscillations in skunk cabbage (Symplocarpus foetidus). Plant Cell Physiol. 2004, 45, 257–264. [Google Scholar] [CrossRef]
- Ito-Inaba, Y. Thermogenesis in skunk cabbage (Symplocarpus renifolius): New insights from the ultrastructure and gene expression profiles. Adv. Hortic. Sci. 2014, 28, 73–78. [Google Scholar]
- Seymour, R.S.; Schultze-Motel, P. Heat-producing flowers. Endeavour 1997, 21, 125–129. [Google Scholar] [CrossRef]
- Seymour, R.S.; White, C.R.; Gibernau, M. Heat reward for insect pollinators. Nature 2003, 426, 243–244. [Google Scholar] [CrossRef] [PubMed]
- Seymour, R.S.; Ito, Y.; Onda, Y.; Ito, K. Effects of floral theromogenesis on pollen function in Asian skunk cabbage Symplocarpus renifolius. Biol. Lett. 2009, 23, 568–570. [Google Scholar] [CrossRef] [PubMed]
- Takeda, S.; Onishi, Y.; Fukui, Y.; Ohsako, T.; Kubo, N. Life cycle and genetic diversity of Symplocarpus nipponicus (Araceae), an endangered species in Japan. Plants 2018, 7, 73. [Google Scholar] [CrossRef] [PubMed]
- Wen, J. Evolution of the eastern Asian and eastern North American disjunct genus Symplocarpus (Araceae): Insights from chloroplast DNA restriction site data. Biochem. Syst. Ecol. 1996, 24, 735–747. [Google Scholar] [CrossRef]
- Kitano, S.; Otsuka, K.; Uesugi, R.; Goka, K. Molecular phylogenetic analysis of the genus Symplocarpus (Araceae) from Japan based on chloroplast DNA sequences. J. Jpn. Bot. 2005, 80, 334–339. [Google Scholar]
- Iwatsubo, Y.; Otsuka, K. Chromosome numbers of Japanese Symplocarpus (Araceae). J. Phytogeogr. Taxon. 2005, 53, 203–205. [Google Scholar]
- Shinozaki, K.; Ohme, M.; Tanaka, M.; Wakasugi, T.; Hayashida, N.; Matsubayashi, T.; Zaita, N.; Chunwonse, J.; Obokata, J.; Yamaguchi-Shinozaki, K.; et al. The complete nucleotide sequence of the tobacco chloroplast genome: Its gene organization and expression. EMBO J. 1986, 5, 2043–2049. [Google Scholar] [CrossRef]
- Jansen, R.K.; Cai, Z.Q.; Raubeson, L.A.; Daniell, H.; de Pamphilis, C.W.; Leebeans-Mack, J.; Müller, K.F.; Guisinger-Bellian, M.; Haberle, R.C.; Hansen, A.K.; et al. Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc. Natl. Acad. Sci. USA 2007, 104, 19369–19374. [Google Scholar] [CrossRef]
- Hollingsworth, P.M.; Graham, S.W.; Little, D.P. Choosing and using a plant DNA barcode. PLoS ONE 2011, 6, e19254. [Google Scholar] [CrossRef]
- Bock, R. Structure, function, and inheritance of plastid genomes. In Cell and Molecular Biology of Plastids; Bock, R., Ed.; Springer: Berlin/Heidelberg, Germany, 2007; Volume 19, pp. 29–63. [Google Scholar]
- Jansen, R.K.; Ruhlman, T.A. Plastid genomes of seed plants. In Genomics of Chloroplasts and Mitochondria. Advances in Photosynthesis and Respiration; Bock, R., Knoop, V., Eds.; Springer: New York, NY, USA, 2012; pp. 103–126. [Google Scholar]
- Ruhlman, T.A.; Jansen, R.K. The plastid genomes of flowering plants. In Chloroplast Biotechnology: Methods and Protocols; Maliga, P., Ed.; Springer Science and Business Media, LLC.: New York, NY, USA, 2014; Volume 1132, pp. 3–38. [Google Scholar]
- Huang, Y.; Li, X.; Yang, Z.; Yang, C.; Yang, J.; Ji, Y. Analysis of complete chloroplast genome sequences improves phylogenetic resolution in Paris (Melanthiaceae). Front. Plant Sci. 2016, 7, 1797. [Google Scholar] [CrossRef]
- Niu, Z.; Zhu, S.; Pan, J.; Li, L.; Sun, J.; Ding, X. Comparative analysis of Dendrobium plastomes and utility of plastomic mutational hotspots. Sci. Rep. 2017, 7, 2073. [Google Scholar]
- Yang, J.Y.; Pak, J.H.; Kim, S.-C. The complete plastome sequence of Rubus takesimensis endemic to Ulleung Island, Korea: Insights into molecular evolution of anagenetically derived species in Rubus (Rosaceae). Gene 2018, 668, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Qi, X.; Chen, J.; Sun, L.; Zhong, Y.; Fang, J.; Hu, C. The complete chloroplast genome sequence of Actinidia arguta using the PacBio RS II platform. PLoS ONE 2018, 13, e0197393. [Google Scholar] [CrossRef] [PubMed]
- Nauheimer, L.; Metzler, D.; Senner, S.S. Global history of the ancient monocot family Araceae inferred with models accounting for past continental positions and previous ranges based on fossils. New Phytol. 2012, 195, 938–950. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, I.; Biggs, P.G.; Mattews, P.J.; Collins, L.J.; Hendy, M.D.; Lockhart, P.J. Mutational dynamics of Aroid chloroplast genomes. Genome Biol. Evol. 2012, 4, 1316–1323. [Google Scholar] [CrossRef]
- Mardanov, A.V.; Ranin, N.V.; Kuznestsov, B.B.; Samigullin, T.H.; Antonov, A.S.; Kolganova, T.V.; Skyabin, K.G. Complete sequence of the Duckweed (Lemna minor) chloroplast genome: Structural organization and phylogenetic relationships to other Angiosperms. J. Mol. Evol. 2008, 66, 555–564. [Google Scholar] [CrossRef]
- Wang, W.; Messing, J. High-throughput sequencing of three Lemnoideae (Duckweeds) chloroplast genomes from total DNA. PLoS ONE 2011, 6, e24670. [Google Scholar] [CrossRef]
- Wang, B.; Han, L. The complete chloroplast genome sequence of Alocasia macrorrhizos. Mitochondrial DNA A DNA Mapp. Seq. Anal. 2016, 27, 3464–3465. [Google Scholar] [CrossRef]
- Wang, B.; Han, L.; Chen, C.; Wang, Z. The complete chloroplast genome sequence of Diffenbachia seguine (Araceae). Mitochondrial DNA A DNA Mapp. Seq. Anal. 2016, 27, 2913–2914. [Google Scholar]
- Liang, J.; Ma, Q.; Yang, Z. The first complete chloroplast genomes of two Alismataceae species, and the phylogenetic relationship under order Alismatales. Mitochondrial DNA B 2018, 4, 122–123. [Google Scholar] [CrossRef]
- Choi, K.S.; Park, K.T.; Park, S. The chloroplast genome of Symplocarpus renifolius: A comparison of chloroplast genome structure in Araceae. Genes 2017, 8, 324. [Google Scholar] [CrossRef] [PubMed]
- Cusimano, N.; Sousa, A.; Renner, S.S. Maximum likelihood inference implies a high, not a low, ancestral haploid chromosome number in Araceae, with a critique of the bias introduced by ‘x’. Ann. Bot. 2012, 109, 681–692. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-H.; Maki, M.; Lepschi, B.; Kim, S.-C. Highly conserved plastomes of the most basal lineage Proto-Araceae in the ancient monocot family Araceae. (unpublished; manuscript in preparation).
- Ravi, V.; Khurana, J.P.; Tyagi, A.K.; Khurana, P. An update on chloroplast genomes. Plant Syst. Evol. 2008, 271, 101–122. [Google Scholar] [CrossRef]
- Pinard, D.; Myburg, A.A.; Mizrachi, E. The plastid and mitochondrial genomes of Eucalyptus grandis. BMC Genom. 2019, 20, 132. [Google Scholar] [CrossRef] [PubMed]
- Rabah, S.O.; Lee, C.; Hajrah, N.H.; Makki, R.M.; Alharby, H.F.; Alhebshi, A.M.; Sabir, J.; Jansen, R.K.; Ruhlman, T.A. Plastome sequencing of ten nonmodel crop species uncovers a large insertion of mitochondrial DNA in cashew. Plant Genome 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Makałowski, W.; Boguski, M.S.; Hughes, A.L.; Yeager, M. Synonymous and nonsynonymous substitution distances are correlated in mouse and rat genes. J. Mol. Evol. 1998, 47, 119–121. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.; Lickey, E.B.; Schilling, E.E.; Small, R.L. Comparison of while chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: The tortoise and the hare III. Am. J. Bot. 2007, 94, 275–288. [Google Scholar] [CrossRef]
- Shaw, J.; Shafer, H.L.; Leonard, R.; Kovach, M.J.; Schorr, M.; Morris, A.B. Chloroplast DNA sequence utility for the lowest phylogenetic and phylogeographic inferences in angiosperms: The tortoise and the hare IV. Am. J. Bot. 2014, 101, 1987–2004. [Google Scholar] [CrossRef]
- Cusimano, N.; Bogner, J.; Mayo, S.J.; Boyce, P.C.; Wong, S.Y.; Hesse, M.; Hetterscheid, W.L.A.; Keating, R.C.; French, J. Relationships within the Araceae: Comparison of morphological patterns with molecular phylogenies. Am. J. Bot. 2011, 98, 654–668. [Google Scholar] [CrossRef]
- Henriquez, C.L.; Arias, T.; Pires, J.C.; Croat, T.B.; Schaal, B.A. Phylogenomics of the plant family Araceae. Mol. Phylogenet. Evol. 2014, 75, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, L.I.; Salazar, G.A.; Chase, M.W.; Mayo, S.J.; Bogner, J.; Dávila, P. Phylogenetic relationships of aroids and duckweeds (Araceae) inferred from coding and noncoding plastid DNA. Am. J. Bot. 2008, 95, 1153–1165. [Google Scholar] [CrossRef] [PubMed]
- Som, A. Causes, consequences and solutions of phylogenetic incongruence. Brief. Bioinform. 2015, 16, 536–548. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kim, S.-H.; Lee, S.; Maki, M.; Otsuka, K.; Kozhevnikov, A.E.; Kozhevnikova, Z.V.; Wen, J.; Kim, S.-C. New insights into the phylogeny and biogeography of subfamily Orontioideae (Araceae). J. Syst. Evol. 2019. [Google Scholar] [CrossRef]
- Kim, S.-H.; Cho, M.-S.; Li, P.; Kim, S.-C. Phylogeography and ecological niche modeling reveal reduced genetic diversity and colonization patterns of skunk cabbage (Symplocarpus foetidus; Araceae) from glacial refugia in eastern North America. Front. Plant Sci. 2018, 9, 648. [Google Scholar] [CrossRef] [PubMed]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Lohse, M.; Drechsel, O.; Bock, R. Organellar genome DRAW (OGDRAW): A tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 2009, 25, 1451–1452. [Google Scholar]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef]
- Brundo, M.; Malde, S.; Poliakov, A.; Do, C.B.; Couronne, O.; Dubchak, I.; Batzoglou, S. Global Alignment: Finding rearrangements during alignment. Bioinformatics (19S1) 2003, 19, i54–i62. [Google Scholar]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP v6: DNA sequence polymorphism analysis of large datasets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.M.; Li, W.H. The codon adaptation index–a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987, 15, 1281–1295. [Google Scholar] [CrossRef]
- Mower, J.P. The PREP suite: Predictive RNA editors for plant mitochondrial genes, chloroplast genes and user-defined alignments. Nucleic Acids Res. 2009, 37, W253–W259. [Google Scholar] [CrossRef]
- Thiel, T.; Michalek, W.; Varshney, R.K.; Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Taxa | S. nipponicus (Korea) | S. nipponicus (Japan) | S. renifolius (Korea) |
|---|---|---|---|
| Accession Number | MK341566 | MK158079 | KY039276 |
| Total cpDNA size (bp)/GC content (%) | 158,508/37.3 | 158,322/37.4 | 158,521/37.3 |
| LSC size (bp)/GC content (%) | 86,595/35.6 | 86,433/34.9 | 86,620/35.6 |
| IR size (bp)/GC content (%) | 25,802/42.8 | 25,809/43.8 | 25,801/42.9 |
| SSC size (bp)/GC content (%) | 20,309/30.9 | 20,271/31.0 | 20,299/31.0 |
| Number of genes | 130 | 130 | 130 |
| Number of protein-coding genes | 85 | 85 | 85 |
| Number of tRNA genes | 37 | 37 | 37 |
| Number of rRNA genes | 8 | 8 | 8 |
| Number of duplicated genes | 17 | 17 | 17 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.-H.; Yang, J.; Park, J.; Yamada, T.; Maki, M.; Kim, S.-C. Comparison of Whole Plastome Sequences between Thermogenic Skunk Cabbage Symplocarpus renifolius and Nonthermogenic S. nipponicus (Orontioideae; Araceae) in East Asia. Int. J. Mol. Sci. 2019, 20, 4678. https://doi.org/10.3390/ijms20194678
Kim S-H, Yang J, Park J, Yamada T, Maki M, Kim S-C. Comparison of Whole Plastome Sequences between Thermogenic Skunk Cabbage Symplocarpus renifolius and Nonthermogenic S. nipponicus (Orontioideae; Araceae) in East Asia. International Journal of Molecular Sciences. 2019; 20(19):4678. https://doi.org/10.3390/ijms20194678
Chicago/Turabian StyleKim, Seon-Hee, JiYoung Yang, Jongsun Park, Takayuki Yamada, Masayuki Maki, and Seung-Chul Kim. 2019. "Comparison of Whole Plastome Sequences between Thermogenic Skunk Cabbage Symplocarpus renifolius and Nonthermogenic S. nipponicus (Orontioideae; Araceae) in East Asia" International Journal of Molecular Sciences 20, no. 19: 4678. https://doi.org/10.3390/ijms20194678
APA StyleKim, S.-H., Yang, J., Park, J., Yamada, T., Maki, M., & Kim, S.-C. (2019). Comparison of Whole Plastome Sequences between Thermogenic Skunk Cabbage Symplocarpus renifolius and Nonthermogenic S. nipponicus (Orontioideae; Araceae) in East Asia. International Journal of Molecular Sciences, 20(19), 4678. https://doi.org/10.3390/ijms20194678