Risperidone Treatment after Transient Ischemia Induces Hypothermia and Provides Neuroprotection in the Gerbil Hippocampus by Decreasing Oxidative Stress

 , ,
, ,  and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Effects of RIS Against TI Injury Under Uncontrolled Body Temperature (UBT)
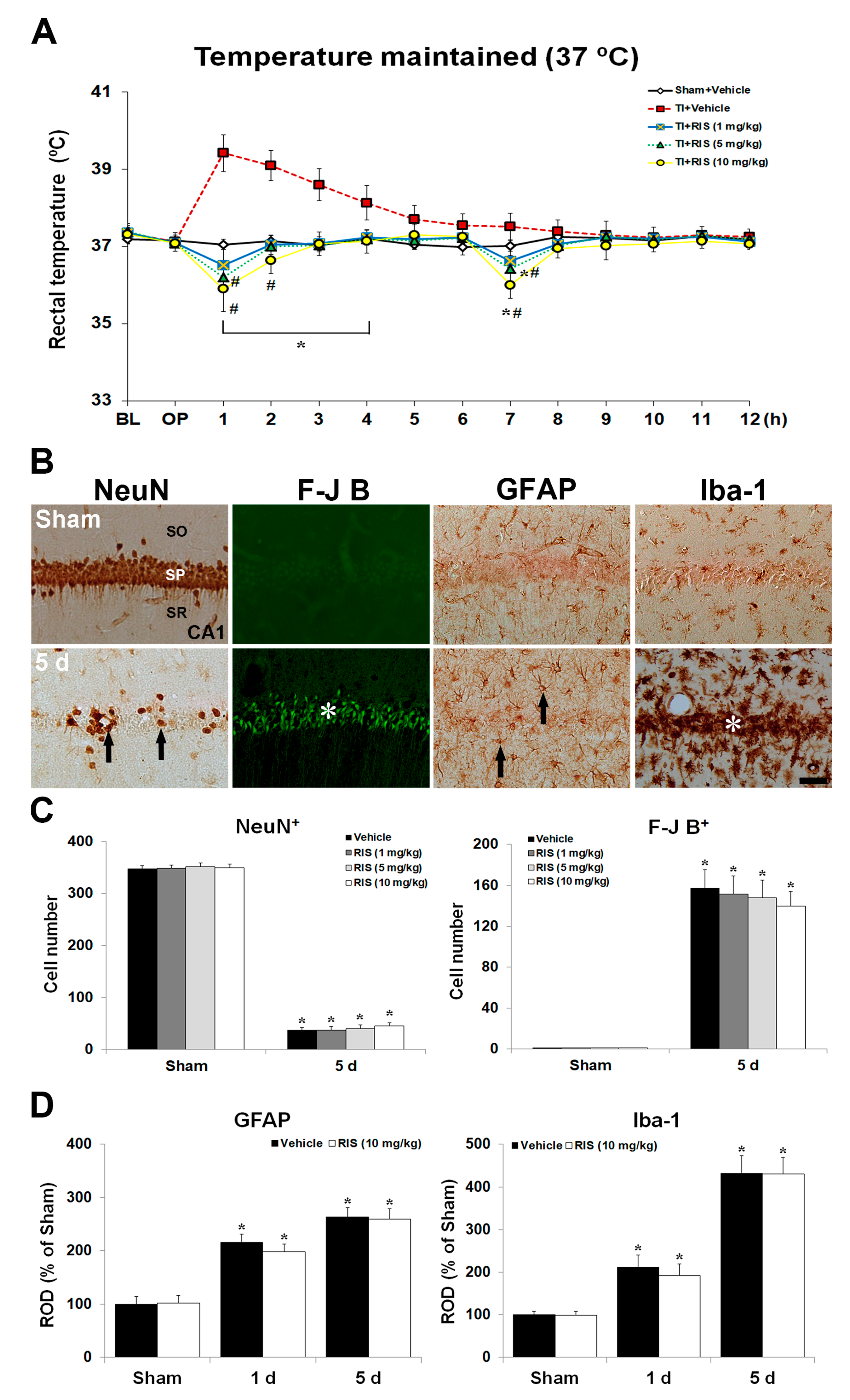
2.1.1. Body Temperature under UBT Condition after TI
2.1.2. Neuronal Nuclear Antigen (NeuN) Positive (+) and Fluoro-Jade B (F-J B)+ Neurons
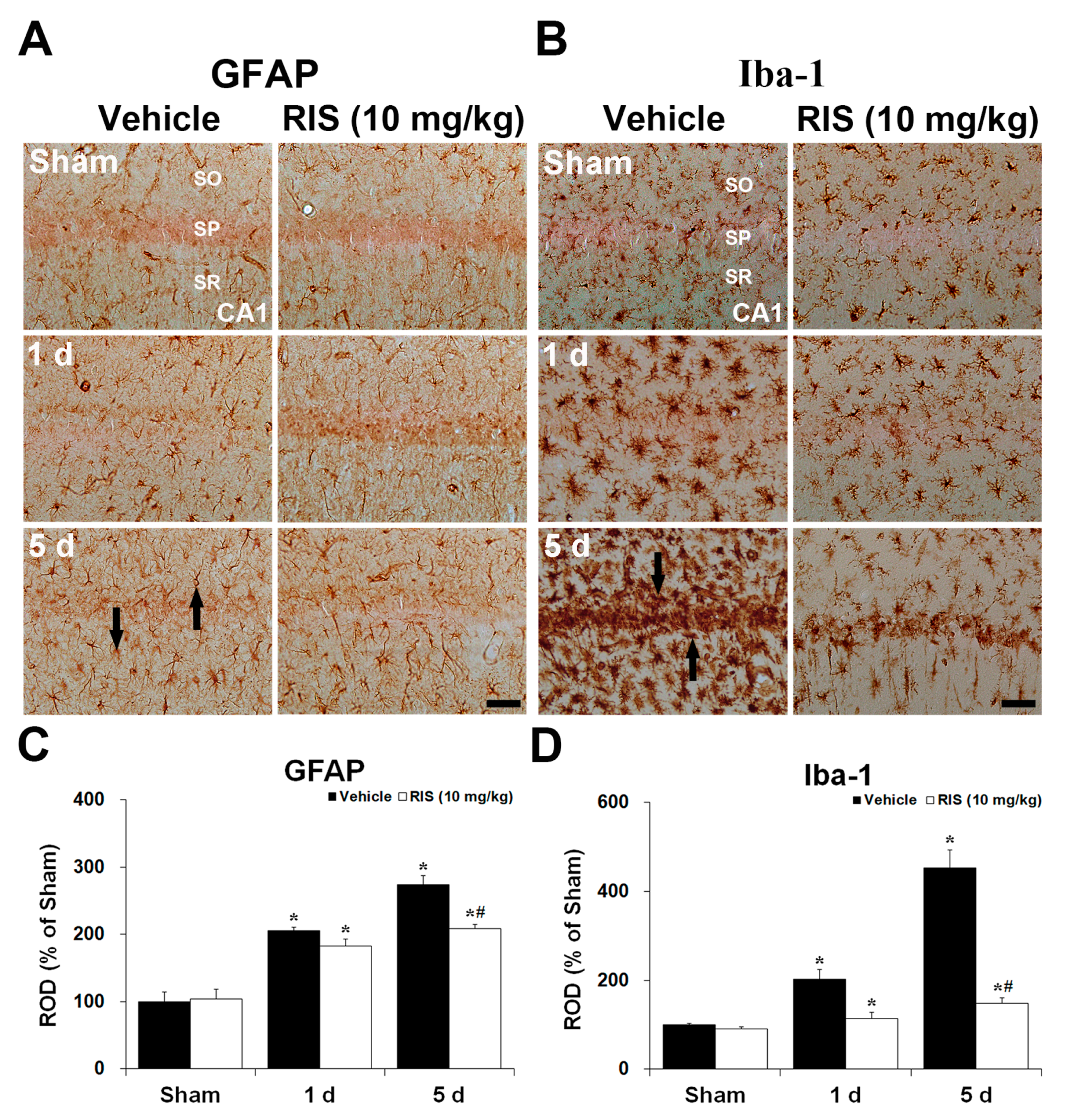
2.1.3. Glial Fibrillary Acidic Protein (GFAP)+ and Ionized Calcium-Binding Adapter Molecule 1 (Iba-1)+ Cells
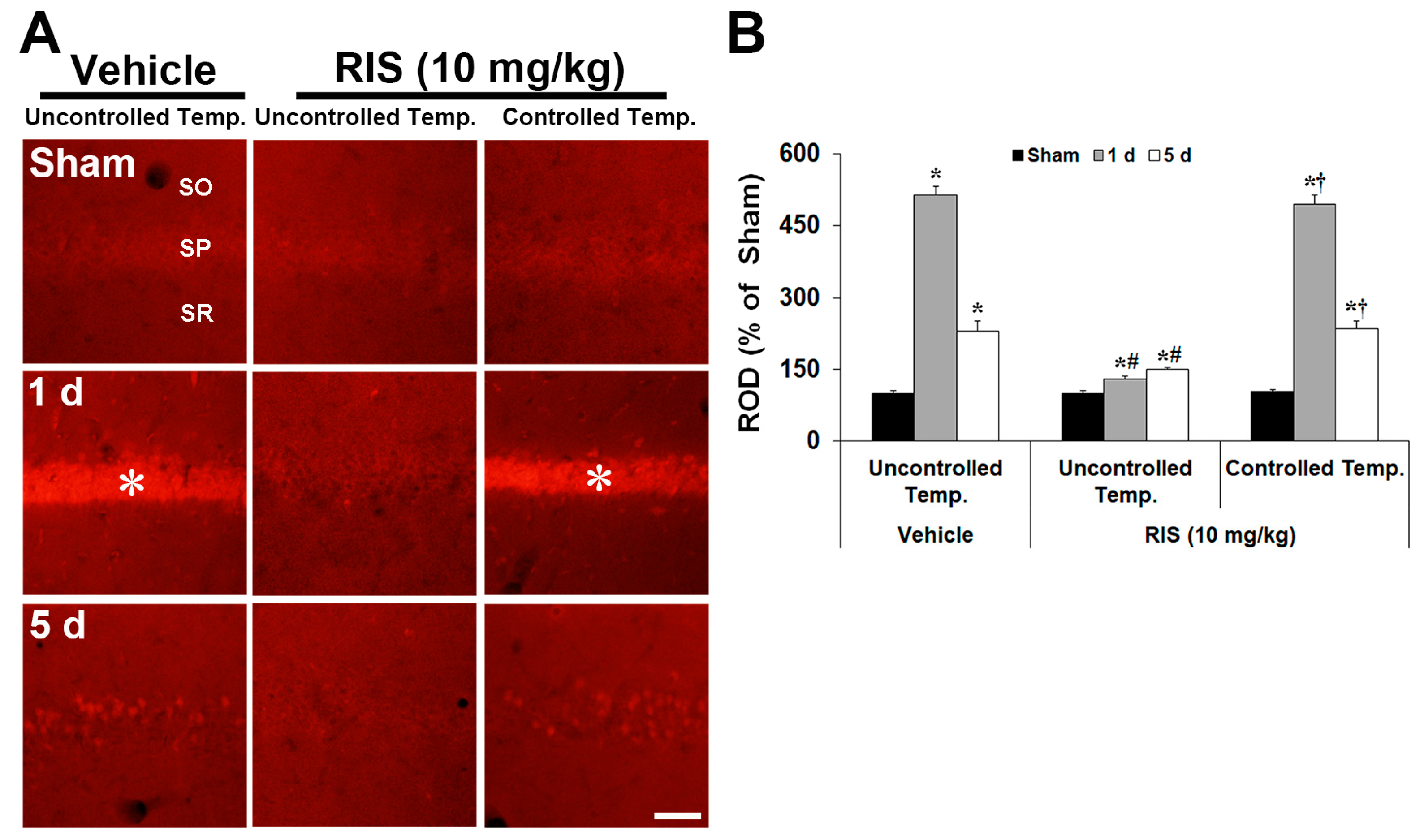
2.2. Abolishment of RIS-Mediated Neuroprotection under Controlled Body Temperature (CBT)
2.2.1. Body Temperature under CBT Condition after TI
2.2.2. NeuN+ Neurons, F-J B+ Cells, GFAP+ Astrocytes, and Iba-1+ Microglia
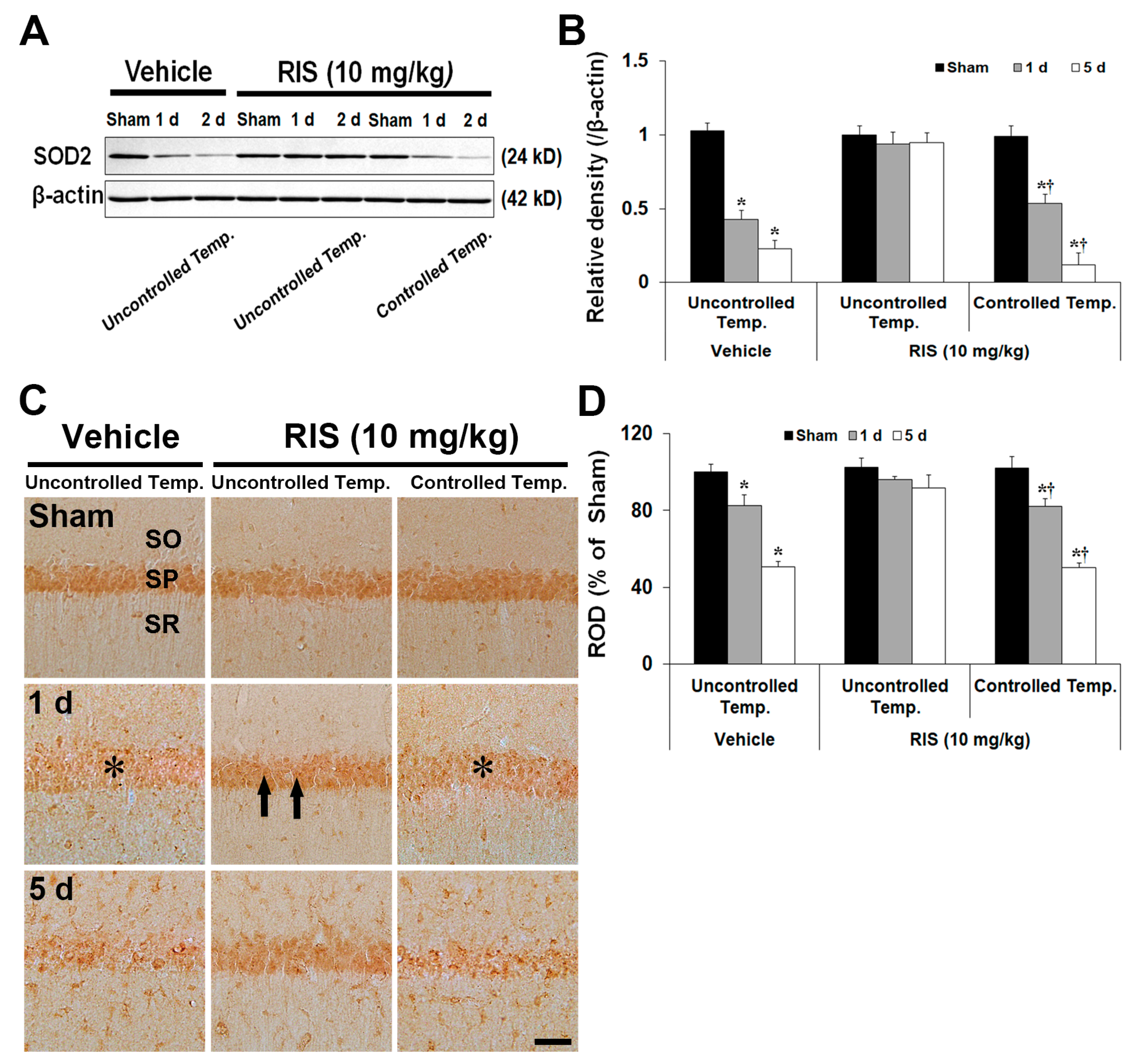
2.3. Effects of RIS Against TI-Induced Oxidative Stress under UBT and CBT Conditions
2.3.1. Superoxide Anion Production
2.3.2. 8-Hydroxy-2′-deoxyguanosine (8-OHdG) and 4-hydroxy-2-nonenal (4-HNE) Immunoreactivity
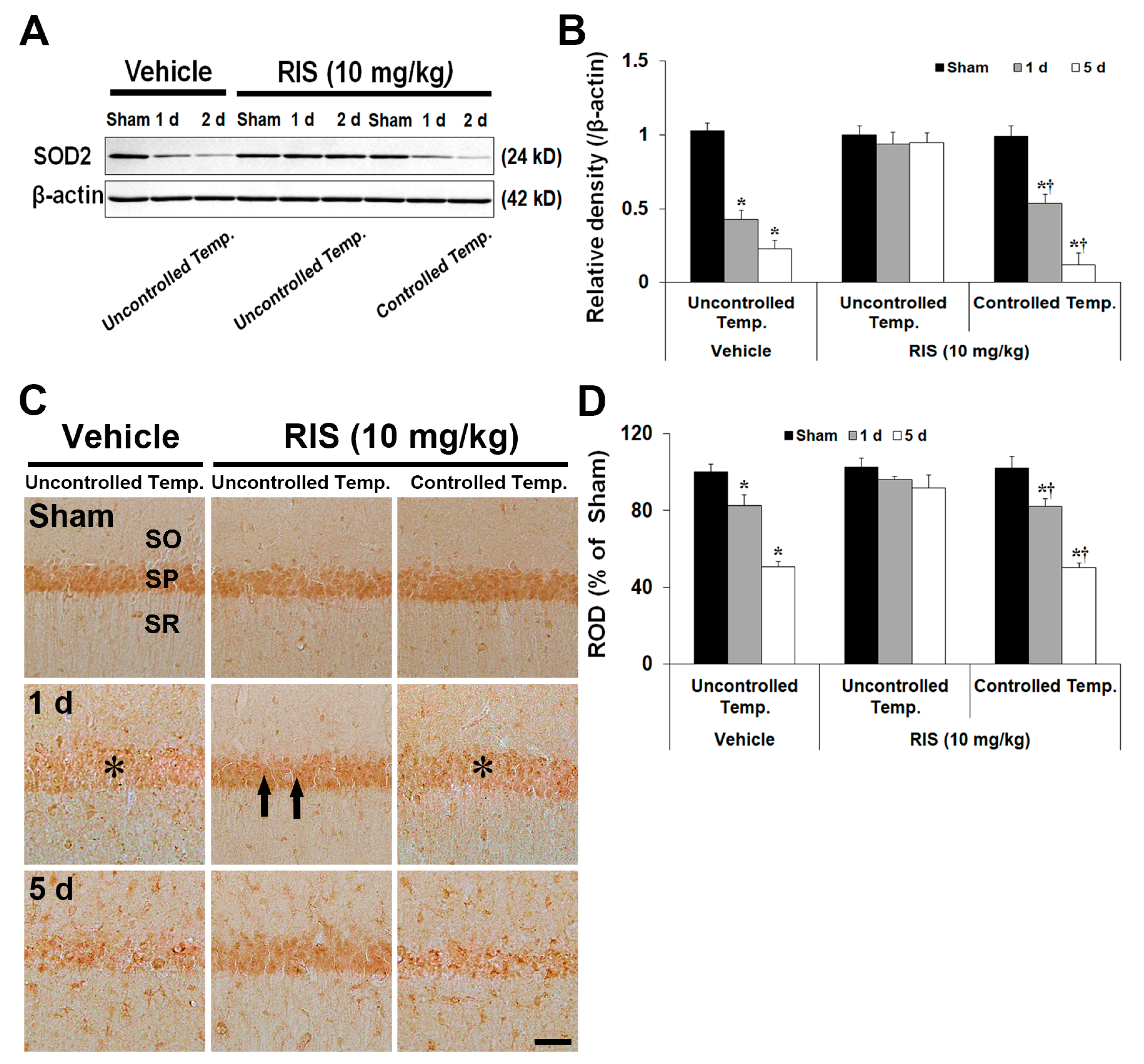
2.3.3. Superoxide Dismutase 2 (SOD2) Level and Immunoreactivity
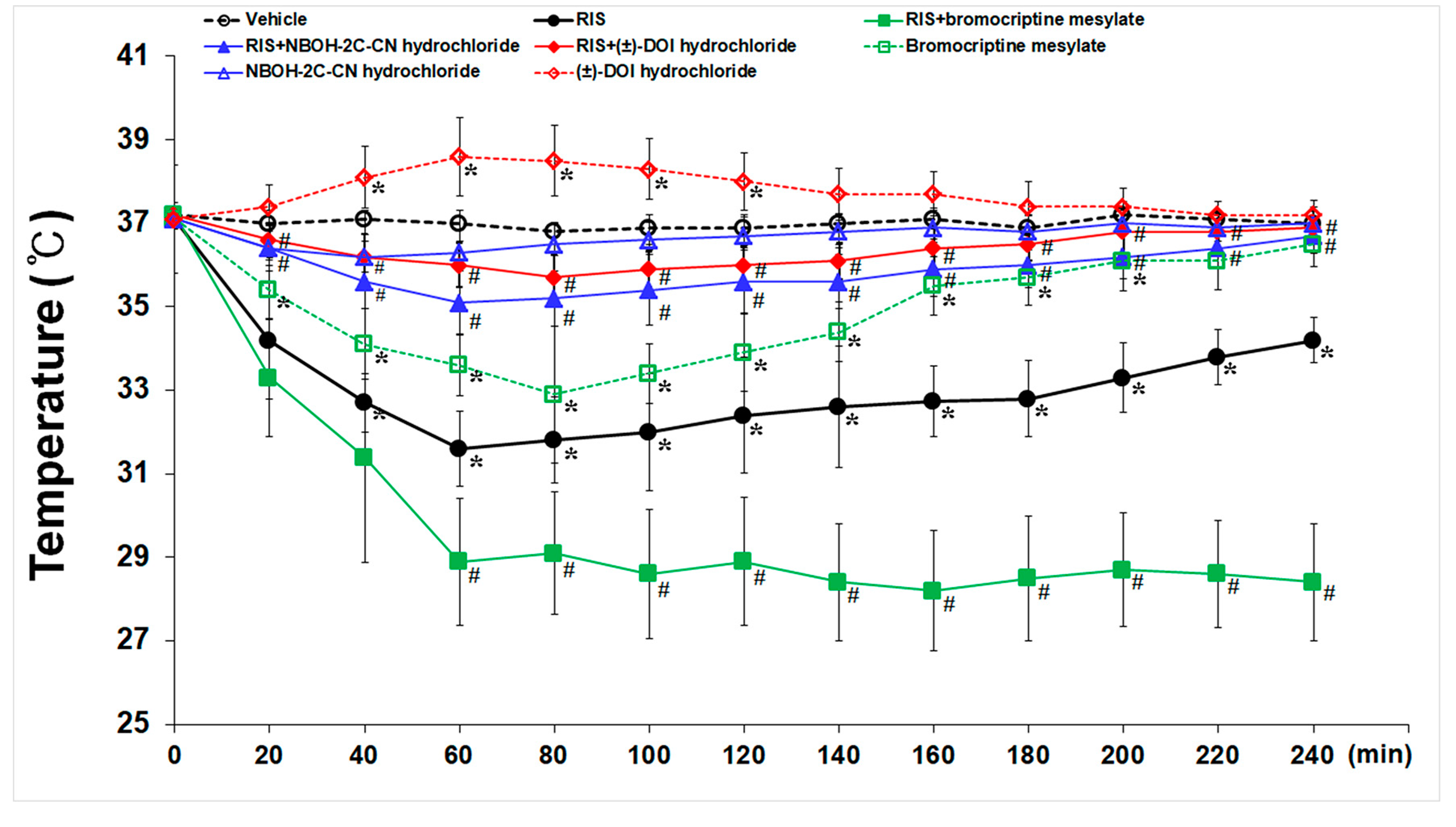
2.4. Effects of 5-HT2A- and D2- Receptors on RIS-Induced Hypothermia
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Experimental Groups, Induction of TI, and RIS Treatment
4.3. Histological Tissue Preparation
4.4. F-J B Histofluorescence Staining
4.5. Superoxide Anion Production Detection
4.6. Immunohistochemistry
4.7. Western Blotting for SOD2
4.8. Effects of 5-HT2A and D2 Agonists against RIS-Induced Hypothermia
4.9. Data Analysis
4.10. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Sutherland, B.A.; Minnerup, J.; Balami, J.S.; Arba, F.; Buchan, A.M.; Kleinschnitz, C. Neuroprotection for ischaemic stroke: Translation from the bench to the bedside. Int. Stroke Soc. 2012, 7, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Busto, R.; Dietrich, W.D.; Globus, M.Y.; Ginsberg, M.D. Postischemic moderate hypothermia inhibits CA1 hippocampal ischemic neuronal injury. Neurosci. Lett. 1989, 101, 299–304. [Google Scholar] [CrossRef]
- Nurse, S.; Corbett, D. Neuroprotection after several days of mild, drug-induced hypothermia. J. Int. Society Cereb. Blood Flow Metab. 1996, 16, 474–480. [Google Scholar] [CrossRef] [PubMed]
- Fisher, M.; Feuerstein, G.; Howells, D.W.; Hurn, P.D.; Kent, T.A.; Savitz, S.I.; Lo, E.H. Update of the stroke therapy academic industry roundtable preclinical recommendations. Stroke 2009, 40, 2244–2250. [Google Scholar] [CrossRef] [PubMed]
- Lyden, P.D.; Krieger, D.; Yenari, M.; Dietrich, W.D. Therapeutic hypothermia for acute stroke. Int. J. Stroke 2006, 1, 9–19. [Google Scholar] [CrossRef]
- van Marum, R.J.; Wegewijs, M.A.; Loonen, A.J.; Beers, E. Hypothermia following antipsychotic drug use. Eur. J. Clin. Pharmacol. 2007, 63, 627–631. [Google Scholar] [CrossRef] [Green Version]
- Zonnenberg, C.; Bueno-de-Mesquita, J.M.; Ramlal, D.; Blom, J.D. Hypothermia due to antipsychotic medication: A systematic review. Front. Psychiatry 2017, 8, 165. [Google Scholar] [CrossRef]
- Schotte, A.; Janssen, P.F.; Gommeren, W.; Luyten, W.H.; Van Gompel, P.; Lesage, A.S.; De Loore, K.; Leysen, J.E. Risperidone compared with new and reference antipsychotic drugs: In vitro and in vivo receptor binding. Psychopharmacology (Berl) 1996, 124, 57–73. [Google Scholar] [CrossRef]
- Corena-McLeod, M. Comparative pharmacology of risperidone and paliperidone. Drugs R D 2015, 15, 163–174. [Google Scholar] [CrossRef]
- Razaq, M.; Samma, M. A case of risperidone-induced hypothermia. Am. J. Ther. 2004, 11, 229–230. [Google Scholar] [CrossRef]
- Brevik, A.; Farver, D. Atypical antipsychotic induced mild hypothermia. S D J. Med. 2003, 56, 67–70. [Google Scholar] [PubMed]
- Colbourne, F.; Li, H.; Buchan, A.M.; Clemens, J.A. Continuing postischemic neuronal death in CA1: Influence of ischemia duration and cytoprotective doses of NBQX and SNX-111 in rats. Stroke 1999, 30, 662–668. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.C.; Park, J.H.; Ahn, J.H.; Kim, I.H.; Park, O.K.; Lee, J.C.; Yoo, K.Y.; Choi, J.H.; Lee, C.H.; Hwang, I.K.; et al. Neuroprotection of posttreatment with risperidone, an atypical antipsychotic drug, in rat and gerbil models of ischemic stroke and the maintenance of antioxidants in a gerbil model of ischemic stroke. J. Neurosci. Res. 2014, 92, 795–807. [Google Scholar] [CrossRef] [PubMed]
- Kouchoukos, N.T.; Daily, B.B.; Wareing, T.H.; Murphy, S.F. Hypothermic circulatory arrest for cerebral protection during combined carotid and cardiac surgery in patients with bilateral carotid artery disease. Ann. Surg. 1994, 219, 699–705. [Google Scholar] [CrossRef] [PubMed]
- Saccani, S.; Beghi, C.; Fragnito, C.; Barboso, G.; Fesani, F. Carotid endarterectomy under hypothermic extracorporeal circulation: A method of brain protection for special patients. J. Cardiovasc. Surg. 1992, 33, 311–314. [Google Scholar]
- Yenari, M.A.; Han, H.S. Neuroprotective mechanisms of hypothermia in brain ischaemia. Nat. Rev. Neurosci. 2012, 13, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Yenari, M.A. Therapeutic hypothermia: Neuroprotective mechanisms. Front. Biosci. 2007, 12, 816–825. [Google Scholar] [CrossRef]
- Yenari, M.; Kitagawa, K.; Lyden, P.; Perez-Pinzon, M. Metabolic downregulation: A key to successful neuroprotection? Stroke 2008, 39, 2910–2917. [Google Scholar] [CrossRef]
- Morrison, L.J.; Deakin, C.D.; Morley, P.T.; Callaway, C.W.; Kerber, R.E.; Kronick, S.L.; Lavonas, E.J.; Link, M.S.; Neumar, R.W.; Otto, C.W.; et al. Part 8: Advanced life support: 2010 international consensus on cardiopulmonary resuscitation and emergency cardiovascular care science with treatment recommendations. Circulation 2010, 122, S345–S421. [Google Scholar] [CrossRef]
- Faridar, A.; Bershad, E.M.; Emiru, T.; Iaizzo, P.A.; Suarez, J.I.; Divani, A.A. Therapeutic hypothermia in stroke and traumatic brain injury. Front. Neurol. 2011, 2, 80. [Google Scholar] [CrossRef]
- Schwab, S.; Georgiadis, D.; Berrouschot, J.; Schellinger, P.D.; Graffagnino, C.; Mayer, S.A. Feasibility and safety of moderate hypothermia after massive hemispheric infarction. Stroke 2001, 32, 2033–2035. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wang, H.; Zhao, J.; Chen, C.; Leak, R.K.; Xu, Y.; Vosler, P.; Chen, J.; Gao, Y.; Zhang, F. Drug-induced hypothermia in stroke models: Does it always protect? CNS Neurol. Disord. Drug Targets 2013, 12, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, T.; Lewen, A.; Noshita, N.; Gasche, Y.; Chan, P.H. Effects of global ischemia duration on neuronal, astroglial, oligodendroglial, and microglial reactions in the vulnerable hippocampal CA1 subregion in rats. J. Neurotrauma 2002, 19, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Stoll, G.; Jander, S.; Schroeter, M. Inflammation and glial responses in ischemic brain lesions. Prog. Neurobiol. 1998, 56, 149–171. [Google Scholar] [CrossRef]
- Muir, K.W.; Tyrrell, P.; Sattar, N.; Warburton, E. Inflammation and ischaemic stroke. Curr. Opin. Neurol. 2007, 20, 334–342. [Google Scholar] [CrossRef]
- Seo, J.W.; Kim, J.H.; Seo, M.; Han, H.S.; Park, J.; Suk, K. Time-dependent effects of hypothermia on microglial activation and migration. J. neuroinflammation 2012, 9, 164. [Google Scholar] [CrossRef] [PubMed]
- Diestel, A.; Troeller, S.; Billecke, N.; Sauer, I.M.; Berger, F.; Schmitt, K.R. Mechanisms of hypothermia-induced cell protection mediated by microglial cells in vitro. Eur. J. Neurosci. 2010, 31, 779–787. [Google Scholar] [CrossRef]
- Matsui, T.; Kida, H.; Iha, T.; Obara, T.; Nomura, S.; Fujimiya, T.; Suzuki, M. Effects of hypothermia on ex vivo microglial production of pro- and anti-inflammatory cytokines and nitric oxide in hypoxic-ischemic brain-injured mice. Folia. Neuropathol. 2014, 52, 151–158. [Google Scholar] [CrossRef]
- George, S.; Bennet, L.; Weaver-Mikaere, L.; Fraser, M.; Bouwmans, J.; Mathai, S.; Skinner, S.J.; Gunn, A.J. White matter protection with insulin-like growth factor 1 and hypothermia is not additive after severe reversible cerebral ischemia in term fetal sheep. Dev. Neurosci. 2011, 33, 280–287. [Google Scholar] [CrossRef]
- Quincozes-Santos, A.; Abib, R.T.; Leite, M.C.; Bobermin, D.; Bambini-Junior, V.; Goncalves, C.A.; Riesgo, R.; Gottfried, C. Effect of the atypical neuroleptic risperidone on morphology and S100B secretion in C6 astroglial lineage cells. Mol. Cell Biochem. 2008, 314, 59–63. [Google Scholar] [CrossRef]
- Kato, R.; Sato, J.; Suzuki, H. Anesthesia for cesarean section in a parturient taking risperidone and haloperidol. Masui. Jpn. J. Anesthesiol. 2005, 54, 301–303. [Google Scholar]
- Chan, P.H. Reactive oxygen radicals in signaling and damage in the ischemic brain. J. Cereb. Blood Flow Metab. 2001, 21, 2–14. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, T.; Chan, P.H. Reactive oxygen radicals and pathogenesis of neuronal death after cerebral ischemia. Antioxid. Redox. Signal. 2003, 5, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Altinkilic, S.; Naziroglu, M.; Uguz, A.C.; Ozcankaya, R. Fish oil and antipsychotic drug risperidone modulate oxidative stress in PC12 cell membranes through regulation of cytosolic calcium ion release and antioxidant system. J. Membr. Biol. 2010, 235, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Stojkovic, T.; Radonjic, N.V.; Velimirovic, M.; Jevtic, G.; Popovic, V.; Doknic, M.; Petronijevic, N.D. Risperidone reverses phencyclidine induced decrease in glutathione levels and alterations of antioxidant defense in rat brain. Prog. Neuropsychopharmacol. Biol. Psychiatry 2012, 39, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.W.; Lewen, A.; Copin, J.; Watson, B.D.; Chan, P.H. The cytosolic antioxidant, copper/zinc superoxide dismutase, attenuates blood-brain barrier disruption and oxidative cellular injury after photothrombotic cortical ischemia in mice. Neuroscience 2001, 105, 1007–1018. [Google Scholar] [CrossRef]
- Yan, B.C.; Park, J.H.; Lee, C.H.; Yoo, K.Y.; Choi, J.H.; Lee, Y.J.; Cho, J.H.; Baek, Y.Y.; Kim, Y.M.; Won, M.H. Increases of antioxidants are related to more delayed neuronal death in the hippocampal CA1 region of the young gerbil induced by transient cerebral ischemia. Brain Res. 2011, 1425, 142–154. [Google Scholar] [CrossRef]
- Mazzola-Pomietto, P.; Aulakh, C.S.; Wozniak, K.M.; Hill, J.L.; Murphy, D.L. Evidence that 1-(2,5-dimethoxy-4-iodophenyl)-2-aminopropane (DOI)-induced hyperthermia in rats is mediated by stimulation of 5-HT2A receptors. Psychopharmacology 1995, 117, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Salmi, P.; Ahlenius, S. Evidence for functional interactions between 5-HT1A and 5-HT2A receptors in rat thermoregulatory mechanisms. Pharmacol. Toxicol. 1998, 82, 122–127. [Google Scholar] [CrossRef]
- Zarrindast, M.R.; Mahmoudi, M. Bromocriptine-induced hypothermia: D-2 receptor involvement. Arch. Int. de Pharmacodyn. et de Ther. 1989, 298, 38–49. [Google Scholar]
- Perera, M.A.L.; Yogaratnam, J. De Novo delayed onset hypothermia secondary to therapeutic doses of risperidone in bipolar affective disorder. Ther. Adv. Psychopharm 2014, 4, 70–74. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, M.J.; Hicks, C.A.; Ward, M.A.; Cardwell, G.P.; Reymann, J.M.; Allain, H.; Bentue-Ferrer, D. Dopamine D2 receptor agonists protect against ischaemia-induced hippocampal neurodegeneration in global cerebral ischaemia. Eur. J. Pharmacol. 1998, 352, 37–46. [Google Scholar] [CrossRef]
- Guide for the Care and Use of Laboratory Animals, 8th ed.; National Academies Press (US): Washington, DC, USA, 2011. Available online: https://www.ncbi.nlm.nih.gov/books/NBK54050/ (accessed on 18 September 2019).
- Lee, J.C.; Park, J.H.; Kim, I.H.; Cho, G.S.; Ahn, J.H.; Tae, H.J.; Choi, S.Y.; Cho, J.H.; Kim, D.W.; Kwon, Y.G.; et al. Neuroprotection of ischemic preconditioning is mediated by thioredoxin 2 in the hippocampal CA1 region following a subsequent transient cerebral ischemia. Brain Pathol. 2017, 27, 276–291. [Google Scholar] [CrossRef] [PubMed]
- Yulug, B.; Yildiz, A.; Guzel, O.; Kilic, E.; Schabitz, W.R. Risperidone attenuates brain damage after focal cerebral ischemia in vivo. Brain Res. Bull. 2006, 69, 656–659. [Google Scholar] [CrossRef] [PubMed]
- Pulsinelli, W.A. Selective neuronal vulnerability: Morphological and molecular characteristics. Prog. brain Res. 1985, 63, 29–37. [Google Scholar] [PubMed]
- Lee, J.C.; Cho, J.H.; Cho, G.S.; Ahn, J.H.; Park, J.H.; Kim, I.H.; Cho, J.H.; Tae, H.J.; Cheon, S.H.; Ahn, J.Y.; et al. Effect of transient cerebral ischemia on the expression of receptor for advanced glycation end products (RAGE) in the gerbil hippocampus proper. Neurochem. Res. 2014, 39, 1553–1563. [Google Scholar] [CrossRef] [PubMed]
- Szocs, K.; Lassegue, B.; Sorescu, D.; Hilenski, L.L.; Valppu, L.; Couse, T.L.; Wilcox, J.N.; Quinn, M.T.; Lambeth, J.D.; Griendling, K.K. Upregulation of Nox-based NAD(P)H oxidases in restenosis after carotid injury. Arter. Thromb. Vasc. Biol. 2002, 22, 21–27. [Google Scholar] [CrossRef] [PubMed]
- McKracken, E.; Graham, D.I.; Nilsen, M.; Stewart, J.; Nicoll, J.A.; Horsburgh, K. 4-Hydroxynonenal immunoreactivity is increased in human hippocampus after global ischemia. Brain Pathol. 2001, 11, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Baik, S.C.; Youn, H.S.; Chung, M.H.; Lee, W.K.; Cho, M.J.; Ko, G.H.; Park, C.K.; Kasai, H.; Rhee, K.H. Increased oxidative DNA damage in Helicobacter pylori-infected human gastric mucosa. Cancer Res. 1996, 56, 1279–1282. [Google Scholar]
- Won, M.H.; Kang, T.C.; Jeon, G.S.; Lee, J.C.; Kim, D.Y.; Choi, E.M.; Lee, K.H.; Choi, C.D.; Chung, M.H.; Cho, S.S. Immunohistochemical detection of oxidative DNA damage induced by ischemia-reperfusion insults in gerbil hippocampus in vivo. Brain Res. 1999, 836, 70–78. [Google Scholar] [CrossRef]
- Radtke-Schuller, S.; Schuller, G.; Angenstein, F.; Grosser, O.S.; Goldschmidt, J.; Budinger, E. Brain atlas of the Mongolian gerbil (Meriones unguiculatus) in CT/MRI-aided stereotaxic coordinates. Brain Struct. Funct. 2016, 221, 1–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozkan, A.; Sen, H.M.; Sehitoglu, I.; Alacam, H.; Guven, M.; Aras, A.B.; Akman, T.; Silan, C.; Cosar, M.; Karaman, H.I. Neuroprotective effect of humic Acid on focal cerebral ischemia injury: An experimental study in rats. Inflammation 2015, 38, 32–39. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, G.E.; Tae, H.-J.; Lee, T.-K.; Park, Y.E.; Cho, J.H.; Kim, D.W.; Park, J.H.; Ahn, J.H.; Ryoo, S.; Kim, Y.-M.; et al. Risperidone Treatment after Transient Ischemia Induces Hypothermia and Provides Neuroprotection in the Gerbil Hippocampus by Decreasing Oxidative Stress. Int. J. Mol. Sci. 2019, 20, 4621. https://doi.org/10.3390/ijms20184621
Yang GE, Tae H-J, Lee T-K, Park YE, Cho JH, Kim DW, Park JH, Ahn JH, Ryoo S, Kim Y-M, et al. Risperidone Treatment after Transient Ischemia Induces Hypothermia and Provides Neuroprotection in the Gerbil Hippocampus by Decreasing Oxidative Stress. International Journal of Molecular Sciences. 2019; 20(18):4621. https://doi.org/10.3390/ijms20184621
Chicago/Turabian StyleYang, Go Eun, Hyun-Jin Tae, Tae-Kyeong Lee, Young Eun Park, Jeong Hwi Cho, Dae Won Kim, Joon Ha Park, Ji Hyeon Ahn, Sungwoo Ryoo, Young-Myeong Kim, and et al. 2019. "Risperidone Treatment after Transient Ischemia Induces Hypothermia and Provides Neuroprotection in the Gerbil Hippocampus by Decreasing Oxidative Stress" International Journal of Molecular Sciences 20, no. 18: 4621. https://doi.org/10.3390/ijms20184621
APA StyleYang, G. E., Tae, H.-J., Lee, T.-K., Park, Y. E., Cho, J. H., Kim, D. W., Park, J. H., Ahn, J. H., Ryoo, S., Kim, Y.-M., Shin, M. C., Cho, J. H., Lee, C.-H., Hwang, I. K., Jin, H., Won, M.-H., & Lee, J.-C. (2019). Risperidone Treatment after Transient Ischemia Induces Hypothermia and Provides Neuroprotection in the Gerbil Hippocampus by Decreasing Oxidative Stress. International Journal of Molecular Sciences, 20(18), 4621. https://doi.org/10.3390/ijms20184621