Chromosomal Density of Cancer Up-Regulated Genes, Aberrant Enhancer Activity and Cancer Fitness Genes Are Associated with Transcriptional Cis-Effects of Broad Copy Number Gains in Colorectal Cancer



Abstract
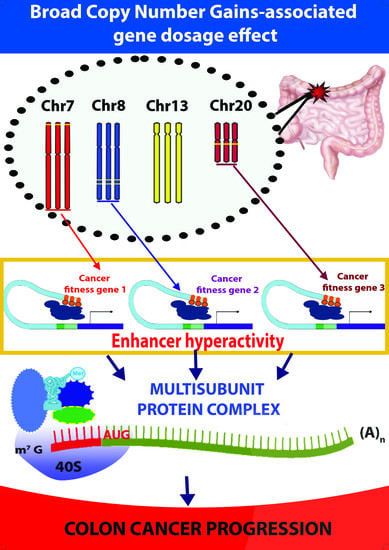
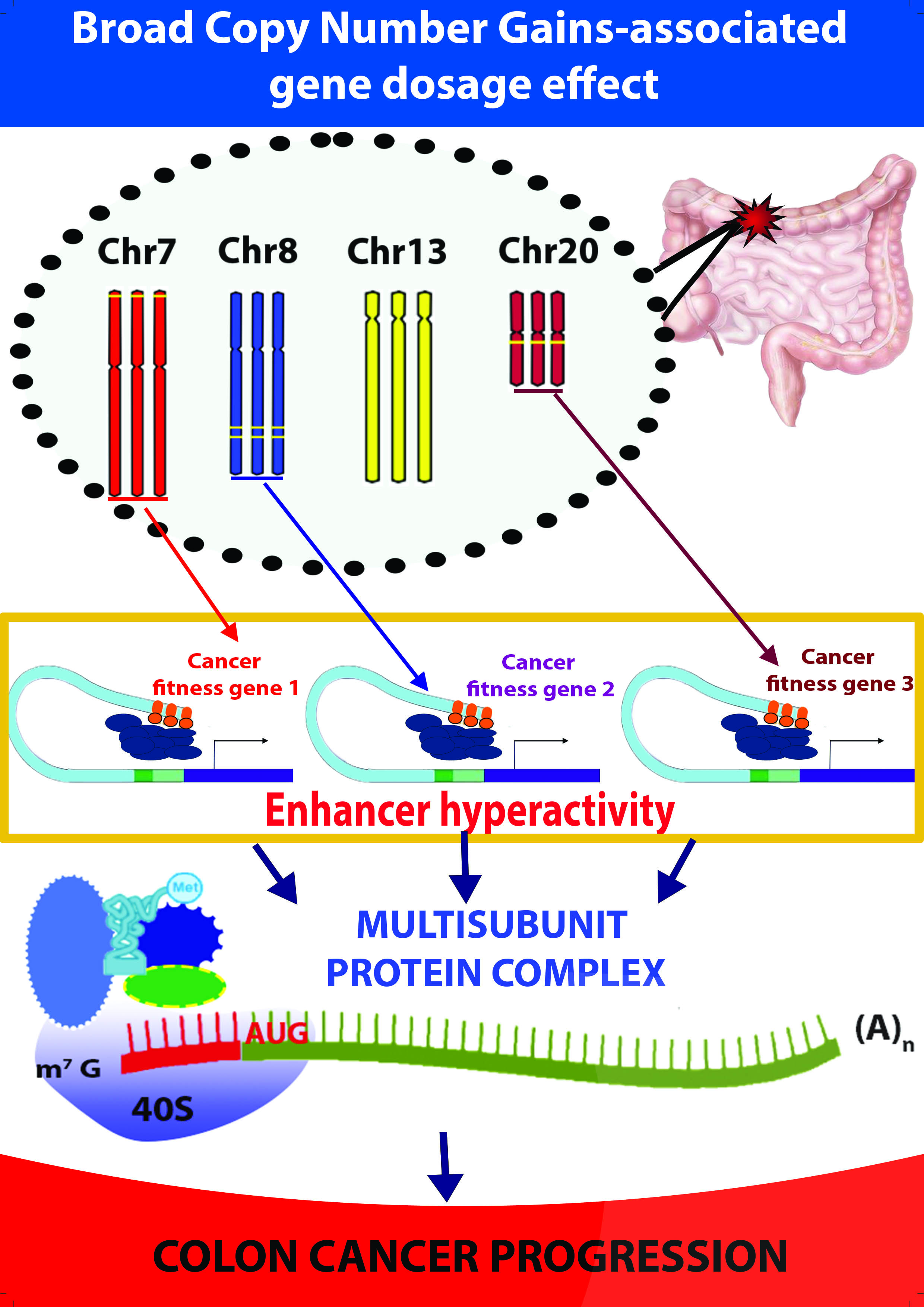
:
1. Introduction
2. Results
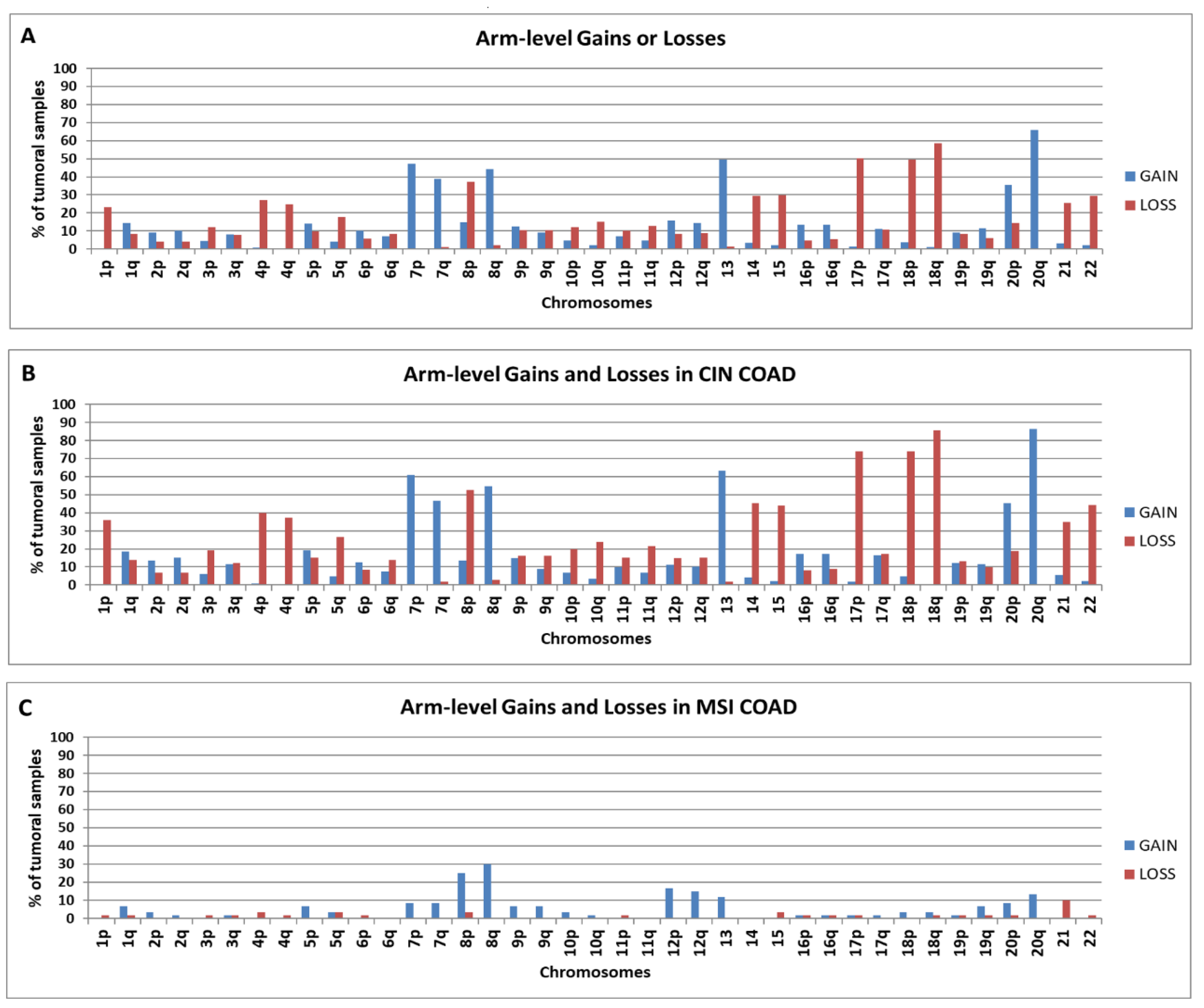
2.1. Chromosomal Distribution of Arm-Level Copy Number Abnormalities in TCGA COAD Samples
2.2. Differential Expression Analysis of RNA-Seq Data in TCGA COAD Samples
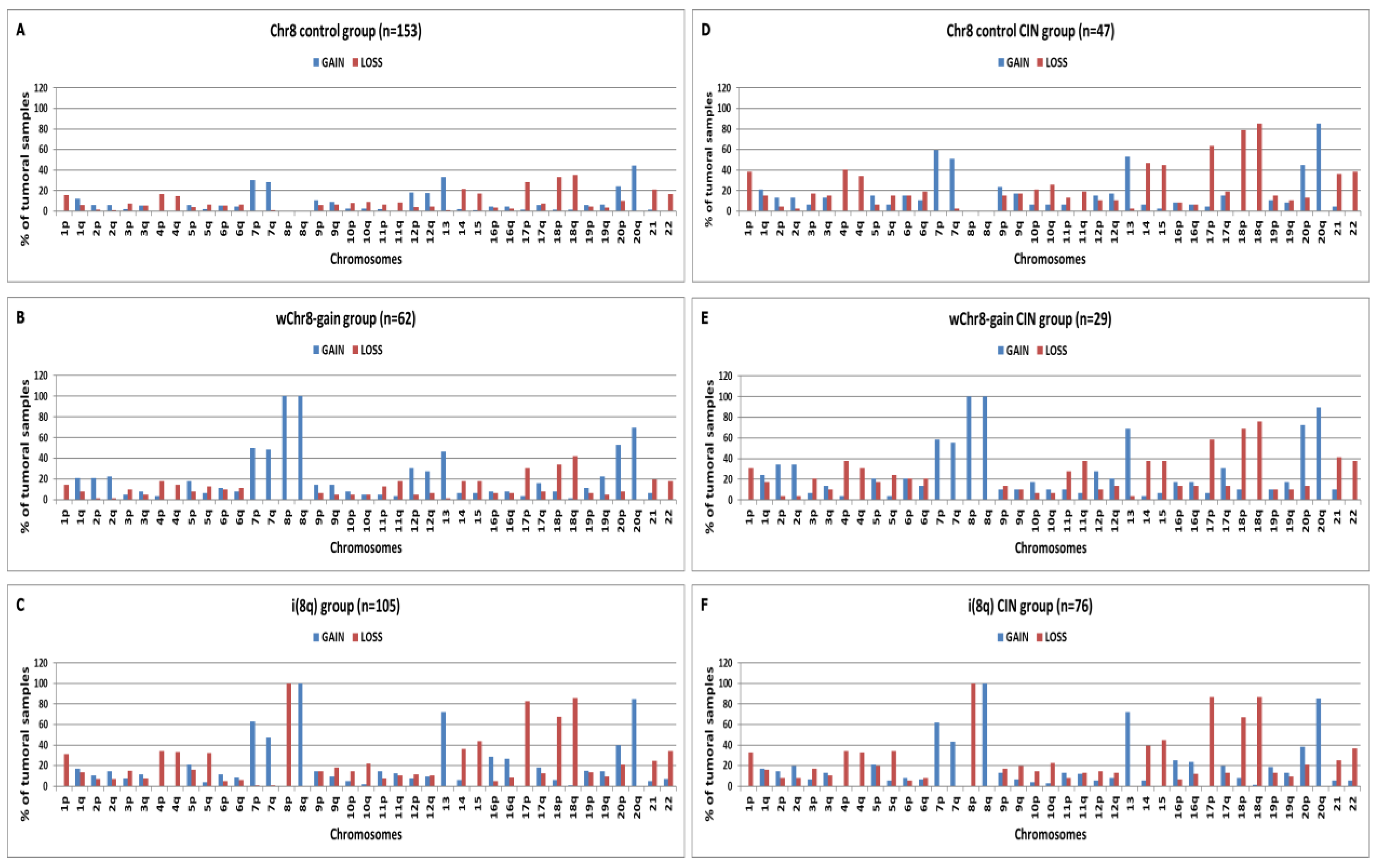
2.3. Chromosomal Distribution of Arm-Level CNAs in “Selected COAD” Groups
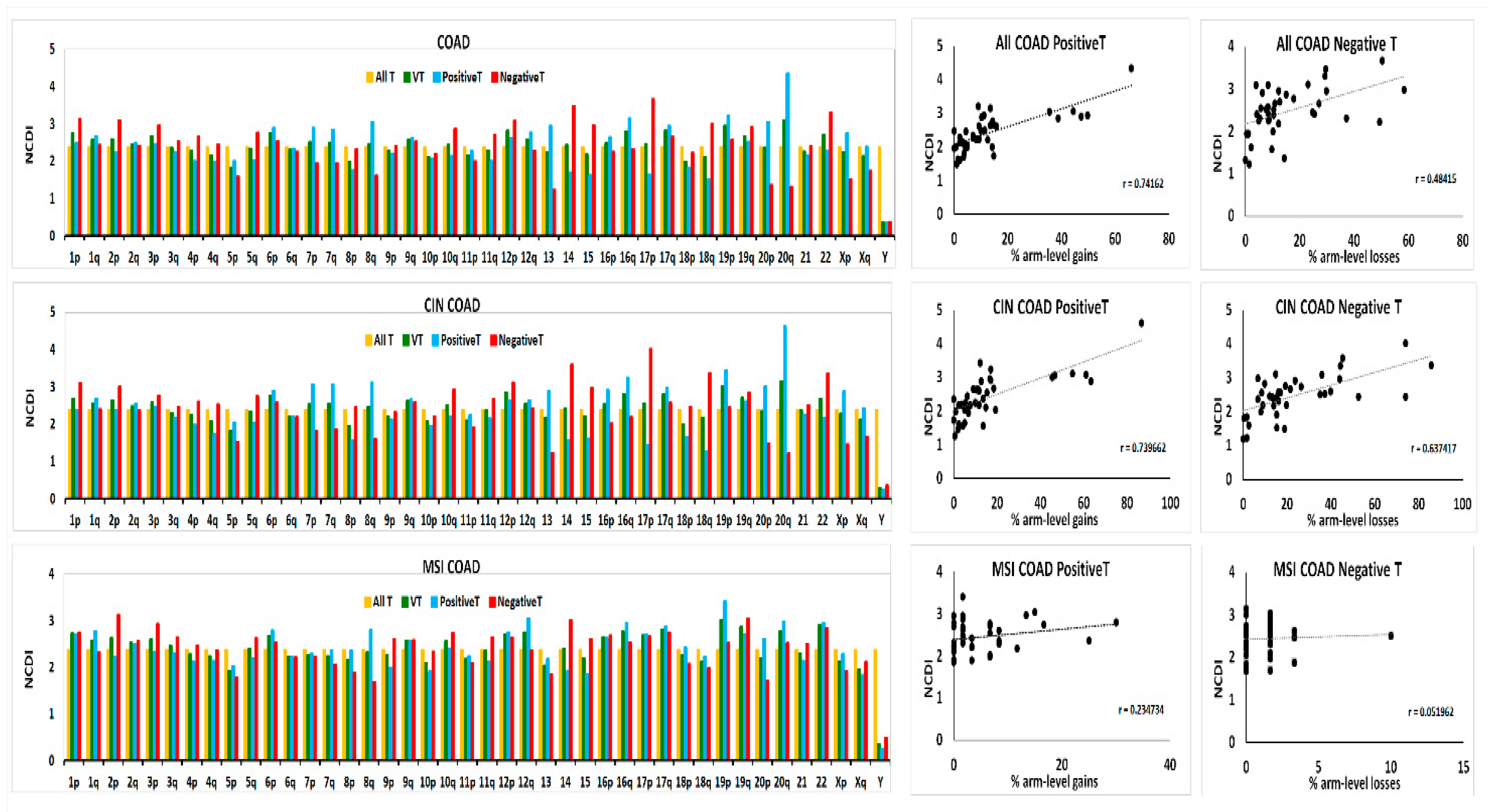
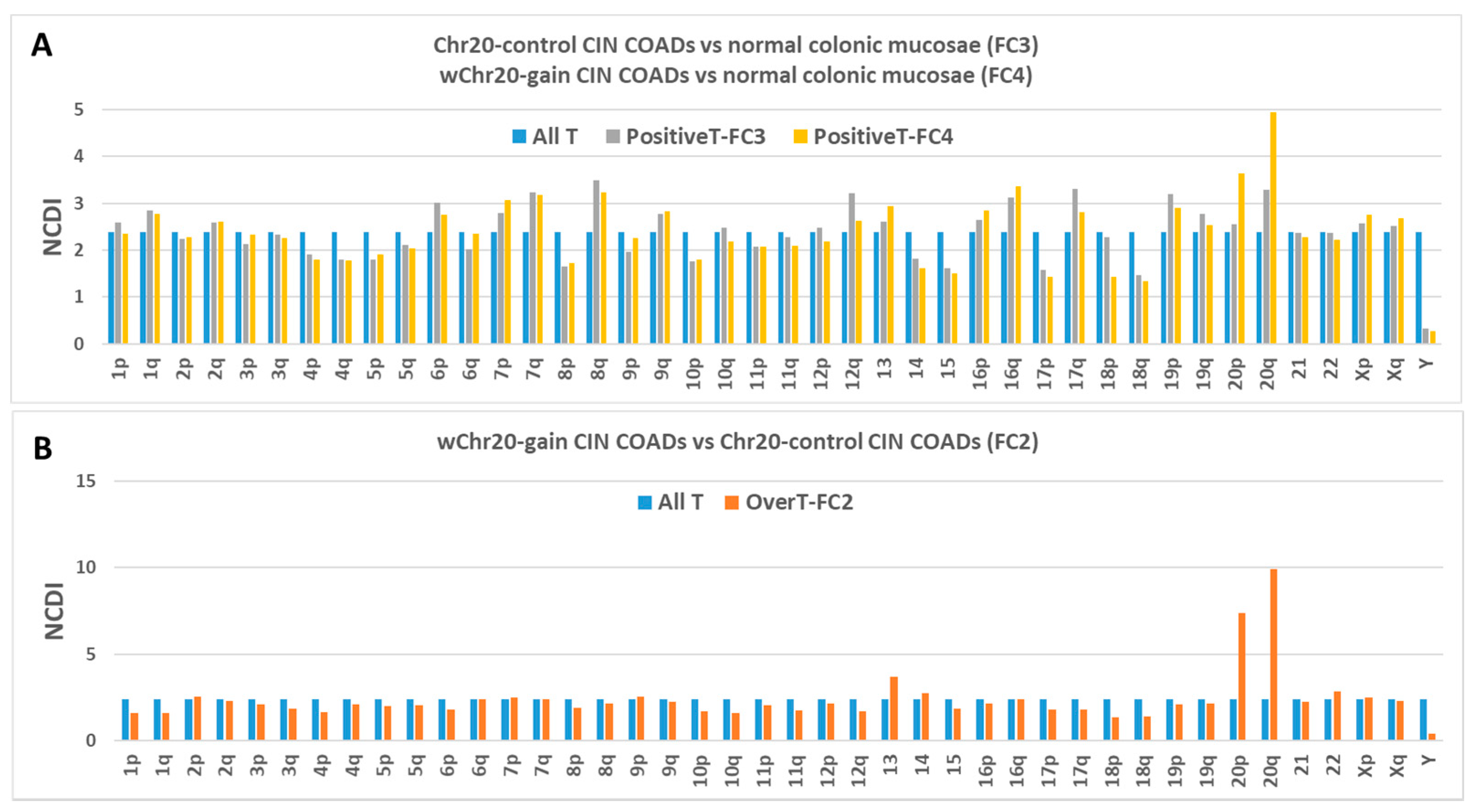
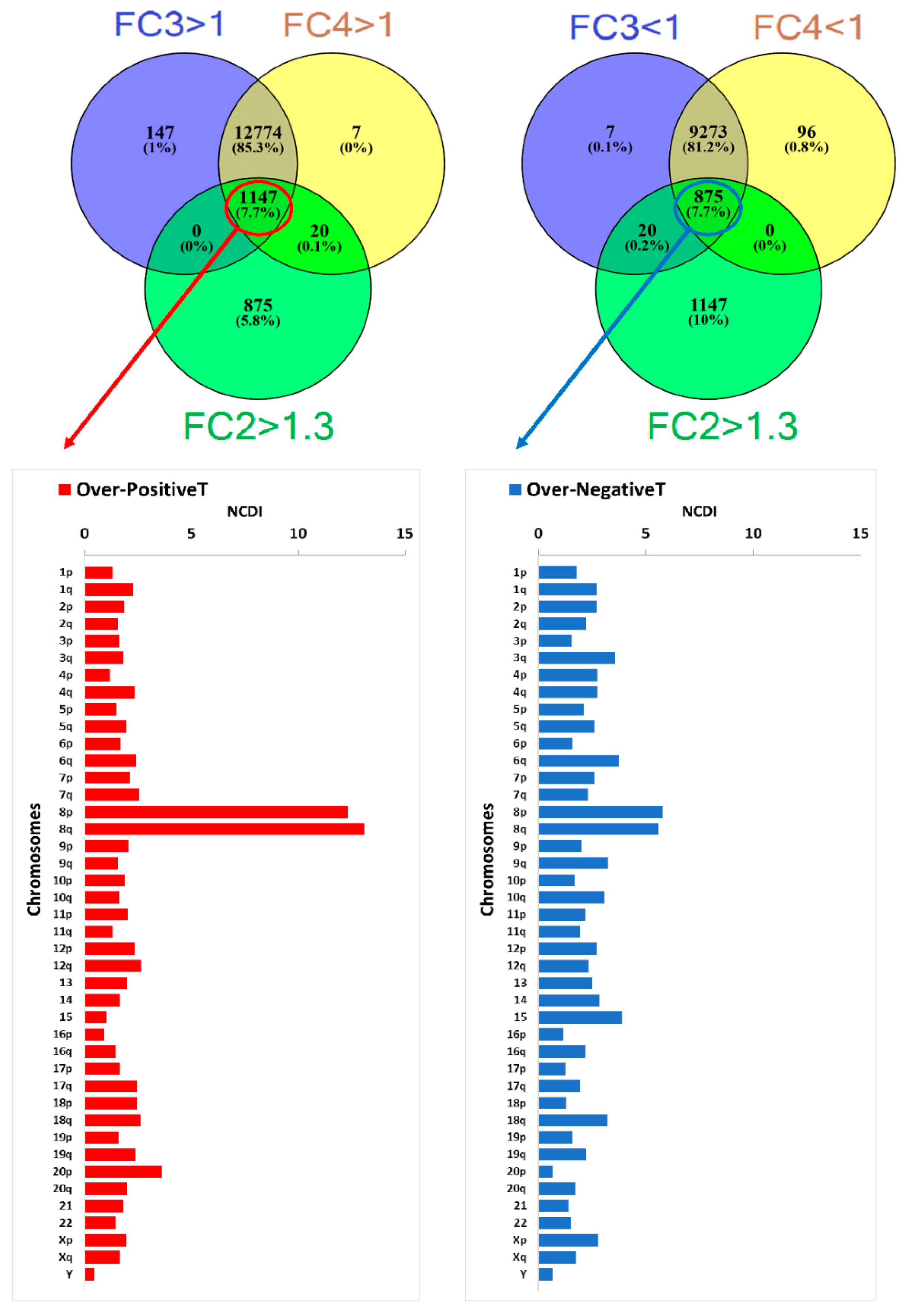
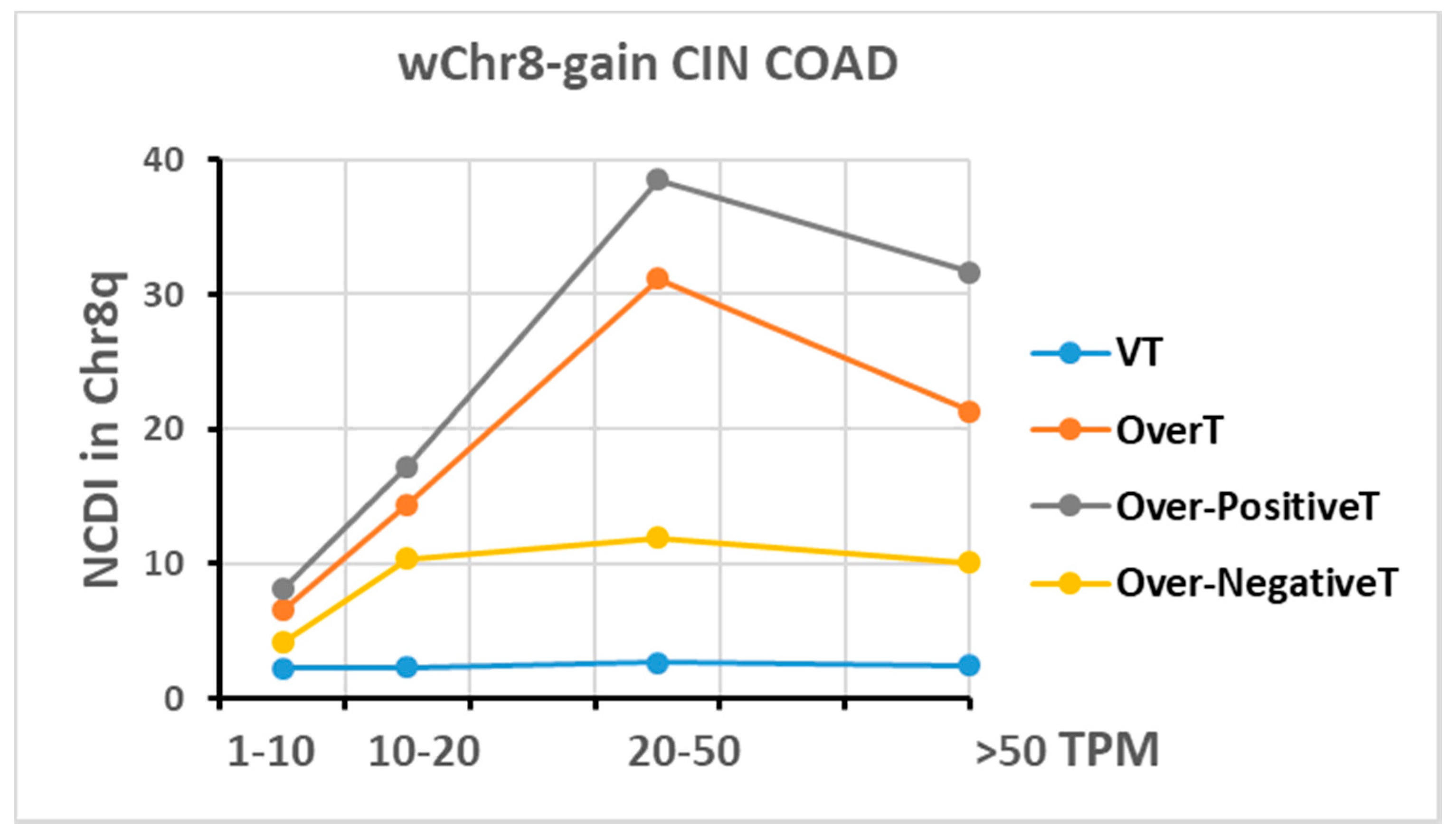
2.4. Transcriptome Analysis in “Selected COAD” Groups
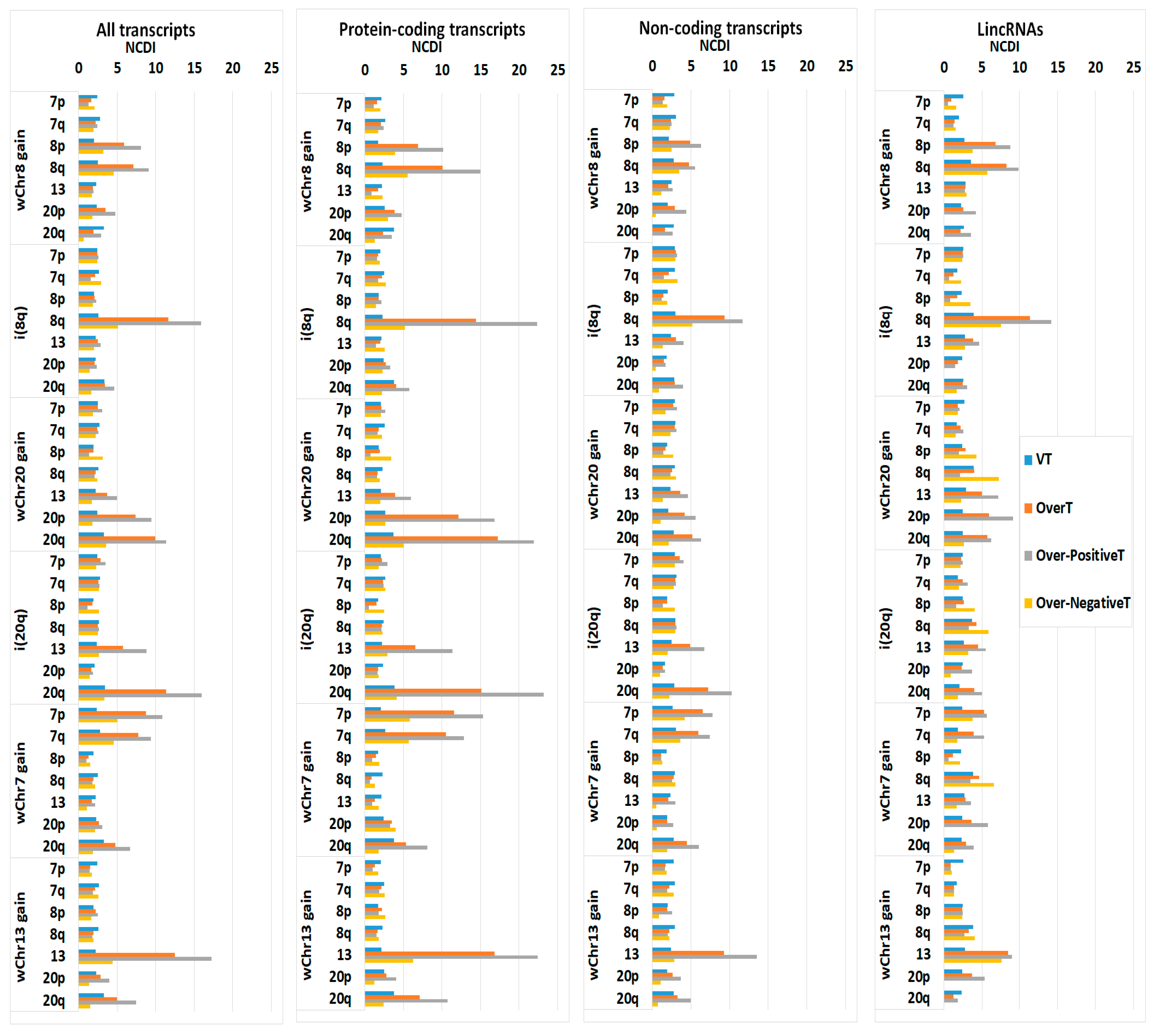
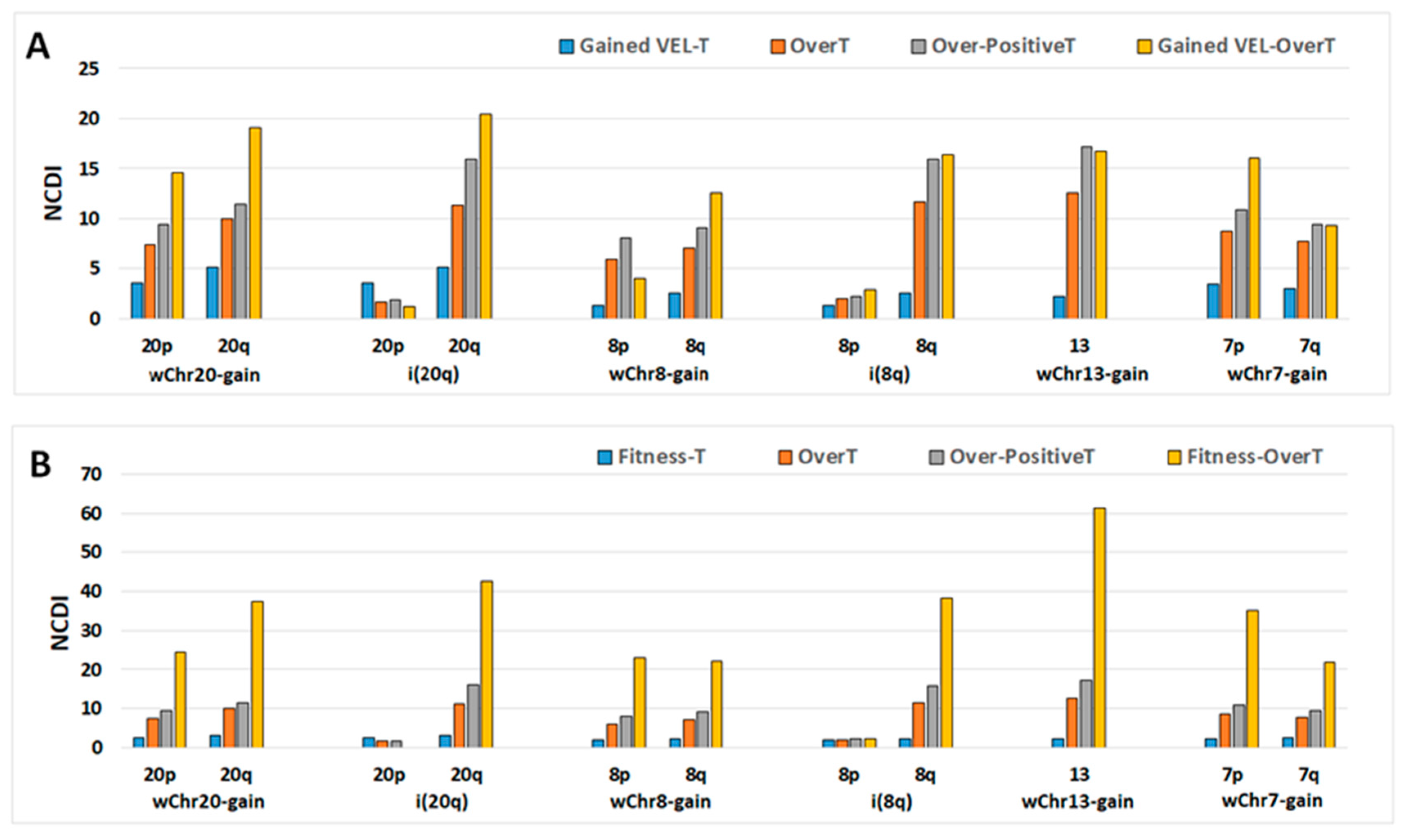
2.5. Recurrent Gained Variant Enhancer Loci
2.6. Cancer Fitness Genes
2.7. Recurrent Focal Amplifications
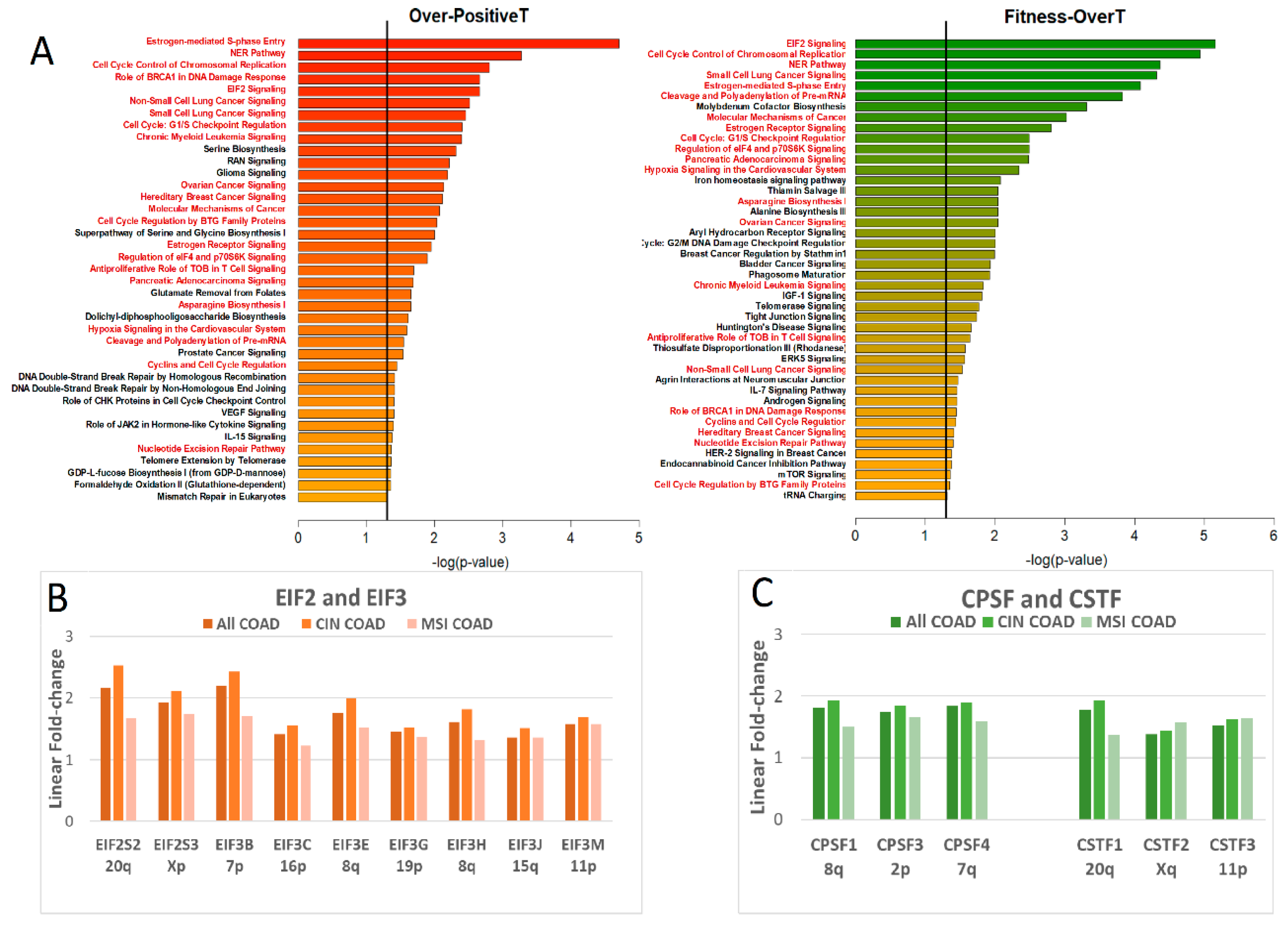
2.8. Ingenuity Pathway Analysis: Over-Positive T and Fitness-OverT
3. Discussion
4. Materials and methods
4.1. RNA-Seq Data Retrieval from GDC Data Portal
4.2. Single-Nucleotide Polymorphism (SNP) Arrays Data Retrieval from Cbioportal
4.3. Statistical Analysis of Differential Expression by RNA-Seq Data
4.4. Normalized Chromosomal Distribution Index (NCDI)
4.5. Bioinformatics and Statistical Tools
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| COAD | COlon ADenocarcinoma |
| CIN | Chromosomal INstability |
| MSI | MicroSatellite Instability |
| BCNGs | Broad Copy Number Gains |
| VT | Variable Transcripts |
| PositiveT | Positive Transcripts |
| NegativeT | Negative Transcripts |
| OverT | Overexpressed Transcripts |
| FC | Fold-Change |
| NCDI | Normalized Chromosomal Distribution Index |
| Over-PositiveT | Overexpressed Positive Transcripts |
| Over-NegativeT | Overexpressed Negative transcripts |
| VELs | Variant Enhancer Loci |
| Gained VEL-T | Transcripts linked to recurrent gained VELs |
References
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018, 173, 371–385. [Google Scholar] [CrossRef]
- Futreal, P.A.; Coin, L.; Marshall, M.; Down, T.; Hubbard, T.; Wooster, R.; Rahman, N.; Stratton, M.R. A census of human cancer genes. Nat. Rev. Cancer 2004, 4, 177–183. [Google Scholar] [CrossRef]
- Mitelman, F.; Johansson, B.; Mertens, F. The impact of translocations and gene fusions on cancer causation. Nat. Rev. Cancer 2007, 7, 233–245. [Google Scholar] [CrossRef]
- Davoli, T.; Xu, A.W.; Mengwasser, K.E.; Sack, L.M.; Yoon, J.C.; Park, P.J.; Elledge, S.J. Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell 2013, 155, 948–962. [Google Scholar] [CrossRef]
- Upender, M.B.; Habermann, J.K.; McShane, L.M.; Korn, E.L.; Barrett, J.C.; Difilippantonio, M.J.; Ried, T. Chromosome transfer induced aneuploidy results in complex dysregulation of the cellular transcriptome in immortalized and cancer cells. Cancer Res. 2004, 64, 6941–6949. [Google Scholar] [CrossRef]
- Nawata, H.; Kashino, G.; Tano, K.; Daino, K.; Shimada, Y.; Kugoh, H.; Oshimura, M.; Watanabe, M. Dysregulation of gene expression in the artificial human trisomy cells of chromosome 8 associated with transformed cell phenotypes. PLoS ONE 2011, 6, e25319. [Google Scholar] [CrossRef]
- Ried, T.; Hu, Y.; Difilippantonio, M.J.; Ghadimi, B.M.; Grade, M.; Camps, J. The consequences of chromosomal aneuploidy on the transcriptome of cancer cells. Biochim. Biophys. Acta 2012, 1819, 784–793. [Google Scholar] [CrossRef] [Green Version]
- Grade, M.; Difilippantonio, M.J.; Camps, J. Patterns of Chromosomal Aberrations in Solid Tumours. Recent Results Cancer Res. 2015, 200, 115–142. [Google Scholar]
- Taylor, A.M.; Shih, J.; Ha, G.; Gao, G.F.; Zhang, X.; Berger, A.C.; Schumacher, S.E.; Wang, C.; Hu, H.; Liu, J.; et al. Genomic and Functional Approaches to Understanding Cancer Aneuploidy. Cancer Cell 2018, 33, 676–689. [Google Scholar] [CrossRef]
- Braun, R.; Ronquist, S.; Wangsa, D.; Chen, H.; Anthuber, L.; Gemoll, T.; Wangsa, D.; Koparde, V.; Hunn, C.; Habermann, J.K.; et al. Single Chromosome Aneuploidy Induces Genome-Wide Perturbation of Nuclear Organization and Gene Expression. Neoplasia 2019, 21, 401–412. [Google Scholar] [CrossRef]
- Sack, L.M.; Davoli, T.; Li, M.Z.; Li, Y.; Xu, Q.; Naxerova, K.; Wooten, E.C.; Bernardi, R.J.; Martin, T.D.; Chen, T.; et al. Profound Tissue Specificity in Proliferation Control Underlies Cancer Drivers and Aneuploidy Patterns. Cell 2018, 173, 499–514. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.; Marcotte, E.M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 2012, 13, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, V.; Akmamedova, O.; Komurov, K. Selective DNA methylation in cancers controls collateral damage induced by large structural variations. Oncotarget 2016, 8, 71385–71392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollack, J.R.; Sørlie, T.; Perou, C.M.; Rees, C.A.; Jeffrey, S.S.; Lonning, P.E.; Tibshirani, R.; Botstein, D.; Børresen-Dale, A.L.; Brown, P.O. Microarray analysis reveals a major direct role of DNA copy number alteration in the transcriptional program of human breast tumours. Proc. Natl. Acad. Sci. USA 2002, 99, 12963–12968. [Google Scholar] [CrossRef] [PubMed]
- Tsafrir, D.; Bacolod, M.; Selvanayagam, Z.; Tsafrir, I.; Shia, J.; Zeng, Z.; Liu, H.; Krier, C.; Stengel, R.F.; Barany, F.; et al. Relationship of gene expression and chromosomal abnormalities in colorectal cancer. Cancer Res. 2006, 66, 2129–2137. [Google Scholar] [CrossRef] [PubMed]
- Grade, M.; Ghadimi, B.M.; Varma, S.; Simon, R.; Wangsa, D.; Barenboim-Stapleton, L.; Liersch, T.; Becker, H.; Ried, T.; Difilippantonio, M.J. Aneuploidy dependent massive deregulation of the cellular transcriptome and apparent divergence of the Wnt/beta-catenin signalling pathway in human rectal carcinomas. Cancer Res. 2006, 66, 267–282. [Google Scholar] [CrossRef] [PubMed]
- Grade, M.; Hörmann, P.; Becker, S.; Hummon, A.B.; Wangsa, D.; Varma, S.; Simon, R.; Liersch, T.; Becker, H.; Difilippantonio, M.J.; et al. Gene expression profiling reveals a massive, aneuploidy-dependent transcriptional deregulation and distinct differences between lymph node-negative and lymph node-positive colon carcinomas. Cancer Res. 2007, 67, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Fehrmann, R.S.; Karjalainen, J.M.; Krajewska, M.; Westra, H.J.; Maloney, D.; Simeonov, A.; Pers, T.H.; Hirschhorn, J.N.; Jansen, R.C.; Schultes, E.A.; et al. Gene expression analysis identifies global gene dosage sensitivity in cancer. Nat. Genet. 2015, 47, 115–125. [Google Scholar] [CrossRef]
- Condorelli, D.F.; Spampinato, G.; Valenti, G.; Musso, N.; Castorina, S.; Barresi, V. Positive Caricature Transcriptomic Effects Associated with Broad Genomic Aberrations in Colorectal Cancer. Sci. Rep. 2018, 8, 14826. [Google Scholar] [CrossRef] [PubMed]
- Hart, T.; Chandrashekhar, M.; Aregger, M.; Steinhart, Z.; Brown, K.R.; MacLeod, G.; Mis, M.; Zimmermann, M.; Fradet-Turcotte, A.; Sun, S.; et al. High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell 2015, 163, 1515–1526. [Google Scholar] [CrossRef] [Green Version]
- Barresi, V.; Castorina, S.; Musso, N.; Capizzi, C.; Luca, T.; Privitera, G.; Condorelli, D.F. Chromosomal instability analysis and regional tumour heterogeneity in colon cancer. Cancer Genet. 2017, 210, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Barbouti, A.; Stankiewicz, P.; Nusbaum, C.; Cuomo, C.; Cook, A.; Höglund, M.; Johansson, B.; Hagemeijer, A.; Park, S.S.; Mitelman, F.; et al. The breakpoint region of the most common isochromosome, i(17q), in human neoplasia is characterized by a complex genomic architecture with large, palindromic, low-copy repeats. Am. J. Hum. Genet. 2004, 74, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sethi, N.S.; Hinoue, T.; Schneider, B.G.; Cherniack, A.D.; Sanchez-Vega, F.; Seoane, J.A.; Farshidfar, F.; Bowlby, R.; Islam, M.; et al. Comparative Molecular Analysis of Gastrointestinal Adenocarcinomas. Cancer Cell 2018, 33, 721–735. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, D.J.; Chen, Y.S.; Smyth, G.K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012, 40, 4288–4297. [Google Scholar] [CrossRef] [Green Version]
- Akhtar-Zaidi, B.; Cowper-Sal-lari, R.; Corradin, O.; Saiakhova, A.; Bartels, C.F.; Balasubramanian, D.; Myeroff, L.; Lutterbaugh, J.; Jarrar, A.; Kalady, M.F.; et al. Epigenomic enhancer profiling defines a signature of colon cancer. Science 2012, 336, 736–739. [Google Scholar] [CrossRef]
- Cohen, A.J.; Saiakhova, A.; Corradin, O.; Luppino, J.M.; Lovrenert, K.; Bartels, C.F.; Morrow, J.J.; Mack, S.C.; Dhillon, G.; Beard, L.; et al. Hotspots of aberrant enhancer activity punctuate the colorectal cancer epigenome. Nat. Commun. 2017, 8, 14400. [Google Scholar] [CrossRef] [Green Version]
- Behan, F.M.; Iorio, F.; Picco, G.; Gonçalves, E.; Beaver, C.M.; Migliardi, G.; Santos, R.; Rao, Y.; Sassi, F.; Pinnelli, M.; et al. Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature 2019, 568, 511–516. [Google Scholar] [CrossRef]
- Mandel, C.R.; Bai, Y.; Tong, L. Protein factors in pre-mRNA 3’-end processing. Cell Mol. Life Sci. 2008, 65, 1099–1122. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Di Giammartino, D.C.; Taylor, D.; Sarkeshik, A.; Rice, W.J.; Yates, J.R., 3rd; Frank, J.; Manley, J.L. Molecular architecture of the human pre-mRNA 3′ processing complex. Mol. Cell 2009, 33, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Tian, B.; Manley, J.L. Alternative polyadenylation of mRNA precursors. Nat. Rev. Mol. Cell. Biol. 2017, 18, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S.; Kranzusch, P.J.; Cate, J.H. eIF3 targets cell-proliferation messenger RNAs for translational activation or repression. Nature 2015, 522, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Pittman, A.M.; Naranjo, S.; Jalava, S.E.; Twiss, P.; Ma, Y.; Olver, B.; Lloyd, A.; Vijayakrishnan, J.; Qureshi, M.; Broderick, P.; et al. Allelic variation at the 8q23.3 colorectal cancer risk locus functions as a cis-acting regulator of EIF3H. PLoS Genet. 2010, 6, e1001126. [Google Scholar] [CrossRef] [PubMed]
- Gandin, V.; Miluzio, A.; Barbieri, A.M.; Beugnet, A.; Kiyokawa, H.; Marchisio, P.C.; Biffo, S. Eukaryotic initiation factor 6 is rate-limiting in translation, growth and transformation. Nature 2008, 455, 684–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Q.; Fan, W.; Zheng, Z.; Lin, S.; Liu, C.; Wang, R.; Li, W.; Zuo, Y.; Sun, Y.; Hu, S.; et al. Cleavage and polyadenylation specific factor 4 promotes colon cancer progression by transcriptionally activating hTERT. Biochim. Biophys. Acta Mol. Cell. Res. 2019, 1866, 1533–1543. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. Cancer Genome Atlas Research Network. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Tomczak, K.; Czerwińska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemp. Oncol. 2015, 19, A68–A77. [Google Scholar] [CrossRef]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef]
- Soneson, C. compcodeR—An R package for benchmarking differential expression methods for RNA-seq data. Bioinformatics 2014, 30, 2517–2518. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Oliveros, J.C. Venny. An Interactive Tool for Comparing Lists with Venn’s Diagrams. 2007. Available online: https://bioinfogp.cnb.csic.es/tools/venny/index.html (accessed on 21 March 2019).










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chr20p | Chr20q | Chr8p | Chr8q | Chr13 | Chr7p | Chr7q | |
|---|---|---|---|---|---|---|---|
| Fold-enrichment of OverT among PositiveT (Hypergeometric p-value) | 1.11 (0.00040) | 1.00 (0.49343) | 1.16 (0.00109) | 1.08 (0.02179) | 1.12 (2.93 × 10−7) | 1.10 (0.02120) | 1.05 (0.14272) |
| Fold-depletion of OverT among NegativeT (Hypergeometric p-value) | −2.69 (4.55 × 10−11) | −1.6 (3.30 × 10−7) | −1.45 (0.00109) | −1.19 (0.01940) | −1.8 (5.94 × 10−11) | −1.38 (0.00331) | −1.3 (0.00653) |
| Fold-enrichment of OverT among Gained VEL-T (Hypergeometric p-value) | 2.14 (2.23 × 10−6) | 1.45 (0.00021) | 1.99 (0.03708) | 3.29 (1.87 × 10−13) | 2.25 (2.14 × 10−7) | 2.33 (1.85 × 10−6) | 1.76 (0.00151) |
| Fold-enrichment of OverT among Fitness-T (Hypergeometric p-value) | 2.43 (5.03 × 10−7) | 2.22 (5.55 × 10−15) | 4.41 (2.31 × 10−13) | 3.14 (1.5 × 10−12) | 3.58 (5.81 × 10−26) | 2.61 (4.88 × 10−6) | 1.65 (0.00929) |
| Fold-enrichment of OverT among AmpT (Hypergeometric p-value) | 9.75 (6.56 × 10−6) | 1.15 (0.109) | 3.73 (0.250) | NC 1 | 2.32 (0.002) | NC 1 | 1.07 (0.523) |
| 1 not computable for the absence of AmpT. | |||||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Condorelli, D.F.; Privitera, A.P.; Barresi, V. Chromosomal Density of Cancer Up-Regulated Genes, Aberrant Enhancer Activity and Cancer Fitness Genes Are Associated with Transcriptional Cis-Effects of Broad Copy Number Gains in Colorectal Cancer. Int. J. Mol. Sci. 2019, 20, 4652. https://doi.org/10.3390/ijms20184652
Condorelli DF, Privitera AP, Barresi V. Chromosomal Density of Cancer Up-Regulated Genes, Aberrant Enhancer Activity and Cancer Fitness Genes Are Associated with Transcriptional Cis-Effects of Broad Copy Number Gains in Colorectal Cancer. International Journal of Molecular Sciences. 2019; 20(18):4652. https://doi.org/10.3390/ijms20184652
Chicago/Turabian StyleCondorelli, Daniele Filippo, Anna Provvidenza Privitera, and Vincenza Barresi. 2019. "Chromosomal Density of Cancer Up-Regulated Genes, Aberrant Enhancer Activity and Cancer Fitness Genes Are Associated with Transcriptional Cis-Effects of Broad Copy Number Gains in Colorectal Cancer" International Journal of Molecular Sciences 20, no. 18: 4652. https://doi.org/10.3390/ijms20184652
APA StyleCondorelli, D. F., Privitera, A. P., & Barresi, V. (2019). Chromosomal Density of Cancer Up-Regulated Genes, Aberrant Enhancer Activity and Cancer Fitness Genes Are Associated with Transcriptional Cis-Effects of Broad Copy Number Gains in Colorectal Cancer. International Journal of Molecular Sciences, 20(18), 4652. https://doi.org/10.3390/ijms20184652