Linkage of Periodontitis and Rheumatoid Arthritis: Current Evidence and Potential Biological Interactions

 ,
,
Abstract
:1. Introduction
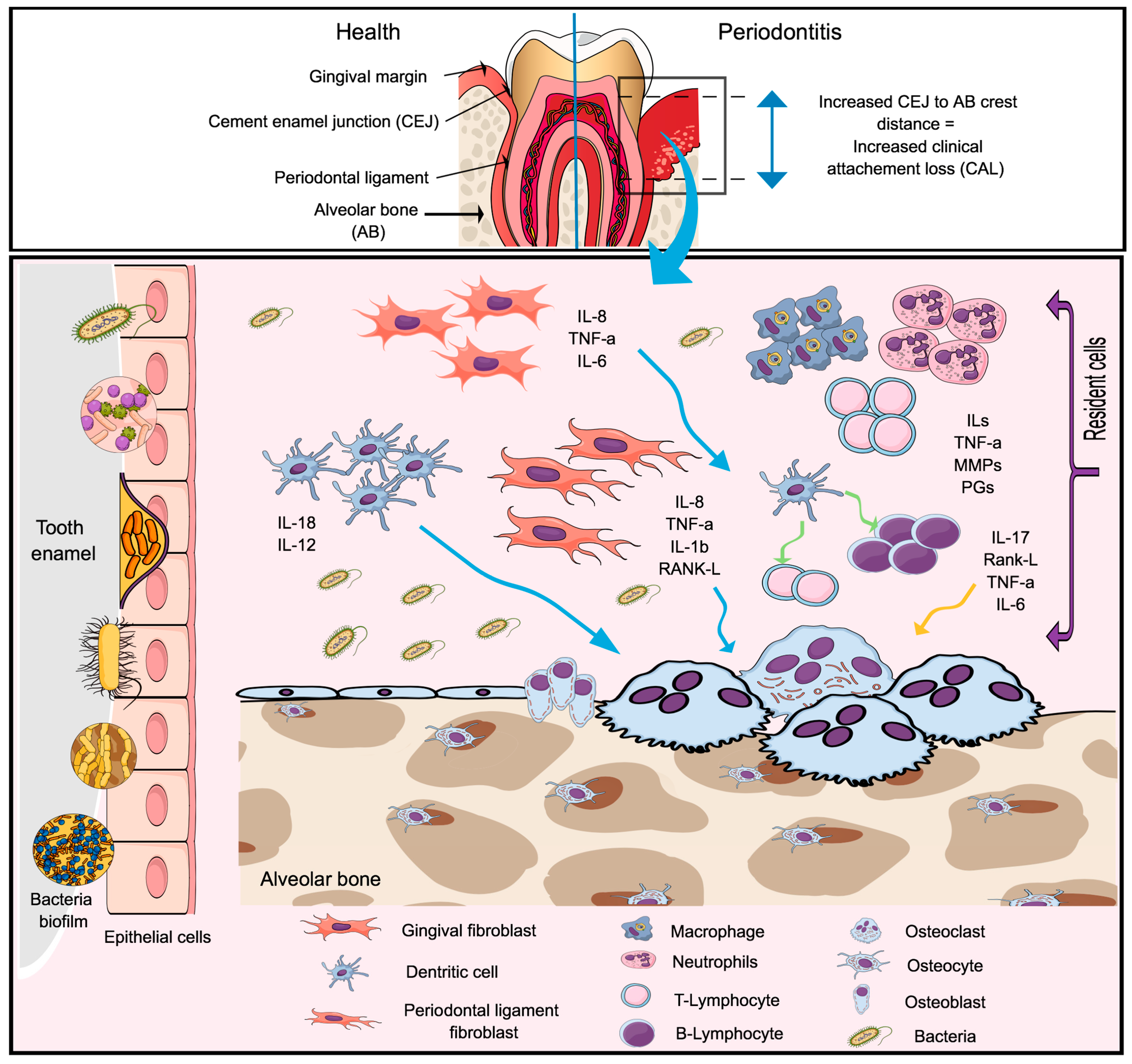
2. Periodontal Disease
3. Rheumatoid Arthritis
4. Mechanistic Studies Linking RA and PD
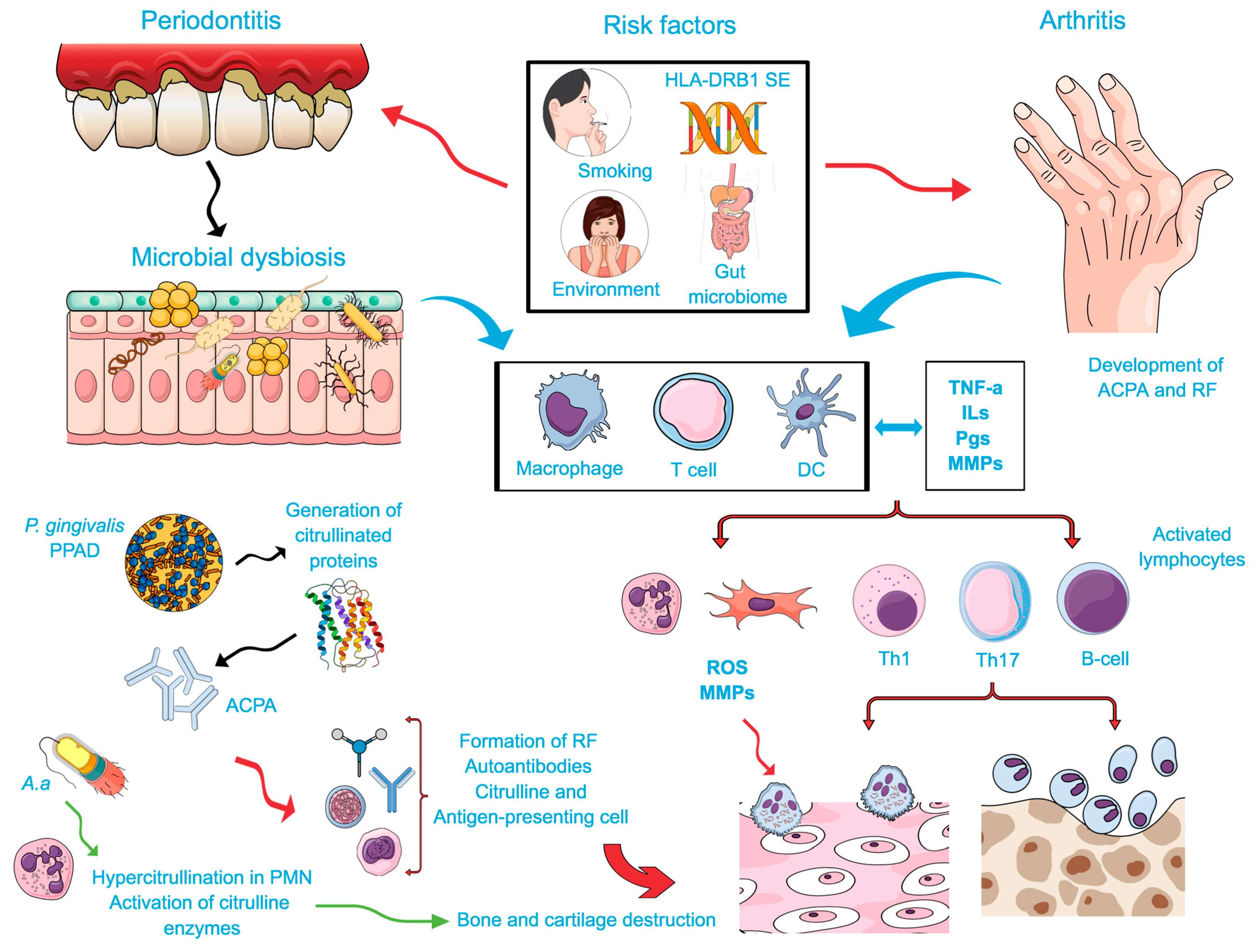
4.1. Two-Hit Model Associating RA and PD
4.2. Genetic Susceptibility
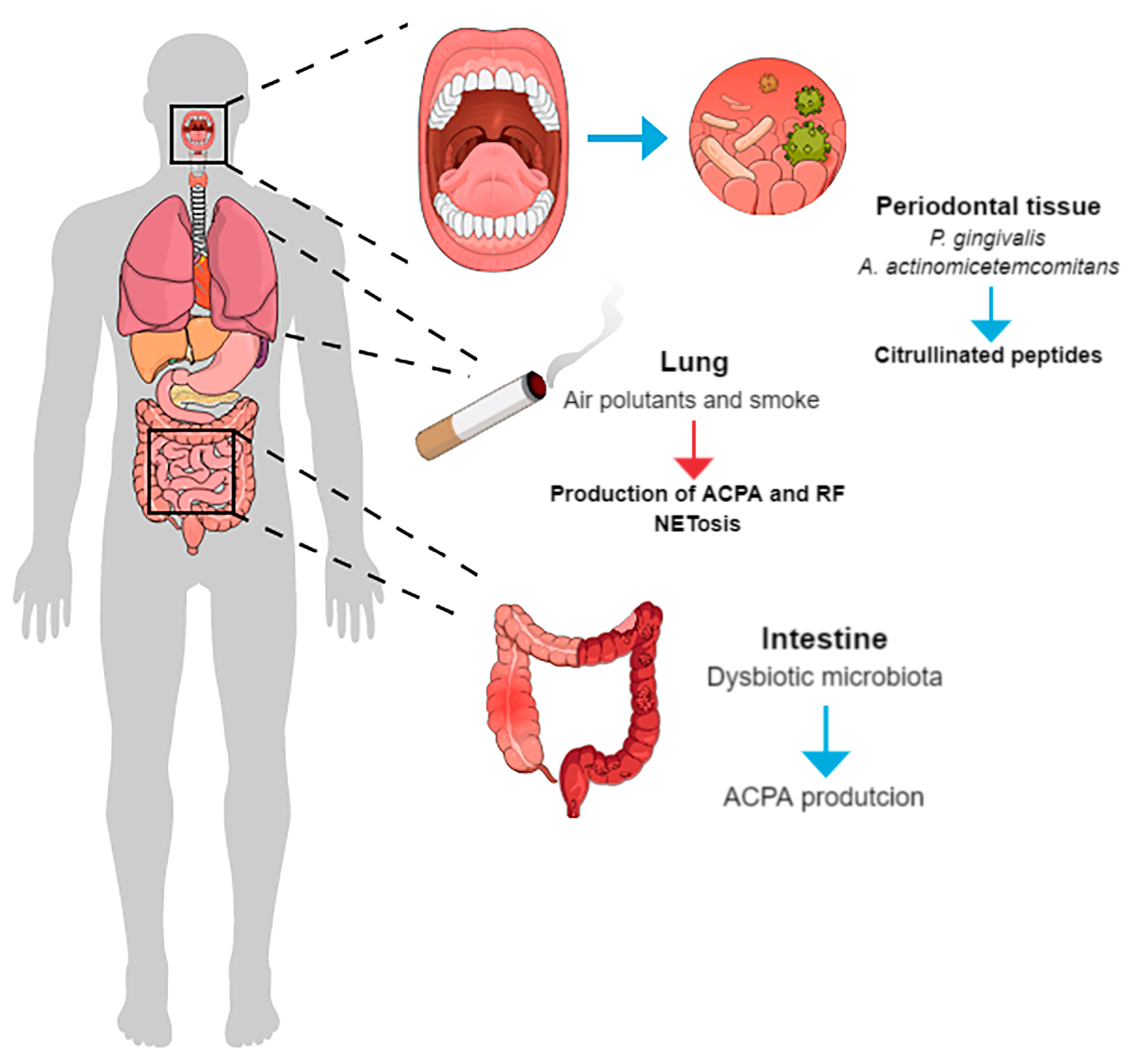
4.3. Bacterial Link Between PD and RA
4.3.1. The Citrullination Process
4.3.2. Modulation of Immune Response by PD-Associated Bacteria
5. Therapeutic Association in RA and PD: Epidemiological Studies
5.1. Effects of Treating RA on PD
5.2. Effects of Treating PD on RA
6. Concluding Remarks
7. Research Agenda
- Current evidence points to a pivotal role of microbiome in the pathogenesis of inflammatory conditions and its imbalance may ultimately result in the disease initiation.
- P. gingivalis and A. actinomycetemcomitans are currently the two most important microorganisms involved in the pathogenesis of PD and RA and are associated with increased citrullination. Evidence suggests that increased citrullination may also participate in tissue destruction associated with periodontitis.
- A bidirectional causal relationship between RA and PD is hypothesized, and citrullination may represent a key mechanism mediating reciprocal influences in this biological intersection.
- Clinical studies suggest that the RA treatment may ameliorate PD. Conversely, there are controversial reports on the benefits of the PD treatment in the improvement of RA. Clinical studies are difficult and limited because of a number of biases, particularly in the approach to control the influence of tobacco use. Studies in never-smokers will provide important information on the reciprocal effects of therapeutic management of RA and PD.
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ABL | Alveolar bone level |
| ACPA | Anti-citrullinated proteins antibodies |
| AIA | Antigen-induced arthritis |
| BOP | Bleeding on probing |
| BSA | Bovine serum albumin |
| CAL | Clinical attachment level |
| CIA | Collagen-induced arthritis |
| CRP | C-reactive protein |
| CT | Computer tomography |
| DAS28 | Disease Activity Score including 28-joint count |
| DMARD | Disease-modifying antirheumatic drugs |
| ESR | Erythrocyte sedimentation rate |
| EULAR | European league against rheumatism |
| GC | Glucocorticoids |
| GCF | Gingival crevicular fluid |
| GI | Gingival index |
| GM-CSF | Granulocyte macrophage colony-stimulating factor |
| IgG | Immunoglobulin G |
| IL-1β | Interleukin-1β |
| IL-17 | Interluekin-17 |
| IL-6 | Interluekin-6 |
| LtxA | Leukotoxin A |
| MAMP | Microbe-associated molecular pattern |
| MMP | Matrix metalloproteinase |
| MRI | Magnetic resonance imaging |
| NHANES | National Health and Nutrition Examination Survey |
| NHS | National Health Service |
| NSAID | Non-steroidal anti-inflammatory drugs |
| NSPT | Non-surgical periodontal treatment |
| PAD | Peptidylarginine deiminase |
| PAMP | Pathogen-associated molecular pattern |
| PD | Periodontal disease |
| PGE2 | Prostaglandin E2 |
| PPD | Probing pocket depth |
| RA | Rheumatoid arthritis |
| RANK-L | Receptor activator of NF-κB ligand |
| RF | Rheumatoid factor |
| ROS | Reactive oxygen species |
| SE | Shared-epitope |
| TGF-β | Transforming growth factor-β |
| TNF-α | Tumor necrosis factor-α |
| VAS | Visual analogue scale |
| WT | Wild-type |
References
- de Pablo, P.; Chapple, I.L.; Buckley, C.D.; Dietrich, T. Periodontitis in systemic rheumatic diseases. Nat. Rev. Rheumatol. 2009, 5, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Farquharson, D.; Butcher, J.P.; Culshaw, S. Periodontitis, Porphyromonas, and the pathogenesis of rheumatoid arthritis. Mucosal Immunol. 2012, 5, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, M.; Brennan, F.M.; Maini, R.N. Rheumatoid arthritis. Cell 1996, 85, 307–310. [Google Scholar] [CrossRef]
- Firestein, G.S. Evolving concepts of rheumatoid arthritis. Nature 2003, 423, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Firestein, G.S.; McInnes, I.B. Immunopathogenesis of Rheumatoid Arthritis. Immunity 2017, 46, 183–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhodes, B.; Furnrohr, B.G.; Vyse, T.J. C-reactive protein in rheumatology: Biology and genetics. Nat. Rev. Rheumatol. 2011, 7, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Engstrom, M.; Eriksson, K.; Lee, L.; Hermansson, M.; Johansson, A.; Nicholas, A.P.; Gerasimcik, N.; Lundberg, K.; Klareskog, L.; Catrina, A.I.; et al. Increased citrullination and expression of peptidylarginine deiminases independently of P. gingivalis and A. actinomycetemcomitans in gingival tissue of patients with periodontitis. J. Transl. Med. 2018, 16, 214. [Google Scholar] [CrossRef] [PubMed]
- Vitkov, L.; Hannig, M.; Minnich, B.; Herrmann, M. Periodontal sources of citrullinated antigens and TLR agonists related to RA. Autoimmunity 2018, 51, 304–309. [Google Scholar] [CrossRef]
- Horta-Baas, G.; Romero-Figueroa, M.D.S.; Montiel-Jarquin, A.J.; Pizano-Zarate, M.L.; Garcia-Mena, J.; Ramirez-Duran, N. Intestinal Dysbiosis and Rheumatoid Arthritis: A Link between Gut Microbiota and the Pathogenesis of Rheumatoid Arthritis. J. Immunol. Res. 2017, 2017, 4835189. [Google Scholar] [CrossRef]
- Jeong, Y.; Kim, J.W.; You, H.J.; Park, S.J.; Lee, J.; Ju, J.H.; Park, M.S.; Jin, H.; Cho, M.L.; Kwon, B.; et al. Gut Microbial Composition and Function are Altered in Patients with Early Rheumatoid Arthritis. J. Clin. Med. 2019, 8, 693. [Google Scholar] [CrossRef]
- Nogueira, A.R.; Shoenfeld, Y. Microbiome and autoimmune diseases: Cause and effect relationship. Curr. Opin. Rheumatol. 2019, 31, 471–474. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S.; Thompson, P.R. Protein Arginine Deiminases (PADs): Biochemistry and Chemical Biology of Protein Citrullination. Acc. Chem. Res. 2019, 52, 818–832. [Google Scholar] [CrossRef] [PubMed]
- Romero, V.; Fert-Bober, J.; Nigrovic, P.A.; Darrah, E.; Haque, U.J.; Lee, D.M.; van Eyk, J.; Rosen, A.; Andrade, F. Immune-mediated pore-forming pathways induce cellular hypercitrullination and generate citrullinated autoantigens in rheumatoid arthritis. Sci. Transl. Med. 2013, 5, 209ra150. [Google Scholar] [CrossRef] [PubMed]
- McGraw, W.T.; Potempa, J.; Farley, D.; Travis, J. Purification, characterization, and sequence analysis of a potential virulence factor from Porphyromonas gingivalis, peptidylarginine deiminase. Infect. Immun. 1999, 67, 3248–3256. [Google Scholar] [PubMed]
- Mangat, P.; Wegner, N.; Venables, P.J.; Potempa, J. Bacterial and human peptidylarginine deiminases: Targets for inhibiting the autoimmune response in rheumatoid arthritis? Arthritis Res. Ther. 2010, 12, 209. [Google Scholar] [CrossRef] [PubMed]
- Konig, M.F.; Abusleme, L.; Reinholdt, J.; Palmer, R.J.; Teles, R.P.; Sampson, K.; Rosen, A.; Nigrovic, P.A.; Sokolove, J.; Giles, J.T.; et al. Aggregatibacter actinomycetemcomitans-induced hypercitrullination links periodontal infection to autoimmunity in rheumatoid arthritis. Sci. Transl. Med. 2016, 8, 369ra176. [Google Scholar] [CrossRef] [PubMed]
- Laugisch, O.; Wong, A.; Sroka, A.; Kantyka, T.; Koziel, J.; Neuhaus, K.; Sculean, A.; Venables, P.J.; Potempa, J.; Moller, B.; et al. Citrullination in the periodontium--a possible link between periodontitis and rheumatoid arthritis. Clin. Oral Investig. 2016, 20, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Mankia, K.; Cheng, Z.; Do, T.; Hunt, L.; Meade, J.; Kang, J.; Clerehugh, V.; Speirs, A.; Tugnait, A.; Hensor, E.M.A.; et al. Prevalence of Periodontal Disease and Periodontopathic Bacteria in Anti-Cyclic Citrullinated Protein Antibody-Positive At-Risk Adults Without Arthritis. JAMA Netw. Open 2019, 2, e195394. [Google Scholar] [CrossRef] [PubMed]
- Loutan, L.; Alpizar-Rodriguez, D.; Courvoisier, D.S.; Finckh, A.; Mombelli, A.; Giannopoulou, C. Periodontal status correlates with anti-citrullinated protein antibodies in first-degree relatives of individuals with rheumatoid arthritis. J. Clin. Periodontol. 2019, 46, 690–698. [Google Scholar] [CrossRef]
- Mercado, F.; Marshall, R.I.; Klestov, A.C.; Bartold, P.M. Is there a relationship between rheumatoid arthritis and periodontal disease? J. Clin. Periodontol. 2000, 27, 267–272. [Google Scholar] [CrossRef]
- Mercado, F.B.; Marshall, R.I.; Bartold, P.M. Inter-relationships between rheumatoid arthritis and periodontal disease. A review. J. Clin. Periodontol. 2003, 30, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Stolt, P.; Bengtsson, C.; Nordmark, B.; Lindblad, S.; Lundberg, I.; Klareskog, L.; Alfredsson, L.; The Other Members of the Eira Study Group. Quantification of the influence of cigarette smoking on rheumatoid arthritis: Results from a population based case-control study, using incident cases. Ann. Rheum. Dis. 2003, 62, 835–841. [Google Scholar] [CrossRef] [PubMed]
- Hedstrom, A.K.; Stawiarz, L.; Klareskog, L.; Alfredsson, L. Smoking and susceptibility to rheumatoid arthritis in a Swedish population-based case-control study. Eur. J. Epidemiol. 2018, 33, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Sundstrom, B.; Johansson, I.; Rantapaa-Dahlqvist, S. Interaction between dietary sodium and smoking increases the risk for rheumatoid arthritis: Results from a nested case-control study. Rheumatology 2015, 54, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Compan, V.; Melguizo-Madrid, E.; Hernandez-Cruz, B.; Santos-Rey, K.; Leyva-Prado, C.; Gonzalez-Martin, C.; Navarro-Sarabia, F.; Gonzalez-Rodriguez, C. Interaction between oxidative stress and smoking is associated with an increased risk of rheumatoid arthritis: A case-control study. Rheumatology 2013, 52, 487–493. [Google Scholar] [CrossRef]
- Leech, M.T.; Bartold, P.M. The association between rheumatoid arthritis and periodontitis. Best Pract. Res. Clin. Rheumatol. 2015, 29, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Catrina, A.I.; Ytterberg, A.J.; Reynisdottir, G.; Malmstrom, V.; Klareskog, L. Lungs, joints and immunity against citrullinated proteins in rheumatoid arthritis. Nat. Rev. Rheumatol. 2014, 10, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Stabholz, A.; Soskolne, W.A.; Shapira, L. Genetic and environmental risk factors for chronic periodontitis and aggressive periodontitis. Periodontology 2000 2010, 53, 138–153. [Google Scholar] [CrossRef] [PubMed]
- Albandar, J.M.; Streckfus, C.F.; Adesanya, M.R.; Winn, D.M. Cigar, pipe, and cigarette smoking as risk factors for periodontal disease and tooth loss. J. Periodontol. 2000, 71, 1874–1881. [Google Scholar] [CrossRef]
- Gonzalez, Y.M.; De Nardin, A.; Grossi, S.G.; Machtei, E.E.; Genco, R.J.; De Nardin, E. Serum cotinine levels, smoking, and periodontal attachment loss. J. Dent. Res. 1996, 75, 796–802. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Canut, P.; Lorca, A.; Magan, R. Smoking and periodontal disease severity. J. Clin. Periodontol. 1995, 22, 743–749. [Google Scholar] [CrossRef] [PubMed]
- Albandar, J.M.; Rams, T.E. Global epidemiology of periodontal diseases: An overview. Periodontology 2000 2002, 29, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Eke, P.I.; Dye, B.A.; Wei, L.; Slade, G.D.; Thornton-Evans, G.O.; Borgnakke, W.S.; Taylor, G.W.; Page, R.C.; Beck, J.D.; Genco, R.J. Update on Prevalence of Periodontitis in Adults in the United States: NHANES 2009 to 2012. J. Periodontol. 2015, 86, 611–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinane, D.F.; Stathopoulou, P.G.; Papapanou, P.N. Periodontal diseases. Nat. Rev. Dis. Primers 2017, 3, 17038. [Google Scholar] [CrossRef] [PubMed]
- Hugoson, A.; Sjodin, B.; Norderyd, O. Trends over 30 years, 1973–2003, in the prevalence and severity of periodontal disease. J. Clin. Periodontol. 2008, 35, 405–414. [Google Scholar] [CrossRef]
- Dye, B.A. Global periodontal disease epidemiology. Periodontology 2000 2012, 58, 10–25. [Google Scholar] [CrossRef]
- Shanbhag, S.; Dahiya, M.; Croucher, R. The impact of periodontal therapy on oral health-related quality of life in adults: A systematic review. J. Clin. Periodontol. 2012, 39, 725–735. [Google Scholar] [CrossRef]
- Listl, S.; Galloway, J.; Mossey, P.A.; Marcenes, W. Global Economic Impact of Dental Diseases. J. Dent. Res. 2015, 94, 1355–1361. [Google Scholar] [CrossRef]
- Cheng, Z.; Meade, J.; Mankia, K.; Emery, P.; Devine, D.A. Periodontal disease and periodontal bacteria as triggers for rheumatoid arthritis. Best Pract. Res. Clin. Rheumatol. 2017, 31, 19–30. [Google Scholar] [CrossRef]
- Lundberg, K.; Wegner, N.; Yucel-Lindberg, T.; Venables, P.J. Periodontitis in RA-the citrullinated enolase connection. Nat. Rev. Rheumatol. 2010, 6, 727–730. [Google Scholar] [CrossRef]
- Genco, R.J.; Van Dyke, T.E. Prevention: Reducing the risk of CVD in patients with periodontitis. Nat. Rev. Cardiol. 2010, 7, 479–480. [Google Scholar] [CrossRef] [PubMed]
- Graves, D.T.; Correa, J.D.; Silva, T.A. The Oral Microbiota Is Modified by Systemic Diseases. J. Dent. Res. 2018, 98, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Correa, J.D.; Fernandes, G.R.; Calderaro, D.C.; Mendonca, S.M.S.; Silva, J.M.; Albiero, M.L.; Cunha, F.Q.; Xiao, E.; Ferreira, G.A.; Teixeira, A.L.; et al. Oral microbial dysbiosis linked to worsened periodontal condition in rheumatoid arthritis patients. Sci. Rep. 2019, 9, 8379. [Google Scholar] [CrossRef] [PubMed]
- Palioto, D.B.; Finoti, L.S.; Kinane, D.F.; Benakanakere, M. Epigenetic and inflammatory events in experimental periodontitis following systemic microbial challenge. J. Clin. Periodontol. 2019, 46, 819–829. [Google Scholar] [CrossRef]
- Hajishengallis, G. Periodontitis: From microbial immune subversion to systemic inflammation. Nat. Rev. Immunol. 2015, 15, 30–44. [Google Scholar] [CrossRef] [PubMed]
- Correa, J.D.; Saraiva, A.M.; Queiroz-Junior, C.M.; Madeira, M.F.; Duarte, P.M.; Teixeira, M.M.; Souza, D.G.; da Silva, T.A. Arthritis-induced alveolar bone loss is associated with changes in the composition of oral microbiota. Anaerobe 2016, 39, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Socransky, S.S.; Smith, C.; Haffajee, A.D. Subgingival microbial profiles in refractory periodontal disease. J. Clin. Periodontol. 2002, 29, 260–268. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, T.; Krauss, J.L.; Abe, T.; Jotwani, R.; Triantafilou, M.; Triantafilou, K.; Hashim, A.; Hoch, S.; Curtis, M.A.; Nussbaum, G.; et al. Porphyromonas gingivalis manipulates complement and TLR signaling to uncouple bacterial clearance from inflammation and promote dysbiosis. Cell Host Microbe 2014, 15, 768–778. [Google Scholar] [CrossRef] [PubMed]
- Di Benedetto, A.; Gigante, I.; Colucci, S.; Grano, M. Periodontal disease: Linking the primary inflammation to bone loss. Clin. Dev. Immunol. 2013, 2013, 503754. [Google Scholar] [CrossRef] [PubMed]
- Potempa, J.; Mydel, P.; Koziel, J. The case for periodontitis in the pathogenesis of rheumatoid arthritis. Nat. Rev. Rheumatol. 2017, 13, 606–620. [Google Scholar] [CrossRef] [PubMed]
- de Molon, R.S.; de Avila, E.D.; Boas Nogueira, A.V.; Chaves de Souza, J.A.; Avila-Campos, M.J.; de Andrade, C.R.; Cirelli, J.A. Evaluation of the host response in various models of induced periodontal disease in mice. J. Periodontol. 2014, 85, 465–477. [Google Scholar] [CrossRef] [PubMed]
- de Molon, R.S.; de Avila, E.D.; Cirelli, J.A. Host responses induced by different animal models of periodontal disease: A literature review. J. Investig. Clin. Dent. 2013, 4, 211–218. [Google Scholar] [CrossRef] [PubMed]
- de Molon, R.S.; Mascarenhas, V.I.; de Avila, E.D.; Finoti, L.S.; Toffoli, G.B.; Spolidorio, D.M.; Scarel-Caminaga, R.M.; Tetradis, S.; Cirelli, J.A. Long-term evaluation of oral gavage with periodontopathogens or ligature induction of experimental periodontal disease in mice. Clin. Oral Investig. 2016, 20, 1203–1216. [Google Scholar] [CrossRef] [PubMed]
- Alencar, V.B.; Bezerra, M.M.; Lima, V.; Abreu, A.L.; Brito, G.A.; Rocha, F.A.; Ribeiro, R.A. Disodium chlodronate prevents bone resorption in experimental periodontitis in rats. J. Periodontol. 2002, 73, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, M.; Miyazawa, K.; Tabuchi, M.; Tanaka, M.; Yoshizako, M.; Minamoto, C.; Torii, Y.; Tamaoka, Y.; Kawatani, M.; Osada, H.; et al. Reveromycin A Administration Prevents Alveolar Bone Loss in Osteoprotegerin Knockout Mice with Periodontal Disease. Sci. Rep. 2015, 5, 16510. [Google Scholar] [CrossRef] [PubMed]
- Soundia, A.; Hadaya, D.; Esfandi, N.; de Molon, R.S.; Bezouglaia, O.; Dry, S.M.; Pirih, F.Q.; Aghaloo, T.; Tetradis, S. Osteonecrosis of the jaws (ONJ) in mice after extraction of teeth with periradicular disease. Bone 2016, 90, 133–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinane, D.F.; Bartold, P.M. Clinical relevance of the host responses of periodontitis. Periodontology 2000 2007, 43, 278–293. [Google Scholar] [CrossRef] [PubMed]
- Trombone, A.P.; Claudino, M.; Colavite, P.; de Assis, G.F.; Avila-Campos, M.J.; Silva, J.S.; Campanelli, A.P.; Ibanez, O.M.; De Franco, M.; Garlet, G.P. Periodontitis and arthritis interaction in mice involves a shared hyper-inflammatory genotype and functional immunological interferences. Genes Immun. 2010, 11, 479–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bingham, C.O., 3rd; Moni, M. Periodontal disease and rheumatoid arthritis: The evidence accumulates for complex pathobiologic interactions. Curr. Opin. Rheumatol. 2013, 25, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Page, R.C.; Eke, P.I. Case definitions for use in population-based surveillance of periodontitis. J. Periodontol. 2007, 78, 1387–1399. [Google Scholar] [CrossRef] [PubMed]
- McInnes, I.B.; Schett, G. The pathogenesis of rheumatoid arthritis. N. Engl. J. Med. 2011, 365, 2205–2219. [Google Scholar] [CrossRef]
- Scott, D.L.; Wolfe, F.; Huizinga, T.W. Rheumatoid arthritis. Lancet 2010, 376, 1094–1108. [Google Scholar] [CrossRef]
- de Molon, R.S.; Hsu, C.; Bezouglaia, O.; Dry, S.M.; Pirih, F.Q.; Soundia, A.; Cunha, F.Q.; Cirelli, J.A.; Aghaloo, T.L.; Tetradis, S. Rheumatoid Arthritis Exacerbates the Severity of Osteonecrosis of the Jaws (ONJ) in Mice. A Randomized, Prospective, Controlled Animal Study. J. Bone Miner. Res. 2016, 31, 1596–1607. [Google Scholar] [CrossRef] [PubMed]
- Edwards, C.J.; Cooper, C. Early environmental factors and rheumatoid arthritis. Clin. Exp. Immunol. 2006, 143, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, D.; Moots, R. Cigarette smoking and severity of rheumatoid arthritis. Rheumatology 2001, 40, 1426–1427. [Google Scholar] [CrossRef] [Green Version]
- Kallberg, H.; Padyukov, L.; Plenge, R.M.; Ronnelid, J.; Gregersen, P.K.; van der Helm-van Mil, A.H.; Toes, R.E.; Huizinga, T.W.; Klareskog, L.; Alfredsson, L.; et al. Gene-gene and gene-environment interactions involving HLA-DRB1, PTPN22, and smoking in two subsets of rheumatoid arthritis. Am. J. Hum. Genet. 2007, 80, 867–875. [Google Scholar] [CrossRef]
- Aggarwal, R.; Ringold, S.; Khanna, D.; Neogi, T.; Johnson, S.R.; Miller, A.; Brunner, H.I.; Ogawa, R.; Felson, D.; Ogdie, A.; et al. Distinctions between diagnostic and classification criteria? Arthritis Care Res. 2015, 67, 891–897. [Google Scholar] [CrossRef]
- Rutger, G.P. Rheumatoid arthritis and periodontitis—Inflammatory and infectious connections. Review of the literature. J. Oral Microbiol. 2012, 4. [Google Scholar] [CrossRef]
- Gaujoux-Viala, C.; Mouterde, G.; Baillet, A.; Claudepierre, P.; Fautrel, B.; Le Loet, X.; Maillefert, J.F. Evaluating disease activity in rheumatoid arthritis: Which composite index is best? A systematic literature analysis of studies comparing the psychometric properties of the DAS, DAS28, SDAI and CDAI. Jt. Bone Spine 2012, 79, 149–155. [Google Scholar] [CrossRef]
- American College of Rheumatology Subcommittee on Rheumatoid Arthritis Guidelines. Guidelines for the management of rheumatoid arthritis: 2002 Update. Arthritis Rheum. 2002, 46, 328–346. [Google Scholar] [CrossRef]
- Singh, J.A.; Saag, K.G.; Bridges, S.L., Jr.; Akl, E.A.; Bannuru, R.R.; Sullivan, M.C.; Vaysbrot, E.; McNaughton, C.; Osani, M.; Shmerling, R.H.; et al. 2015 American College of Rheumatology Guideline for the Treatment of Rheumatoid Arthritis. Arthritis Rheumatol. 2016, 68, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Weyand, C.M. New insights into the pathogenesis of rheumatoid arthritis. Rheumatology 2000, 39, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araujo, V.M.; Melo, I.M.; Lima, V. Relationship between Periodontitis and Rheumatoid Arthritis: Review of the Literature. Mediat. Inflamm. 2015, 2015, 259074. [Google Scholar] [CrossRef] [PubMed]
- McInnes, I.B.; Schett, G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat. Rev. Immunol. 2007, 7, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Roeleveld, D.M.; Koenders, M.I. The role of the Th17 cytokines IL-17 and IL-22 in Rheumatoid Arthritis pathogenesis and developments in cytokine immunotherapy. Cytokine 2015, 74, 101–107. [Google Scholar] [CrossRef]
- Nielen, M.M.; van Schaardenburg, D.; Reesink, H.W.; van de Stadt, R.J.; van der Horst-Bruinsma, I.E.; de Koning, M.H.; Habibuw, M.R.; Vandenbroucke, J.P.; Dijkmans, B.A. Specific autoantibodies precede the symptoms of rheumatoid arthritis: A study of serial measurements in blood donors. Arthritis Rheum. 2004, 50, 380–386. [Google Scholar] [CrossRef] [PubMed]
- van de Stadt, L.A.; de Koning, M.H.; van de Stadt, R.J.; Wolbink, G.; Dijkmans, B.A.; Hamann, D.; van Schaardenburg, D. Development of the anti-citrullinated protein antibody repertoire prior to the onset of rheumatoid arthritis. Arthritis Rheum. 2011, 63, 3226–3233. [Google Scholar] [CrossRef]
- Trouw, L.A.; Huizinga, T.W.; Toes, R.E. Autoimmunity in rheumatoid arthritis: Different antigens—Common principles. Ann. Rheum. Dis. 2013, 72, ii132–ii136. [Google Scholar] [CrossRef]
- Avouac, J.; Gossec, L.; Dougados, M. Diagnostic and predictive value of anti-cyclic citrullinated protein antibodies in rheumatoid arthritis: A systematic literature review. Ann. Rheum. Dis. 2006, 65, 845–851. [Google Scholar] [CrossRef]
- Besada, E.; Nikolaissen, C.; Nossent, H. Should rheumatoid factor in rheumatoid arthritis be sent to Davy Jones’s Locker? Scand. J. Rheumatol. 2012, 41, 85–88. [Google Scholar] [CrossRef]
- van der Linden, M.P.; van der Woude, D.; Ioan-Facsinay, A.; Levarht, E.W.; Stoeken-Rijsbergen, G.; Huizinga, T.W.; Toes, R.E.; van der Helm-van Mil, A.H. Value of anti-modified citrullinated vimentin and third-generation anti-cyclic citrullinated peptide compared with second-generation anti-cyclic citrullinated peptide and rheumatoid factor in predicting disease outcome in undifferentiated arthritis and rheumatoid arthritis. Arthritis Rheum. 2009, 60, 2232–2241. [Google Scholar] [PubMed]
- Abdollahi-Roodsaz, S.; Abramson, S.B.; Scher, J.U. The metabolic role of the gut microbiota in health and rheumatic disease: Mechanisms and interventions. Nat. Rev. Rheumatol. 2016, 12, 446–455. [Google Scholar] [CrossRef] [PubMed]
- Scher, J.U.; Littman, D.R.; Abramson, S.B. Microbiome in Inflammatory Arthritis and Human Rheumatic Diseases. Arthritis Rheumatol. 2016, 68, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Evans-Marin, H.; Rogier, R.; Koralov, S.B.; Manasson, J.; Roeleveld, D.; van der Kraan, P.M.; Scher, J.U.; Koenders, M.I.; Abdollahi-Roodsaz, S. Microbiota-Dependent Involvement of Th17 Cells in Murine Models of Inflammatory Arthritis. Arthritis Rheumatol. 2018, 70, 1971–1983. [Google Scholar] [CrossRef] [PubMed]
- Rogier, R.; Evans-Marin, H.; Manasson, J.; van der Kraan, P.M.; Walgreen, B.; Helsen, M.M.; van den Bersselaar, L.A.; van de Loo, F.A.; van Lent, P.L.; Abramson, S.B.; et al. Alteration of the intestinal microbiome characterizes preclinical inflammatory arthritis in mice and its modulation attenuates established arthritis. Sci. Rep. 2017, 7, 15613. [Google Scholar] [CrossRef] [PubMed]
- Rogier, R.; Koenders, M.I.; Abdollahi-Roodsaz, S. Toll-like receptor mediated modulation of T cell response by commensal intestinal microbiota as a trigger for autoimmune arthritis. J. Immunol. Res. 2015, 2015, 527696. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.J.; Ivanov, I.I.; Darce, J.; Hattori, K.; Shima, T.; Umesaki, Y.; Littman, D.R.; Benoist, C.; Mathis, D. Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity 2010, 32, 815–827. [Google Scholar] [CrossRef]
- Abdollahi-Roodsaz, S.; Joosten, L.A.; Koenders, M.I.; Devesa, I.; Roelofs, M.F.; Radstake, T.R.; Heuvelmans-Jacobs, M.; Akira, S.; Nicklin, M.J.; Ribeiro-Dias, F.; et al. Stimulation of TLR2 and TLR4 differentially skews the balance of T cells in a mouse model of arthritis. J. Clin. Investig. 2008, 118, 205–216. [Google Scholar] [CrossRef] [Green Version]
- Rosenbaum, J.T.; Asquith, M.J. The Microbiome: A Revolution in Treatment for Rheumatic Diseases? Curr. Rheumatol. Rep. 2016, 18, 62. [Google Scholar] [CrossRef]
- Gan, R.W.; Trouw, L.A.; Shi, J.; Toes, R.E.; Huizinga, T.W.; Demoruelle, M.K.; Kolfenbach, J.R.; Zerbe, G.O.; Deane, K.D.; Edison, J.D.; et al. Anti-carbamylated protein antibodies are present prior to rheumatoid arthritis and are associated with its future diagnosis. J. Rheumatol. 2015, 42, 572–579. [Google Scholar] [CrossRef]
- McLean, M.H.; Dieguez, D., Jr.; Miller, L.M.; Young, H.A. Does the microbiota play a role in the pathogenesis of autoimmune diseases? Gut 2015, 64, 332–341. [Google Scholar] [CrossRef] [PubMed]
- Mikuls, T.R.; Payne, J.B.; Deane, K.D.; Thiele, G.M. Autoimmunity of the lung and oral mucosa in a multisystem inflammatory disease: The spark that lights the fire in rheumatoid arthritis? J. Allergy Clin. Immunol. 2016, 137, 28–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- England, B.R.; Thiele, G.M.; Mikuls, T.R. Anticitrullinated protein antibodies: Origin and role in the pathogenesis of rheumatoid arthritis. Curr. Opin. Rheumatol. 2017, 29, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Yeoh, N.; Burton, J.P.; Suppiah, P.; Reid, G.; Stebbings, S. The role of the microbiome in rheumatic diseases. Curr. Rheumatol. Rep. 2013, 15, 314. [Google Scholar] [CrossRef] [PubMed]
- Paster, B.J.; Olsen, I.; Aas, J.A.; Dewhirst, F.E. The breadth of bacterial diversity in the human periodontal pocket and other oral sites. Periodontology 2000 2006, 42, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Forner, L.; Larsen, T.; Kilian, M.; Holmstrup, P. Incidence of bacteremia after chewing, tooth brushing and scaling in individuals with periodontal inflammation. J. Clin. Periodontol. 2006, 33, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Kempsell, K.E.; Cox, C.J.; Hurle, M.; Wong, A.; Wilkie, S.; Zanders, E.D.; Gaston, J.S.; Crowe, J.S. Reverse transcriptase-PCR analysis of bacterial rRNA for detection and characterization of bacterial species in arthritis synovial tissue. Infect. Immun. 2000, 68, 6012–6026. [Google Scholar] [CrossRef]
- Martinez-Martinez, R.E.; Abud-Mendoza, C.; Patino-Marin, N.; Rizo-Rodriguez, J.C.; Little, J.W.; Loyola-Rodriguez, J.P. Detection of periodontal bacterial DNA in serum and synovial fluid in refractory rheumatoid arthritis patients. J. Clin. Periodontol. 2009, 36, 1004–1010. [Google Scholar] [CrossRef]
- Moen, K.; Brun, J.G.; Valen, M.; Skartveit, L.; Eribe, E.K.; Olsen, I.; Jonsson, R. Synovial inflammation in active rheumatoid arthritis and psoriatic arthritis facilitates trapping of a variety of oral bacterial DNAs. Clin. Exp. Rheumatol. 2006, 24, 656–663. [Google Scholar]
- Temoin, S.; Chakaki, A.; Askari, A.; El-Halaby, A.; Fitzgerald, S.; Marcus, R.E.; Han, Y.W.; Bissada, N.F. Identification of oral bacterial DNA in synovial fluid of patients with arthritis with native and failed prosthetic joints. J. Clin. Rheumatol. 2012, 18, 117–121. [Google Scholar] [CrossRef]
- Brusca, S.B.; Abramson, S.B.; Scher, J.U. Microbiome and mucosal inflammation as extra-articular triggers for rheumatoid arthritis and autoimmunity. Curr. Opin. Rheumatol. 2014, 26, 101–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barra, L.; Scinocca, M.; Saunders, S.; Bhayana, R.; Rohekar, S.; Racape, M.; Coles, R.; Cairns, E.; Bell, D.A. Anti-citrullinated protein antibodies in unaffected first-degree relatives of rheumatoid arthritis patients. Arthritis Rheum. 2013, 65, 1439–1447. [Google Scholar] [CrossRef] [PubMed]
- Kinslow, J.D.; Blum, L.K.; Deane, K.D.; Demoruelle, M.K.; Okamoto, Y.; Parish, M.C.; Kongpachith, S.; Lahey, L.J.; Norris, J.M.; Robinson, W.H.; et al. Elevated IgA Plasmablast Levels in Subjects at Risk of Developing Rheumatoid Arthritis. Arthritis Rheumatol. 2016, 68, 2372–2383. [Google Scholar] [CrossRef] [PubMed]
- Demoruelle, M.K.; Bowers, E.; Lahey, L.J.; Sokolove, J.; Purmalek, M.; Seto, N.L.; Weisman, M.H.; Norris, J.M.; Kaplan, M.J.; Holers, V.M.; et al. Antibody Responses to Citrullinated and Noncitrullinated Antigens in the Sputum of Subjects with Rheumatoid Arthritis and Subjects at Risk for Development of Rheumatoid Arthritis. Arthritis Rheumatol. 2018, 70, 516–527. [Google Scholar] [CrossRef] [PubMed]
- Demoruelle, M.K.; Harrall, K.K.; Ho, L.; Purmalek, M.M.; Seto, N.L.; Rothfuss, H.M.; Weisman, M.H.; Solomon, J.J.; Fischer, A.; Okamoto, Y.; et al. Anti-Citrullinated Protein Antibodies Are Associated with Neutrophil Extracellular Traps in the Sputum in Relatives of Rheumatoid Arthritis Patients. Arthritis Rheumatol. 2017, 69, 1165–1175. [Google Scholar] [CrossRef]
- Willis, V.C.; Demoruelle, M.K.; Derber, L.A.; Chartier-Logan, C.J.; Parish, M.C.; Pedraza, I.F.; Weisman, M.H.; Norris, J.M.; Holers, V.M.; Deane, K.D. Sputum autoantibodies in patients with established rheumatoid arthritis and subjects at risk of future clinically apparent disease. Arthritis Rheum. 2013, 65, 2545–2554. [Google Scholar]
- Holers, V.M.; Demoruelle, M.K.; Kuhn, K.A.; Buckner, J.H.; Robinson, W.H.; Okamoto, Y.; Norris, J.M.; Deane, K.D. Rheumatoid arthritis and the mucosal origins hypothesis: Protection turns to destruction. Nat. Rev. Rheumatol. 2018, 14, 542–557. [Google Scholar] [CrossRef]
- Lucchino, B.; Spinelli, F.R.; Iannuccelli, C.; Guzzo, M.P.; Conti, F.; Di Franco, M. Mucosa-Environment Interactions in the Pathogenesis of Rheumatoid Arthritis. Cells 2019, 8, 700. [Google Scholar] [CrossRef]
- Golub, L.M.; Payne, J.B.; Reinhardt, R.A.; Nieman, G. Can systemic diseases co-induce (not just exacerbate) periodontitis? A hypothetical “two-hit” model. J. Dent. Res. 2006, 85, 102–105. [Google Scholar] [CrossRef]
- Kaur, S.; White, S.; Bartold, M. Periodontal Disease as a Risk Factor for Rheumatoid Arthritis: A Systematic Review. JBI Libr. Syst. Rev. 2012, 10, 1–12. [Google Scholar] [CrossRef]
- Wegner, N.; Wait, R.; Sroka, A.; Eick, S.; Nguyen, K.A.; Lundberg, K.; Kinloch, A.; Culshaw, S.; Potempa, J.; Venables, P.J. Peptidylarginine deiminase from Porphyromonas gingivalis citrullinates human fibrinogen and alpha-enolase: Implications for autoimmunity in rheumatoid arthritis. Arthritis Rheum. 2010, 62, 2662–2672. [Google Scholar] [CrossRef] [PubMed]
- Gehlot, P.; Volk, S.L.; Rios, H.F.; Jepsen, K.J.; Holoshitz, J. Spontaneous destructive periodontitis and skeletal bone damage in transgenic mice carrying a human shared epitope-coding HLA-DRB1 allele. RMD Open 2016, 2, e000349. [Google Scholar] [CrossRef] [PubMed]
- van der Woude, D.; Houwing-Duistermaat, J.J.; Toes, R.E.; Huizinga, T.W.; Thomson, W.; Worthington, J.; van der Helm-van Mil, A.H.; de Vries, R.R. Quantitative heritability of anti-citrullinated protein antibody-positive and anti-citrullinated protein antibody-negative rheumatoid arthritis. Arthritis Rheum. 2009, 60, 916–923. [Google Scholar] [CrossRef] [PubMed]
- Smolen, J.S.; Aletaha, D.; Koeller, M.; Weisman, M.H.; Emery, P. New therapies for treatment of rheumatoid arthritis. Lancet 2007, 370, 1861–1874. [Google Scholar] [CrossRef]
- James, E.A.; Moustakas, A.K.; Bui, J.; Papadopoulos, G.K.; Bondinas, G.; Buckner, J.H.; Kwok, W.W. HLA-DR1001 presents “altered-self” peptides derived from joint-associated proteins by accepting citrulline in three of its binding pockets. Arthritis Rheum. 2010, 62, 2909–2918. [Google Scholar] [CrossRef] [PubMed]
- Cooles, F.A.; Isaacs, J.D. Pathophysiology of rheumatoid arthritis. Curr. Opin. Rheumatol. 2011, 23, 233–240. [Google Scholar] [CrossRef]
- Kharlamova, N.; Jiang, X.; Sherina, N.; Potempa, B.; Israelsson, L.; Quirke, A.M.; Eriksson, K.; Yucel-Lindberg, T.; Venables, P.J.; Potempa, J.; et al. Antibodies to Porphyromonas gingivalis Indicate Interaction Between Oral Infection, Smoking, and Risk Genes in Rheumatoid Arthritis Etiology. Arthritis Rheumatol. 2016, 68, 604–613. [Google Scholar] [CrossRef]
- Gregersen, P.K.; Silver, J.; Winchester, R.J. The shared epitope hypothesis. An approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum. 1987, 30, 1205–1213. [Google Scholar] [CrossRef]
- Bonfil, J.J.; Dillier, F.L.; Mercier, P.; Reviron, D.; Foti, B.; Sambuc, R.; Brodeur, J.M.; Sedarat, C. A “case control” study on the role of HLA DR4 in severe periodontitis and rapidly progressive periodontitis. Identification of types and subtypes using molecular biology (PCR.SSO). J. Clin. Periodontol. 1999, 26, 77–84. [Google Scholar] [CrossRef]
- Marotte, H.; Farge, P.; Gaudin, P.; Alexandre, C.; Mougin, B.; Miossec, P. The association between periodontal disease and joint destruction in rheumatoid arthritis extends the link between the HLA-DR shared epitope and severity of bone destruction. Ann. Rheum. Dis. 2006, 65, 905–909. [Google Scholar] [CrossRef]
- Fu, J.; Ling, S.; Liu, Y.; Yang, J.; Naveh, S.; Hannah, M.; Gilon, C.; Zhang, Y.; Holoshitz, J. A small shared epitope-mimetic compound potently accelerates osteoclast-mediated bone damage in autoimmune arthritis. J. Immunol. 2013, 191, 2096–2103. [Google Scholar] [CrossRef] [PubMed]
- De Almeida, D.E.; Ling, S.; Pi, X.; Hartmann-Scruggs, A.M.; Pumpens, P.; Holoshitz, J. Immune dysregulation by the rheumatoid arthritis shared epitope. J. Immunol. 2010, 185, 1927–1934. [Google Scholar] [CrossRef] [PubMed]
- Burger, D.R.; Vandenbark, A.A.; Finke, P.; Nolte, J.E.; Vetto, R.M. Human transfer factor: Effects on lymphocyte transformation. J. Immunol. 1976, 117, 782–788. [Google Scholar] [PubMed]
- Sandal, I.; Karydis, A.; Luo, J.; Prislovsky, A.; Whittington, K.B.; Rosloniec, E.F.; Dong, C.; Novack, D.V.; Mydel, P.; Zheng, S.G.; et al. Bone loss and aggravated autoimmune arthritis in HLA-DRbeta1-bearing humanized mice following oral challenge with Porphyromonas gingivalis. Arthritis Res. Ther. 2016, 18, 249. [Google Scholar] [CrossRef] [PubMed]
- Mikuls, T.R.; Walker, C.; Qiu, F.; Yu, F.; Thiele, G.M.; Alfant, B.; Li, E.C.; Zhao, L.Y.; Wang, G.P.; Datta, S.; et al. The subgingival microbiome in patients with established rheumatoid arthritis. Rheumatology 2018, 57, 1162–1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenstein, E.D.; Greenwald, R.A.; Kushner, L.J.; Weissmann, G. Hypothesis: The humoral immune response to oral bacteria provides a stimulus for the development of rheumatoid arthritis. Inflammation 2004, 28, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Routsias, J.G.; Goules, J.D.; Goules, A.; Charalampakis, G.; Pikazis, D. Autopathogenic correlation of periodontitis and rheumatoid arthritis. Rheumatology 2011, 50, 1189–1193. [Google Scholar] [CrossRef] [Green Version]
- Scannapieco, F.A.; Cantos, A. Oral inflammation and infection, and chronic medical diseases: Implications for the elderly. Periodontology 2000 2016, 72, 153–175. [Google Scholar] [CrossRef]
- Darrah, E.; Andrade, F. Rheumatoid arthritis and citrullination. Curr. Opin. Rheumatol. 2018, 30, 72–78. [Google Scholar] [CrossRef]
- Maresz, K.J.; Hellvard, A.; Sroka, A.; Adamowicz, K.; Bielecka, E.; Koziel, J.; Gawron, K.; Mizgalska, D.; Marcinska, K.A.; Benedyk, M.; et al. Porphyromonas gingivalis facilitates the development and progression of destructive arthritis through its unique bacterial peptidylarginine deiminase (PAD). PLoS Pathog. 2013, 9, e1003627. [Google Scholar] [CrossRef]
- Willis, V.C.; Gizinski, A.M.; Banda, N.K.; Causey, C.P.; Knuckley, B.; Cordova, K.N.; Luo, Y.; Levitt, B.; Glogowska, M.; Chandra, P.; et al. N-alpha-benzoyl-N5-(2-chloro-1-iminoethyl)-l-ornithine amide, a protein arginine deiminase inhibitor, reduces the severity of murine collagen-induced arthritis. J. Immunol. 2011, 186, 4396–4404. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, J.R.; Groeger, S.; Johansson, A.; Meyle, J. T helper cells from aggressive periodontitis patients produce higher levels of interleukin-1 beta and interleukin-6 in interaction with Porphyromonas gingivalis. Clin. Oral Investig. 2014, 18, 1835–1843. [Google Scholar] [CrossRef] [PubMed]
- Marchesan, J.T.; Gerow, E.A.; Schaff, R.; Taut, A.D.; Shin, S.Y.; Sugai, J.; Brand, D.; Burberry, A.; Jorns, J.; Lundy, S.K.; et al. Porphyromonas gingivalis oral infection exacerbates the development and severity of collagen-induced arthritis. Arthritis Res. Ther. 2013, 15, R186. [Google Scholar] [CrossRef] [PubMed]
- Mysak, J.; Podzimek, S.; Sommerova, P.; Lyuya-Mi, Y.; Bartova, J.; Janatova, T.; Prochazkova, J.; Duskova, J. Porphyromonas gingivalis: Major periodontopathic pathogen overview. J. Immunol. Res. 2014, 2014, 476068. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, K.; Kinloch, A.; Fisher, B.A.; Wegner, N.; Wait, R.; Charles, P.; Mikuls, T.R.; Venables, P.J. Antibodies to citrullinated alpha-enolase peptide 1 are specific for rheumatoid arthritis and cross-react with bacterial enolase. Arthritis Rheum. 2008, 58, 3009–3019. [Google Scholar] [CrossRef]
- Pischon, N.; Rohner, E.; Hocke, A.; N’Guessan, P.; Muller, H.C.; Matziolis, G.; Kanitz, V.; Purucker, P.; Kleber, B.M.; Bernimoulin, J.P.; et al. Effects of Porphyromonas gingivalis on cell cycle progression and apoptosis of primary human chondrocytes. Ann. Rheum. Dis. 2009, 68, 1902–1907. [Google Scholar] [CrossRef] [PubMed]
- Mikuls, T.R.; Payne, J.B.; Yu, F.; Thiele, G.M.; Reynolds, R.J.; Cannon, G.W.; Markt, J.; McGowan, D.; Kerr, G.S.; Redman, R.S.; et al. Periodontitis and Porphyromonas gingivalis in patients with rheumatoid arthritis. Arthritis Rheumatol. 2014, 66, 1090–1100. [Google Scholar] [CrossRef]
- Fuggle, N.R.; Smith, T.O.; Kaul, A.; Sofat, N. Hand to Mouth: A Systematic Review and Meta-Analysis of the Association between Rheumatoid Arthritis and Periodontitis. Front. Immunol. 2016, 7, 80. [Google Scholar] [CrossRef] [Green Version]
- Lappin, D.F.; Apatzidou, D.; Quirke, A.M.; Oliver-Bell, J.; Butcher, J.P.; Kinane, D.F.; Riggio, M.P.; Venables, P.; McInnes, I.B.; Culshaw, S. Influence of periodontal disease, Porphyromonas gingivalis and cigarette smoking on systemic anti-citrullinated peptide antibody titres. J. Clin. Periodontol. 2013, 40, 907–915. [Google Scholar] [CrossRef]
- Hitchon, C.A.; Chandad, F.; Ferucci, E.D.; Willemze, A.; Ioan-Facsinay, A.; van der Woude, D.; Markland, J.; Robinson, D.; Elias, B.; Newkirk, M.; et al. Antibodies to porphyromonas gingivalis are associated with anticitrullinated protein antibodies in patients with rheumatoid arthritis and their relatives. J. Rheumatol. 2010, 37, 1105–1112. [Google Scholar] [CrossRef]
- Mukherjee, A.; Jantsch, V.; Khan, R.; Hartung, W.; Fischer, R.; Jantsch, J.; Ehrenstein, B.; Konig, M.F.; Andrade, F. Rheumatoid Arthritis-Associated Autoimmunity Due to Aggregatibacter actinomycetemcomitans and Its Resolution With Antibiotic Therapy. Front. Immunol. 2018, 9, 2352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwenzer, A.; Quirke, A.M.; Marzeda, A.M.; Wong, A.; Montgomery, A.B.; Sayles, H.R.; Eick, S.; Gawron, K.; Chomyszyn-Gajewska, M.; Lazarz-Bartyzel, K.; et al. Association of Distinct Fine Specificities of Anti-Citrullinated Peptide Antibodies With Elevated Immune Responses to Prevotella intermedia in a Subgroup of Patients With Rheumatoid Arthritis and Periodontitis. Arthritis Rheumatol. 2017, 69, 2303–2313. [Google Scholar] [CrossRef] [PubMed]
- Courbon, G.; Rinaudo-Gaujous, M.; Blasco-Baque, V.; Auger, I.; Caire, R.; Mijola, L.; Vico, L.; Paul, S.; Marotte, H. Porphyromonas gingivalis experimentally induces periodontis and an anti-CCP2-associated arthritis in the rat. Ann. Rheum. Dis. 2019, 78, 594–599. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Takahashi, N.; Kato, T.; Matsuda, Y.; Yokoji, M.; Yamada, M.; Nakajima, T.; Kondo, N.; Endo, N.; Yamamoto, R.; et al. Aggravation of collagen-induced arthritis by orally administered Porphyromonas gingivalis through modulation of the gut microbiota and gut immune system. Sci. Rep. 2017, 7, 6955. [Google Scholar] [CrossRef]
- Jung, H.; Jung, S.M.; Rim, Y.A.; Park, N.; Nam, Y.; Lee, J.; Park, S.H.; Ju, J.H. Arthritic role of Porphyromonas gingivalis in collagen-induced arthritis mice. PLoS ONE 2017, 12, e0188698. [Google Scholar] [CrossRef]
- de Aquino, S.G.; Abdollahi-Roodsaz, S.; Koenders, M.I.; van de Loo, F.A.; Pruijn, G.J.; Marijnissen, R.J.; Walgreen, B.; Helsen, M.M.; van den Bersselaar, L.A.; de Molon, R.S.; et al. Periodontal pathogens directly promote autoimmune experimental arthritis by inducing a TLR2- and IL-1-driven Th17 response. J. Immunol. 2014, 192, 4103–4111. [Google Scholar] [CrossRef] [PubMed]
- de Aquino, S.G.; Talbot, J.; Sonego, F.; Turato, W.M.; Grespan, R.; Avila-Campos, M.J.; Cunha, F.Q.; Cirelli, J.A. The aggravation of arthritis by periodontitis is dependent of IL-17 receptor A activation. J. Clin. Periodontol. 2017, 44, 881–891. [Google Scholar] [CrossRef]
- Bunte, K.; Beikler, T. Th17 Cells and the IL-23/IL-17 Axis in the Pathogenesis of Periodontitis and Immune-Mediated Inflammatory Diseases. Int. J. Mol. Sci. 2019, 20, 3394. [Google Scholar] [CrossRef]
- Chukkapalli, S.; Rivera-Kweh, M.; Gehlot, P.; Velsko, I.; Bhattacharyya, I.; Calise, S.J.; Satoh, M.; Chan, E.K.; Holoshitz, J.; Kesavalu, L. Periodontal bacterial colonization in synovial tissues exacerbates collagen-induced arthritis in B10.RIII mice. Arthritis Res. Ther. 2016, 18, 161. [Google Scholar] [CrossRef]
- Bartold, P.M.; Marino, V.; Cantley, M.; Haynes, D.R. Effect of Porphyromonas gingivalis-induced inflammation on the development of rheumatoid arthritis. J. Clin. Periodontol. 2010, 37, 405–411. [Google Scholar] [CrossRef]
- Queiroz-Junior, C.M.; Madeira, M.F.; Coelho, F.M.; de Oliveira, C.R.; Candido, L.C.; Garlet, G.P.; Teixeira, M.M.; de Souza Dda, G.; Silva, T.A. Experimental arthritis exacerbates Aggregatibacter actinomycetemcomitans-induced periodontitis in mice. J. Clin. Periodontol. 2012, 39, 608–616. [Google Scholar] [CrossRef] [PubMed]
- Queiroz-Junior, C.M.; Madeira, M.F.; Coelho, F.M.; Costa, V.V.; Bessoni, R.L.; Sousa, L.F.; Garlet, G.P.; Souza Dda, G.; Teixeira, M.M.; Silva, T.A. Experimental arthritis triggers periodontal disease in mice: Involvement of TNF-alpha and the oral Microbiota. J. Immunol. 2011, 187, 3821–3830. [Google Scholar] [CrossRef]
- Cantley, M.D.; Haynes, D.R.; Marino, V.; Bartold, P.M. Pre-existing periodontitis exacerbates experimental arthritis in a mouse model. J. Clin. Periodontol. 2011, 38, 532–541. [Google Scholar] [CrossRef] [PubMed]
- de Pablo, P.; Dietrich, T.; McAlindon, T.E. Association of periodontal disease and tooth loss with rheumatoid arthritis in the US population. J. Rheumatol. 2008, 35, 70–76. [Google Scholar] [PubMed]
- Ayravainen, L.; Leirisalo-Repo, M.; Kuuliala, A.; Ahola, K.; Koivuniemi, R.; Meurman, J.H.; Heikkinen, A.M. Periodontitis in early and chronic rheumatoid arthritis: A prospective follow-up study in Finnish population. BMJ Open 2017, 7, e011916. [Google Scholar] [CrossRef] [PubMed]
- Unriza-Puin, S.; Bautista-Molano, W.; Lafaurie, G.I.; Valle-Onate, R.; Chalem, P.; Chila-Moreno, L.; Bello-Gualtero, J.M.; Romero-Sanchez, C. Are obesity, ACPAs and periodontitis conditions that influence the risk of developing rheumatoid arthritis in first-degree relatives? Clin. Rheumatol. 2017, 36, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, K.; Nise, L.; Kats, A.; Luttropp, E.; Catrina, A.I.; Askling, J.; Jansson, L.; Alfredsson, L.; Klareskog, L.; Lundberg, K.; et al. Prevalence of Periodontitis in Patients with Established Rheumatoid Arthritis: A Swedish Population Based Case-Control Study. PLoS ONE 2016, 11, e0155956. [Google Scholar] [CrossRef] [PubMed]
- Ouedraogo, D.D.; Tiendrebeogo, J.; Guiguimde, P.L.; Nikiema, P.I.; Ouedraogo, D.; Kabore, F.; Zongo, E.; Hayem, G. Periodontal disease in patients with rheumatoid arthritis in Sub-Saharan Africa: A case-control study. Jt. Bone Spine 2017, 84, 113–114. [Google Scholar] [CrossRef]
- Chou, Y.Y.; Lai, K.L.; Chen, D.Y.; Lin, C.H.; Chen, H.H. Rheumatoid Arthritis Risk Associated with Periodontitis Exposure: A Nationwide, Population-Based Cohort Study. PLoS ONE 2015, 10, e0139693. [Google Scholar] [CrossRef]
- Wolff, B.; Berger, T.; Frese, C.; Max, R.; Blank, N.; Lorenz, H.M.; Wolff, D. Oral status in patients with early rheumatoid arthritis: A prospective, case-control study. Rheumatology 2014, 53, 526–531. [Google Scholar] [CrossRef]
- Bartold, P.M.; Marshall, R.I.; Haynes, D.R. Periodontitis and rheumatoid arthritis: A review. J. Periodontol. 2005, 76, 2066–2074. [Google Scholar] [CrossRef] [PubMed]
- Detert, J.; Pischon, N.; Burmester, G.R.; Buttgereit, F. The association between rheumatoid arthritis and periodontal disease. Arthritis Res. Ther. 2010, 12, 218. [Google Scholar] [CrossRef] [PubMed]
- Pischon, N.; Pischon, T.; Kroger, J.; Gulmez, E.; Kleber, B.M.; Bernimoulin, J.P.; Landau, H.; Brinkmann, P.G.; Schlattmann, P.; Zernicke, J.; et al. Association among rheumatoid arthritis, oral hygiene, and periodontitis. J. Periodontol. 2008, 79, 979–986. [Google Scholar] [CrossRef] [PubMed]
- Mercado, F.B.; Marshall, R.I.; Klestov, A.C.; Bartold, P.M. Relationship between rheumatoid arthritis and periodontitis. J. Periodontol. 2001, 72, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Erciyas, K.; Sezer, U.; Ustun, K.; Pehlivan, Y.; Kisacik, B.; Senyurt, S.Z.; Tarakcioglu, M.; Onat, A.M. Effects of periodontal therapy on disease activity and systemic inflammation in rheumatoid arthritis patients. Oral Dis. 2013, 19, 394–400. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, P.; Bissada, N.F.; Palomo, L.; Han, Y.W.; Al-Zahrani, M.S.; Panneerselvam, A.; Askari, A. Periodontal therapy reduces the severity of active rheumatoid arthritis in patients treated with or without tumor necrosis factor inhibitors. J. Periodontol. 2009, 80, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Al-Katma, M.K.; Bissada, N.F.; Bordeaux, J.M.; Sue, J.; Askari, A.D. Control of periodontal infection reduces the severity of active rheumatoid arthritis. J. Clin. Rheumatol. 2007, 13, 134–137. [Google Scholar] [CrossRef]
- Cutolo, M.; Spies, C.M.; Buttgereit, F.; Paolino, S.; Pizzorni, C. The supplementary therapeutic DMARD role of low-dose glucocorticoids in rheumatoid arthritis. Arthritis Res. Ther. 2014, 16, S1. [Google Scholar] [CrossRef]
- Corbett, M.; Chehadah, F.; Biswas, M.; Moe-Byrne, T.; Palmer, S.; Soares, M.; Walton, M.; Harden, M.; Ho, P.; Woolacott, N.; et al. Certolizumab pegol and secukinumab for treating active psoriatic arthritis following inadequate response to disease-modifying antirheumatic drugs: A systematic review and economic evaluation. Health Technol. Assess. 2017, 21, 1–326. [Google Scholar] [CrossRef]
- Kavanaugh, A.; Husni, M.E.; Harrison, D.D.; Kim, L.; Lo, K.H.; Leu, J.H.; Hsia, E.C. Safety and Efficacy of Intravenous Golimumab in Patients With Active Psoriatic Arthritis: Results Through Week Twenty-Four of the GO-VIBRANT Study. Arthritis Rheumatol. 2017, 69, 2151–2161. [Google Scholar] [CrossRef]
- Smolen, J.S.; Burmester, G.R.; Combe, B.; Curtis, J.R.; Hall, S.; Haraoui, B.; van Vollenhoven, R.; Cioffi, C.; Ecoffet, C.; Gervitz, L.; et al. Head-to-head comparison of certolizumab pegol versus adalimumab in rheumatoid arthritis: 2-year efficacy and safety results from the randomised EXXELERATE study. Lancet 2016, 388, 2763–2774. [Google Scholar] [CrossRef]
- Smolen, J.S.; Landewe, R.; Bijlsma, J.; Burmester, G.; Chatzidionysiou, K.; Dougados, M.; Nam, J.; Ramiro, S.; Voshaar, M.; van Vollenhoven, R.; et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2016 update. Ann. Rheum. Dis. 2017, 76, 960–977. [Google Scholar] [CrossRef] [PubMed]
- Weinblatt, M.E.; Bingham, C.O., 3rd; Burmester, G.R.; Bykerk, V.P.; Furst, D.E.; Mariette, X.; van der Heijde, D.; van Vollenhoven, R.; VanLunen, B.; Ecoffet, C.; et al. A Phase III Study Evaluating Continuation, Tapering, and Withdrawal of Certolizumab Pegol after One Year of Therapy in Patients with Early Rheumatoid Arthritis. Arthritis Rheumatol. 2017, 69, 1937–1948. [Google Scholar] [CrossRef] [PubMed]
- Horneff, G.; Schulz, A.C.; Klotsche, J.; Hospach, A.; Minden, K.; Foeldvari, I.; Trauzeddel, R.; Ganser, G.; Weller-Heinemann, F.; Haas, J.P. Experience with etanercept, tocilizumab and interleukin-1 inhibitors in systemic onset juvenile idiopathic arthritis patients from the BIKER registry. Arthritis Res. Ther. 2017, 19, 256. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, J.; Canete, J.D. Anakinra for the treatment of rheumatoid arthritis: A safety evaluation. Expert Opin. Drug Saf. 2018, 17, 727–732. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, H.; Hsu, J.C.; Lu, P.; Fettner, S.; Zhang, X.; Douglass, W.; Bao, M.; Rowell, L.; Burmester, G.R.; Kivitz, A. Pharmacokinetic and Pharmacodynamic Analysis of Subcutaneous Tocilizumab in Patients With Rheumatoid Arthritis From 2 Randomized, Controlled Trials: SUMMACTA and BREVACTA. J. Clin. Pharmacol. 2017, 57, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Ito, S.; Murasawa, A.; Ishikawa, H.; Yoshie, H. Effects of tofacitinib on the clinical features of periodontitis in patients with rheumatoid arthritis: Two case reports. BMC Rheumatol. 2019, 3, 13. [Google Scholar] [CrossRef] [PubMed]
- Fleischmann, R.; Mysler, E.; Hall, S.; Kivitz, A.J.; Moots, R.J.; Luo, Z.; DeMasi, R.; Soma, K.; Zhang, R.; Takiya, L.; et al. Efficacy and safety of tofacitinib monotherapy, tofacitinib with methotrexate, and adalimumab with methotrexate in patients with rheumatoid arthritis (ORAL Strategy): A phase 3b/4, double-blind, head-to-head, randomised controlled trial. Lancet 2017, 390, 457–468. [Google Scholar] [CrossRef]
- Schwartz, D.M.; Kanno, Y.; Villarino, A.; Ward, M.; Gadina, M.; O’Shea, J.J. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat. Rev. Drug Discov. 2017, 16, 843–862. [Google Scholar] [CrossRef]
- Lee, E.B.; Fleischmann, R.; Hall, S.; Wilkinson, B.; Bradley, J.D.; Gruben, D.; Koncz, T.; Krishnaswami, S.; Wallenstein, G.V.; Zang, C.; et al. Tofacitinib versus methotrexate in rheumatoid arthritis. N. Engl. J. Med. 2014, 370, 2377–2386. [Google Scholar] [CrossRef]
- Koenders, M.I.; van den Berg, W.B. Novel therapeutic targets in rheumatoid arthritis. Trends Pharmacol. Sci. 2015, 36, 189–195. [Google Scholar] [CrossRef]
- Gualtierotti, R.; Marzano, A.V.; Spadari, F.; Cugno, M. Main Oral Manifestations in Immune-Mediated and Inflammatory Rheumatic Diseases. J. Clin. Med. 2018, 8, 21. [Google Scholar] [CrossRef] [PubMed]
- Holzhausen, M.; Rossa Junior, C.; Marcantonio Junior, E.; Nassar, P.O.; Spolidorio, D.M.; Spolidorio, L.C. Effect of selective cyclooxygenase-2 inhibition on the development of ligature-induced periodontitis in rats. J. Periodontol. 2002, 73, 1030–1036. [Google Scholar] [CrossRef] [PubMed]
- Nassar, C.A.; Nassar, P.O.; Abi Rached, R.S.; Holzhausen, M.; Marcantonio, E., Jr.; Spolidorio, L.C. Effect of cyclosporin A on alveolar bone homeostasis in a rat periodontitis model. J. Periodontal Res. 2004, 39, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Vogel, R.I.; Copper, S.A.; Schneider, L.G.; Goteiner, D. The effects of topical steroidal and systemic nonsteroidal anti-inflammatory drugs on experimental gingivitis in man. J. Periodontol. 1984, 55, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Feldman, R.S.; Szeto, B.; Chauncey, H.H.; Goldhaber, P. Non-steroidal anti-inflammatory drugs in the reduction of human alveolar bone loss. J. Clin. Periodontol. 1983, 10, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Spolidorio, L.C.; Spolidorio, D.M.; Nassar, P.O.; Nassar, C.A.; Holzhausen, M.; Almeida, O.P. Influence of age on combined effects of cyclosporin and nifedipine on rat alveolar bone. J. Periodontol. 2004, 75, 268–272. [Google Scholar] [CrossRef]
- Schmalz, G.; Berisha, L.; Wendorff, H.; Widmer, F.; Marcinkowski, A.; Teschler, H.; Sommerwerck, U.; Haak, R.; Kollmar, O.; Ziebolz, D. Association of time under immunosuppression and different immunosuppressive medication on periodontal parameters and selected bacteria of patients after solid organ transplantation. Med. Oral Patol. Oral Cir. Bucal 2018, 23, e326–e334. [Google Scholar] [CrossRef] [PubMed]
- Groenewegen, H.; Bierman, W.F.W.; Delli, K.; Dijkstra, P.U.; Nesse, W.; Vissink, A.; Spijkervet, F.K.L. Severe periodontitis is more common in HIV- infected patients. J. Infect. 2019, 78, 171–177. [Google Scholar] [CrossRef]
- Deeming, G.M.; Collingwood, J.; Pemberton, M.N. Methotrexate and oral ulceration. Br. Dent. J. 2005, 198, 83–85. [Google Scholar] [CrossRef] [Green Version]
- Beeraka, S.S.; Natarajan, K.; Patil, R.; Manne, R.K.; Prathi, V.S.; Kolaparthi, V.S. Clinical and radiological assessment of effects of long-term corticosteroid therapy on oral health. Dent. Res. J. 2013, 10, 666–673. [Google Scholar]
- Payne, J.B.; Golub, L.M.; Thiele, G.M.; Mikuls, T.R. The Link Between Periodontitis and Rheumatoid Arthritis: A Periodontist’s Perspective. Curr. Oral Health Rep. 2015, 2, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Jung, G.U.; Han, J.Y.; Hwang, K.G.; Park, C.J.; Stathopoulou, P.G.; Fiorellini, J.P. Effects of Conventional Synthetic Disease-Modifying Antirheumatic Drugs on Response to Periodontal Treatment in Patients with Rheumatoid Arthritis. Biomed. Res. Int. 2018, 2018, 1465402. [Google Scholar] [CrossRef] [PubMed]
- Ziebolz, D.; Rupprecht, A.; Schmickler, J.; Bothmann, L.; Kramer, J.; Patschan, D.; Muller, G.A.; Mausberg, R.F.; Schmidt, J.; Schmalz, G.; et al. Association of different immunosuppressive medications with periodontal condition in patients with rheumatoid arthritis: Results from a cross-sectional study. J. Periodontol. 2018, 89, 1310–1317. [Google Scholar] [CrossRef] [PubMed]
- Romero-Sanchez, C.; Rodriguez, C.; Santos-Moreno, P.; Mesa, A.M.; Lafaurie, G.I.; Giraldo, Q.S.; De-Avila, J.; Castillo, D.M.; Duran, M.; Chalem, P.C.; et al. Is the Treatment with Biological or Non-biological DMARDS a Modifier of Periodontal Condition in Patients with Rheumatoid Arthritis? Curr. Rheumatol. Rev. 2017, 13, 139–151. [Google Scholar] [CrossRef]
- Kobayashi, T.; Ito, S.; Kobayashi, D.; Kojima, A.; Shimada, A.; Narita, I.; Murasawa, A.; Nakazono, K.; Yoshie, H. Interleukin-6 receptor inhibitor tocilizumab ameliorates periodontal inflammation in patients with rheumatoid arthritis and periodontitis as well as tumor necrosis factor inhibitors. Clin. Exp. Dent. Res. 2015, 1, 63–73. [Google Scholar] [CrossRef]
- Coat, J.; Demoersman, J.; Beuzit, S.; Cornec, D.; Devauchelle-Pensec, V.; Saraux, A.; Pers, J.O. Anti-B lymphocyte immunotherapy is associated with improvement of periodontal status in subjects with rheumatoid arthritis. J. Clin. Periodontol. 2015, 42, 817–823. [Google Scholar] [CrossRef]
- Kobayashi, T.; Okada, M.; Ito, S.; Kobayashi, D.; Ishida, K.; Kojima, A.; Narita, I.; Murasawa, A.; Yoshie, H. Assessment of interleukin-6 receptor inhibition therapy on periodontal condition in patients with rheumatoid arthritis and chronic periodontitis. J. Periodontol. 2014, 85, 57–67. [Google Scholar] [CrossRef]
- Ustun, K.; Erciyas, K.; Kisacik, B.; Sezer, U.; Pehlivan, Y.; Oztuzcu, S.; Gundogar, H.; Onat, A.M. Host modulation in rheumatoid arthritis patients with TNF blockers significantly decreases biochemical parameters in periodontitis. Inflammation 2013, 36, 1171–1177. [Google Scholar] [CrossRef]
- Mayer, Y.; Elimelech, R.; Balbir-Gurman, A.; Braun-Moscovici, Y.; Machtei, E.E. Periodontal condition of patients with autoimmune diseases and the effect of anti-tumor necrosis factor-alpha therapy. J. Periodontol. 2013, 84, 136–142. [Google Scholar] [CrossRef]
- Savioli, C.; Ribeiro, A.C.; Fabri, G.M.; Calich, A.L.; Carvalho, J.; Silva, C.A.; Viana, V.S.; Bonfa, E.; Siqueira, J.T. Persistent periodontal disease hampers anti-tumor necrosis factor treatment response in rheumatoid arthritis. J. Clin. Rheumatol. 2012, 18, 180–184. [Google Scholar] [CrossRef] [PubMed]
- Mayer, Y.; Balbir-Gurman, A.; Machtei, E.E. Anti-tumor necrosis factor-alpha therapy and periodontal parameters in patients with rheumatoid arthritis. J. Periodontol. 2009, 80, 1414–1420. [Google Scholar] [CrossRef] [PubMed]
- Pers, J.O.; Saraux, A.; Pierre, R.; Youinou, P. Anti-TNF-alpha immunotherapy is associated with increased gingival inflammation without clinical attachment loss in subjects with rheumatoid arthritis. J. Periodontol. 2008, 79, 1645–1651. [Google Scholar] [CrossRef] [PubMed]
- Miranda, L.A.; Islabao, A.G.; Fischer, R.G.; Figueredo, C.M.; Oppermann, R.V.; Gustafsson, A. Decreased interleukin-1beta and elastase in the gingival crevicular fluid of individuals undergoing anti-inflammatory treatment for rheumatoid arthritis. J. Periodontol. 2007, 78, 1612–1619. [Google Scholar] [CrossRef] [PubMed]
- Han, J.Y.; Reynolds, M.A. Effect of anti-rheumatic agents on periodontal parameters and biomarkers of inflammation: A systematic review and meta-analysis. J. Periodontal Implant Sci. 2012, 42, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Yokoyama, T.; Ito, S.; Kobayashi, D.; Yamagata, A.; Okada, M.; Oofusa, K.; Narita, I.; Murasawa, A.; Nakazono, K.; et al. Periodontal and serum protein profiles in patients with rheumatoid arthritis treated with tumor necrosis factor inhibitor adalimumab. J. Periodontol. 2014, 85, 1480–1488. [Google Scholar] [CrossRef] [PubMed]
- McGowan, K.; McGowan, T.; Ivanovski, S. Optimal dose and duration of amoxicillin-plus-metronidazole as an adjunct to non-surgical periodontal therapy: A systematic review and meta-analysis of randomized, placebo-controlled trials. J. Clin. Periodontol. 2018, 45, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Silvestre, F.J.; Silvestre-Rangil, J.; Bagan, L.; Bagan, J.V. Effect of nonsurgical periodontal treatment in patients with periodontitis and rheumatoid arthritis: A systematic review. Med. Oral Patol. Oral Cir. Bucal 2016, 21, e349–e354. [Google Scholar] [CrossRef]
- Cosgarea, R.; Tristiu, R.; Dumitru, R.B.; Arweiler, N.B.; Rednic, S.; Sirbu, C.I.; Lascu, L.; Sculean, A.; Eick, S. Effects of non-surgical periodontal therapy on periodontal laboratory and clinical data as well as on disease activity in patients with rheumatoid arthritis. Clin. Oral Investig. 2019, 23, 141–151. [Google Scholar] [CrossRef]
- Kaushal, S.; Singh, A.K.; Lal, N.; Das, S.K.; Mahdi, A.A. Effect of periodontal therapy on disease activity in patients of rheumatoid arthritis with chronic periodontitis. J. Oral Biol. Craniofac. Res. 2019, 9, 128–132. [Google Scholar] [CrossRef]
- Monsarrat, P.; de Grado, G.F.; Constantin, A.; Willmann, C.; Nabet, C.; Sixou, M.; Cantagrel, A.; Barnetche, T.; Mehsen, N.; Schaeverbeke, T.; et al. The effect of periodontal treatment on patients with rheumatoid arthritis: The ESPERA randomised controlled trial. Jt. Bone Spine 2019, in press. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Liu, Z.; Shu, D.; Xiong, Y.; He, M.; Xu, S.; Si, S.; Guo, B. Association of Periodontitis with Rheumatoid Arthritis and the Effect of Non-Surgical Periodontal Treatment on Disease Activity in Patients with Rheumatoid Arthritis. Med. Sci. Monit. 2018, 24, 5802–5810. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.Y.; Wang, C.Y.; Chyuan, I.T.; Wu, K.J.; Tu, Y.K.; Chang, C.W.; Hsu, P.N.; Kuo, M.Y.; Chen, Y.W. Significant association of rheumatoid arthritis-related inflammatory markers with non-surgical periodontal therapy. J. Formos. Med. Assoc. 2018, 117, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Balci Yuce, H.; Gokturk, O.; Aydemir Turkal, H.; Inanir, A.; Benli, I.; Demir, O. Assessment of local and systemic 25-hydroxy-vitamin D, RANKL, OPG, and TNF levels in patients with rheumatoid arthritis and periodontitis. J. Oral Sci. 2017, 59, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Kurgan, S.; Onder, C.; Balci, N.; Fentoglu, O.; Eser, F.; Balseven, M.; Serdar, M.A.; Tatakis, D.N.; Gunhan, M. Gingival crevicular fluid tissue/blood vessel-type plasminogen activator and plasminogen activator inhibitor-2 levels in patients with rheumatoid arthritis: Effects of nonsurgical periodontal therapy. J. Periodontal Res. 2017, 52, 574–581. [Google Scholar] [CrossRef]
- Kurgan, S.; Fentoglu, O.; Onder, C.; Serdar, M.; Eser, F.; Tatakis, D.N.; Gunhan, M. The effects of periodontal therapy on gingival crevicular fluid matrix metalloproteinase-8, interleukin-6 and prostaglandin E2 levels in patients with rheumatoid arthritis. J. Periodontal Res. 2016, 51, 586–595. [Google Scholar] [CrossRef] [PubMed]
- Biyikoglu, B.; Buduneli, N.; Aksu, K.; Nalbantsoy, A.; Lappin, D.F.; Evrenosoglu, E.; Kinane, D.F. Periodontal therapy in chronic periodontitis lowers gingival crevicular fluid interleukin-1beta and DAS28 in rheumatoid arthritis patients. Rheumatol. Int. 2013, 33, 2607–2616. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Kobayashi, T.; Ito, S.; Yokoyama, T.; Abe, A.; Murasawa, A.; Yoshie, H. Periodontal treatment decreases levels of antibodies to Porphyromonas gingivalis and citrulline in patients with rheumatoid arthritis and periodontitis. J. Periodontol. 2013, 84, e74–e84. [Google Scholar] [CrossRef] [PubMed]
- Pinho Mde, N.; Oliveira, R.D.; Novaes, A.B., Jr.; Voltarelli, J.C. Relationship between periodontitis and rheumatoid arthritis and the effect of non-surgical periodontal treatment. Braz. Dent. J. 2009, 20, 355–364. [Google Scholar] [CrossRef] [Green Version]
- Kaur, S.; Bright, R.; Proudman, S.M.; Bartold, P.M. Does periodontal treatment influence clinical and biochemical measures for rheumatoid arthritis? A systematic review and meta-analysis. Semin. Arthritis Rheum. 2014, 44, 113–122. [Google Scholar] [CrossRef]
- Havemose-Poulsen, A.; Sorensen, L.K.; Stoltze, K.; Bendtzen, K.; Holmstrup, P. Cytokine profiles in peripheral blood and whole blood cell cultures associated with aggressive periodontitis, juvenile idiopathic arthritis, and rheumatoid arthritis. J. Periodontol. 2005, 76, 2276–2285. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Murasawa, A.; Komatsu, Y.; Yokoyama, T.; Ishida, K.; Abe, A.; Yamamoto, K.; Yoshie, H. Serum cytokine and periodontal profiles in relation to disease activity of rheumatoid arthritis in Japanese adults. J. Periodontol. 2010, 81, 650–657. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, M.; Kopp, S. Gingivitis and periodontitis are related to repeated high levels of circulating tumor necrosis factor-alpha in patients with rheumatoid arthritis. J. Periodontol. 2008, 79, 1689–1696. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | Country | Patient Number | Objective | Study Design | Findings | Conclusions |
|---|---|---|---|---|---|---|
| Jung et al. (2018) [193] | Korea | 64 | To evaluate the adjunctive effect of DMARDs in response to NSPT in RA patients. | Prospective clinical trial. All patients received NSPT and only the RA-PD group received DMARDs. Periodontitis indices (probing depth, CAL, GI, and BOP) were evaluated at the baseline and four weeks later. | Four weeks after NSPT, the periodontal indices (probing depth reduction, and CAL gain) were significantly different in the RA group treated with DMARDs compared to the systemically healthy patients. | The study provides clinical evidence that DMARDs may have an adjunctive effect on response to NSPT in patients with RA. |
| Ziebolz et al. (2018) [194] | Germany | 168 | To investigate clinical periodontal findings in patients with RA under immunosuppressive rheumatic medications. | Cross-sectional study. Patients with RA treated with different immunosuppressive medications were involved. Periodontal parameters (probing depth, BOP and CAL) was measured. | RA medication was associated with periodontal inflammation, without differences in PD severity. | Based upon their mechanisms of action and efficacy in the reduction of systemic inflammation associated with RA-related medications, they have varying effects on periodontal inflammation. |
| Romero-Sanches et al. (2017) [195] | Colombia | 177 | To evaluate the effects of conventional drug treatment and anti-TNF therapy in patients with RA on microbiological and periodontal condition. | Prospective clinical trial. RA patients under anti-TNF therapy and under DMARD were involved. Periodontal evaluation (BOP, CAL, probing depth) and rheumatologic markers (ACPA, RF, DAS28, ESR and CRP) were measured. | The anti-TNF therapy with methotrexate resulted in lower extension of CAL. Increased ACPAs titers were associated with the presence of periodontal pathogens. BOP was associated with elevated CRP levels, and ESR was associated with a greater probing depth. | RA treatment affect the clinical condition and subgingival microbiota. |
| Ayravainen et al. (2017) [155] | Finland | 124 | To evaluate the role of antirheumatic medication in the periodontal health. | Prospective follow up clinical trial. RA patients treated with synthetic DMARD; patients with chronic RA treated with biological DMARDs. Degree of PD (probing depth, BOP and CAL) and clinical RA status (DAS28) were measured. | Periodontal status in patients with RA was worse compared to the population controls. Almost 80% of patients with synthetic DMARDs and 85% of patients with biological DMARDs suffered from PD compared to 40% of the controls. | There was no association between antirheumatic treatment and periodontal parameters. |
| Kobayashi et al. (2015) [196] | Japan | 60 | To compare the periodontal condition in patients with RA and PD before and after treatment with the anti-human IL-6 receptor (IL-6R) monoclonal antibody (Tocilizumab—TCZ) and anti-TNF therapy. | Longitudinal case control study. Patients with RA-PD treated with TCZ and patients with RA-PD who received the anti-TNF were involved. Clinical periodontal (GI, CAL, BOP and probing depth) and rheumatologic assessments (DAS28 and CRP) were assessed at the baseline and three and six months later. | Decreased levels of GI, BOP, and probing depth in patients with RA-PD after medication with anti-IL6 and anti-TNF were observed. Both therapies decreased DAS28, CRP, the number of tender and swollen joints, and serum levels of ACPA, RF, CRP, and MMP-3. | Anti-IL6 therapy significantly decreased the levels of periodontal inflammation in patients with RA-PD. |
| Coat et al. (2015) [197] | France | 21 | To evaluate the efficacy of rituximab in the periodontal parameters of patients with RA. | A cross-sectional and longitudinal study. Patients were divided in two groups: Group 1 received two doses of rituximab and group 2 received more than two courses of rituximab. The periodontal status (BOP, GI, CAL and probing depth) were measured. | Significant decrease in the probing depth and CAL were observed after six months of treatment with rituximab in group 1. Patients from group 2 presented better periodontal status than patients from group 1 before treatment with rituximab. | Anti-IL6 therapy could be beneficial to improve PD. |
| Kobayashi et al. (2014) [198] | Japan | 20 | To assess the effect of the anti-TNF inhibitor (adalimumab), on the periodontal condition of patients with RA and to compare the serum protein profiles before and after therapy. | Prospective clinical trial. Patients with RA under the adalimumab treatment were included. Periodontal indices (GI, BOP, CAL and probing depth) and rheumatologic scores (DAS28-CRP) were measured. | A significant decrease in GI, BOP, probing depth, DAS28-CRP, and serum levels of TNF-α and IL-6 after adalimumab therapy were evidenced. | These findings might suggest a promising effect of adalimumab therapy on the periodontal condition of patients with RA. |
| Ustun et al. (2013) [199] | Turkey | 16 | To evaluate the effects of host modulation with the anti-TNF therapy in periodontal tissues of patients with RA. | Longitudinal clinical trial. RA individuals were included, and periodontal indices (BOP, CAL, GI, and probing depth), GCF samples of IL-1β, IL-8 and MCP-1, and arthritis parameters (DAS28, CRP, and ESR) were measured at the baseline and 30 days after. | A decrease in the GCF volume, as well as IL-1β, IL-8, and MCP-1 levels in RA patients on the anti-TNF therapy was observed compared to the baseline. Probing depth and CAL of all patients remained unchanged. After 30 days of the anti-TNF therapy, CRP, ESR and DAS28 values were significantly lower compared to the baseline. | Host modulation might alter biochemical parameters of the periodontium in PD patients even without NSPT. |
| Mayer et al. (2013) [200] | Israel | 58 | To evaluate the effect of autoimmune diseases (AD) treated with anti-TNF on the clinical and immunologic parameters of the periodontium. | Observational clinical trial. Patients with AD were enrolled (12 RA; 12 psoriatic arthritis; 12 systemic sclerosis patients). Ten RA patients were at the anti-TNF therapy (RA+) and 12 were systemically health individuals (H). the periodontal indices (GI, BOP, CAL and probing depth) and TNF-α levels were measured. | No differences were found among the AD groups in clinical and immunologic parameters. GI was increased in the AD patients compared to the H and RA+ groups. Significantly more BOP and decreased probing depth in the SD groups were observed compared to H and RA+. Increased levels of TNF-α in the AD groups were seen compared to H and RA+. | Patients with AD diseases presented with worse PD and higher TNF-α levels than the H controls. Anti-TNF-α treatment appears to hinder this scenario. |
| Savioli et al. (2012) [201] | Brazil | 18 | To evaluate the influence and the evolution of PD in RA patients treated with anti-TNF-α. | Longitudinal and prospective clinical. RA patients on the anti-TNF treatment were included. Periodontal assessment (GI, BOP, CAL and probing depth) and rheumatologic evaluation (DAS28, ESR, and CRP) were measured at the baseline and six months later. | Eight out of 18 patients were diagnosed with PD. Periodontal indices were stable in the entire group throughout the experimental period. Significant improvement in all rheumatologic parameters were evidenced after six months of treatment with anti-TNF. This improvement was restricted to the individuals without PD. | PD patients did not improve rheumatologic parameters. Underlying PD may affect TNF blockers efficacy in patients with RA. |
| Mayer et al. (2009) [202] | Israel | 30 | To investigate the influence of the anti-TNF-α therapy on the clinical and immunologic parameters of the periodontium. | Longitudinal clinical study including 10 subjects with RA receiving anti-TNF-α; 10 RA patients without biological DMARD and 10 health control. Periodontal parameters (GI, BOP, CAL and probing depth) were measured as well as levels of TNF-α in GCF and DAS28. | The anti-TNF- α therapy decreased the GCF levels of TNF-α and lead to milder PD (decreased probing depth and CAL) compared to the RA patients who did not receive this medication. Rheumatologic markers (DAS28, CCP and RF) were similar between groups receiving or not receiving the medication. | Anti-TNF-α agents may halt periodontal inflammation and bone resorption. |
| Pers et al. (2008) [203] | France | 40 | To investigate the beneficial effects of anti-TNF-α in RA patients with coexisting PD. | Cross sectional and longitudinal study. Patients were divided into: RA patients that had already started treatment at the time of periodontal examination and RA patients that were evaluated before treatment. | A significant decrease in CAL was observed in RA patients receiving anti-TNF-α. On the other hand, increased gingival inflammation was evidenced in patients under the infliximab therapy with the coexisting PD. | Blocking the TNF-α activity may help in the treatment of PD. |
| Miranda et al. (2007) [204] | Brazil | 34 | Aiming at comparing the inflammatory activity in the GCF of RA patients and to evaluate the effect of the RA treatment on PD | Cross sectional observational study. Seventeen patients were RA positive and the other half were health control. IL-1β, -18 and the elastase activity were measured. RA patients were under NSAID medication. | Significantly lower amounts of IL-1β and elastase activity in RA individuals were observed when compared to the health control. | The NSAID treatment taken by RA patients might influence the PD status by decreasing inflammatory mediators commonly seen during the PD progression. |
| Study | Country | Patient Number | Objective | Study Design | Findings | Conclusions |
|---|---|---|---|---|---|---|
| Cosgarea et al. (2019) [209] | Romania | 36 | To evaluate the effect of NSPT in patients with RA and PD. | Prospective, case-controlled trial. The RA-PD group and the PD group were treated with scale and root planning (SRP). At the baseline and at three and six months after SRP the periodontal status and RA disease activity were measured. | RA patients presented a statistically significant decrease in the serum-CRP at three months. At all time-points, levels of inflammatory markers in GCF were higher in the RA-PD than in PD patients. | Only tendencies to reduction of DAS28 were observed after three and six months after NSPT. |
| Kaushal et al. (2019) [210] | India | 40 | To evaluate the effects of NSPT on the RA disease activity. | Prospective clinical trial. PD and RA parameters were examined at the baseline and eight weeks following NSPT. | Significant reduction in PI, GI, PPD, CAL and DAS28 scores were observed in patients that received NSPT compared to the untreated patients. The serum levels of ACPA, RF and CRP were not different between groups. | NSPT improved the RA disease activity and periodontal clinical conditions. |
| Monsarrat et al. (2019) [211] | France | 22 | To assess the effects of NSPT on the clinical and biochemical parameters of the RA disease activity and quality of life. | Open-label randomized clinical trial. Patients were allocated to immediate and delayed NSPT. The DAS28-ESR and health assessment questionnaire were employed. The PD and RA parameters were examined three months following NSPT. | NSPT did not lead to a significant reduction of the DAS28-ESR scores in RA patients with PD. Improvement in all periodontal parameters were evidenced after NSPT. | No improvement of the quality of life after NSPT was noted. No beneficial effect of NSPT was observed on patients with active RA. |
| Zhao et al. (2018) [212] | China | 64 | To investigate the effects of NSPT on RA. | Prospective clinical study. Patients were divided into four groups: PD patients, RA, RA-PD, and healthy controls. PD and RA parameters were examined at the baseline and one month following NSPT. | The RA-PD group had significantly higher levels of CRP, ACPA, ESR, and DAS28 than those in the RA group. | NSPT lead to improvement of rheumatologic parameters in RA-PD patients. RA showed little effect on accelerating the development of PD. |
| Yang et al. (2018) [213] | Taiwan | 31 | Aiming at investigating the effect of NSPT on the serum levels of RA-related inflammatory markers in patients with PD. | Prospective clinical trial. Patients were treated with NSPT and the serum levels of ACPA, RF, TNF-α, CRP, IL-1β, and IL-6 were measured at the end of the treatment. | NSPT significantly reduced the levels of ACPA and TNF-α in the serum of PD patients. A positive correlation was noted between the number of extracted teeth and the reduction of ACPA and IL-1β after NSPT. | RA-clinical parameter might be improved after NSPT. |
| Balci Yuce et al. (2017) [214] | Turkey | 53 | To evaluate proinflammatory cytokine and vitamin D levels in RA and PD patients before and after NSPT. | Controlled, parallel-group clinical trial. Patients were treated with NSPT and levels of vitamin D, TNF-α, OPG, and RANKL in GCF and serum were measured. | After NSPT, the levels of 25-hydroxy-vitamin D were reduced in RA-PD patients. RANKL and TNF-α levels in RA patients decreased after NSPT. | Significant improvements in clinical parameters after NSPT in both RA and PD patients were observed. |
| Kurgan et al. (2017) [215] | Turkey | 45 | To evaluate the effect of NSPT on clinical parameters and GCF levels of t-PA and PAI-2 in patients with PD, with or without RA. | Prospective clinical trial evaluating T-PA, PAI-2, CRP, DAS28, ESR and periodontal parameters were measured at the baseline and three months after NSPT. | All periodontal clinical parameters were significantly higher in the RA-PD and PD groups compared with the control group. NSPT significantly reduced the GCF t-PA levels in the RA-PD group. | NSPT significantly improves clinical periodontal parameters both in RA-PD and in the PD patients. |
| Kurgan et al. (2016) [216] | Turkey | 66 | To evaluate whether NSPT influences the levels of MMP-8, IL-6 and PGE2 in the GCF, and serum levels of RA biomarkers in patients with RA-PD. | Observational clinical trial. Patients were evaluated at the baseline and after three months of NSPT. | The GCF levels of MMP-8, PGE2 and IL-6 were higher in all groups than the control. After NSPT, there were significant decreases in the GCF levels of MMP-8, PGE2 and IL-6 from patients with RA-PD. | NSPT may provide beneficial effects on local inflammatory mediators via decreases in the GCF of inflammatory biomarkers. |
| Bıyıkoglu et al. (2013) [217] | Turkey | 30 | To evaluate clinical and biochemical outcomes of NSPT on the serum and GCF in PD patients with or without RA. | Single-centered interventional study. Clinical and biochemical periodontal (IL-1β, and TNF-α) and RA (DAS28) parameters were obtained at the baseline, one, three, and six months after NSPT. | The DAS28 decreased significantly after NSPT in the RA-PD group. The serum TNF-α of the PD group were significantly higher than those of RA-PD. After NSPT, no changes were noted in the levels of these cytokines. The GCF of the IL-1β levels decreased in both groups after NSPT. At six-months, the GCF of the IL-1β levels were significantly lower than the baseline. | NSPT might be beneficial in decreasing local inflammatory markers of PD and RA. |
| Okada et al. (2013) [218] | Japan | 55 | To evaluate whether NSPT affect the serum antibodies to P. gingivalis and citrulline levels in relation to the disease activity of RA. | Interventional and prospective clinical trial. Periodontal and rheumatologic parameters and serum levels of cytokine and inflammatory markers citrulline and IgG to P. gingivalis were examined at the baseline and eight weeks later. | The NSPT group exhibited a significantly greater decrease in DAS28-CRP, the serum levels of IgG to P. gingivalis, and citrulline than the control group. The serum levels of IgG to P. gingivalis were positively correlated with those of the ACPA antibodies. | The findings suggest that NSPT decreases DAS28-CRP and the serum levels of IgG to P. gingivalis and citrulline in the RA patients and may reflect a role of P. gingivalis in the protein citrullination. |
| Erciyas et al. (2013) [165] | Turkey | 60 | Aiming at evaluating the effects of NSPT on clinical periodontal measurements and systemic inflammatory mediator levels in RA-PD patients. | Observational prospective cohort study. Thirty patients were RA-PD with a moderate to high DAS28 score and the others were RA-PD with a low DAS28 score. The ESR, CRP, TNF-α levels in serum, DAS28 and periodontal parameters were evaluated at the baseline and after three months of NSPT. | The ESR, CRP, TNF-α levels in serum, DAS28 and periodontal parameters exhibited similar and significant reduction three months after the NSPT. | These findings might indicate beneficial effects of NSPT in reducing RA severity as measured by a significant decrease in inflammatory markers in the serum and DAS28 score in low or moderate to highly active RA patients with PD. |
| Pinho et al. (2009) [219] | Brazil | 75 | To evaluate the effects of NSPT on clinical and laboratory parameters in patients with RA and PD. | Clinical and interventional trial. Patients were assigned to five groups according to the presence or absence of RA and PD and with or without NSPT. Clinical periodontal indices, DAS28, CRP, ESR and alpha-1 acid glycoprotein (AAG) were measured at the baseline, three and six months after NSPT. | Significant reduction of periodontal clinical parameters was observed in three and six months after NSPT. A significant decrease in the DAS28 scores of patients with RA-PD were observed when compared to the RA patients that underwent NSPT at the baseline and after three months, but no differences were found after six months. | NSPT might be considered an adjunctive approach to reduce the levels of DAS28 in the RA patients. No other parameters for RA (CRP, ESR and AAG) were significantly affected by NSPT. |
| Ortiz et al. (2009) [166] | USA | 40 | To investigate the effect of NSPT on the signs and symptoms of RA in patients treated with or without anti-TNF-α. | Clinical and interventional trial. RA-PD patients under the RA treatment were enrolled. Half of them received NSPT and the other half did not. Clinical periodontal parameters and RA disease activity levels (DAS28 and ESR) were measured at the baseline and six weeks later. | NSPT lead to a significant decrease in the mean DAS28, ESR, and serum TNF-α levels. No significant decrease in these parameters were observed in the untreated control patients. The anti-TNF-α therapy decreased the clinical signs of periodontitis characterized by a reduction in CAL, BOP, probing depth and GI. | The control of PD and inflammation by means of NSPT might contribute to a reduction in the signs and symptoms of active RA. |
| Al Katma et al. (2007) [167] | USA | 29 | To evaluate the impact of NSPT on the activity of RA. | Prospective clinical trial. Seventeen RA-PD patients received NSPT and 12 did not. Patients were under the DMARD medication. RA measurements (DAS28 and ESR) and PD indices (CAL, probing depth, BOP, GI) were measured at the baseline and eight weeks after. | Significant decrease in the DAS28 and ESR levels were observed in patients under the NSPT treatment compared to the untreated control. NSPT led to a significant improvement in all periodontal clinical parameters including PI, GI, BOP, and probing depth. | NSPT might reduce the severity of periodontal patients with active RA. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Molon, R.S.; Rossa Jr., C.; Thurlings, R.M.; Cirelli, J.A.; Koenders, M.I. Linkage of Periodontitis and Rheumatoid Arthritis: Current Evidence and Potential Biological Interactions. Int. J. Mol. Sci. 2019, 20, 4541. https://doi.org/10.3390/ijms20184541
de Molon RS, Rossa Jr. C, Thurlings RM, Cirelli JA, Koenders MI. Linkage of Periodontitis and Rheumatoid Arthritis: Current Evidence and Potential Biological Interactions. International Journal of Molecular Sciences. 2019; 20(18):4541. https://doi.org/10.3390/ijms20184541
Chicago/Turabian Stylede Molon, Rafael Scaf, Carlos Rossa Jr., Rogier M. Thurlings, Joni Augusto Cirelli, and Marije I. Koenders. 2019. "Linkage of Periodontitis and Rheumatoid Arthritis: Current Evidence and Potential Biological Interactions" International Journal of Molecular Sciences 20, no. 18: 4541. https://doi.org/10.3390/ijms20184541