A Model for the Homotypic Interaction between Na+,K+-ATPase β1 Subunits Reveals the Role of Extracellular Residues 221–229 in Its Ig-Like Domain

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
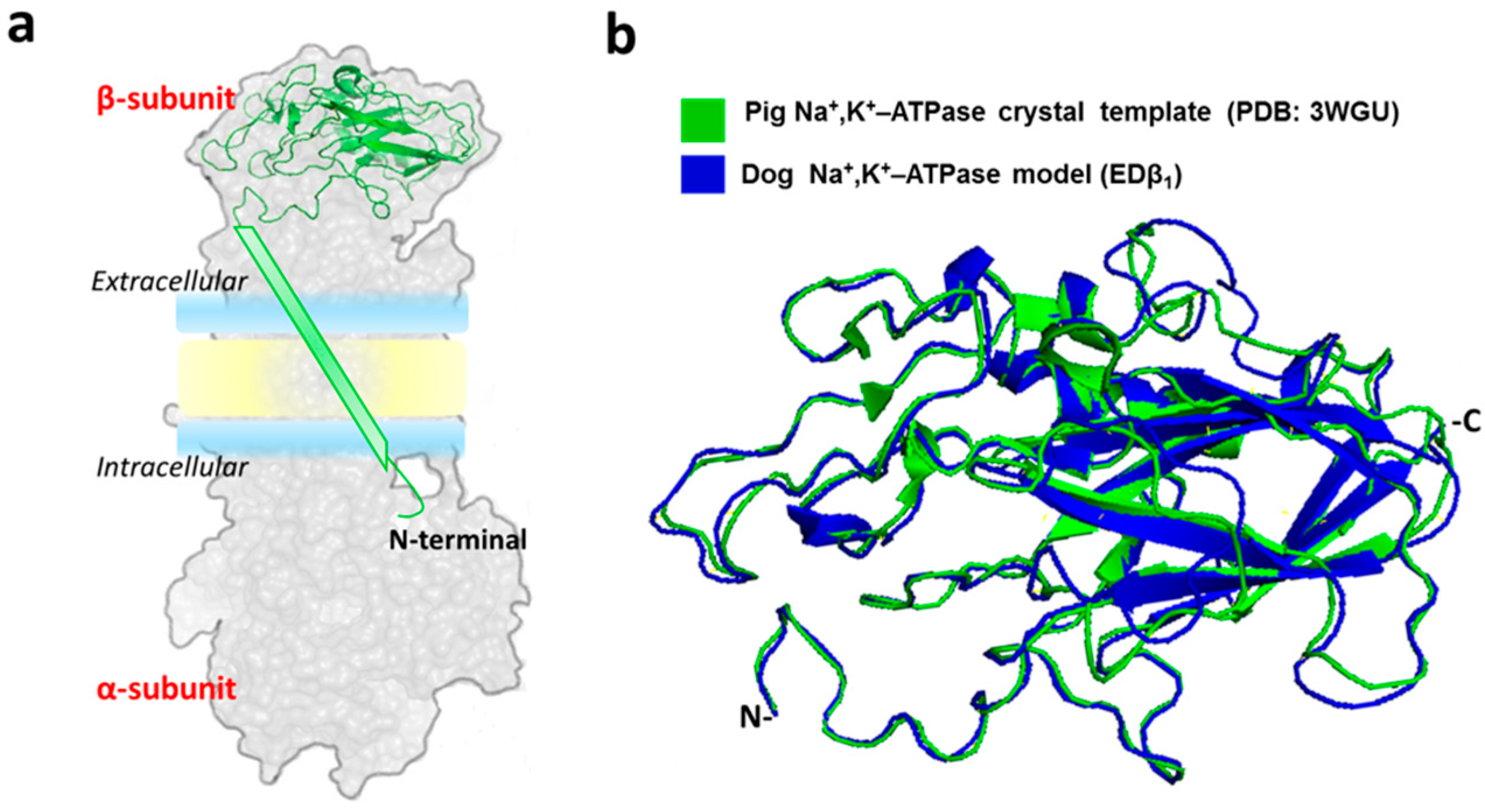
2.1. Generation of the Dog Β1 Subunit Extracellular Domain Model
2.2. Molecular Dynamics Simulations of the EDβ1
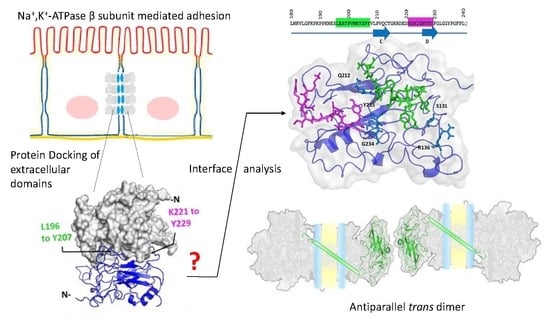
2.3. Protein-Protein Docking Predicts that Na+, K+-Atpase Β1 Subunit Dimer Is Formed by Regions Leu196–Tyr207 and Lys221–Tyr229
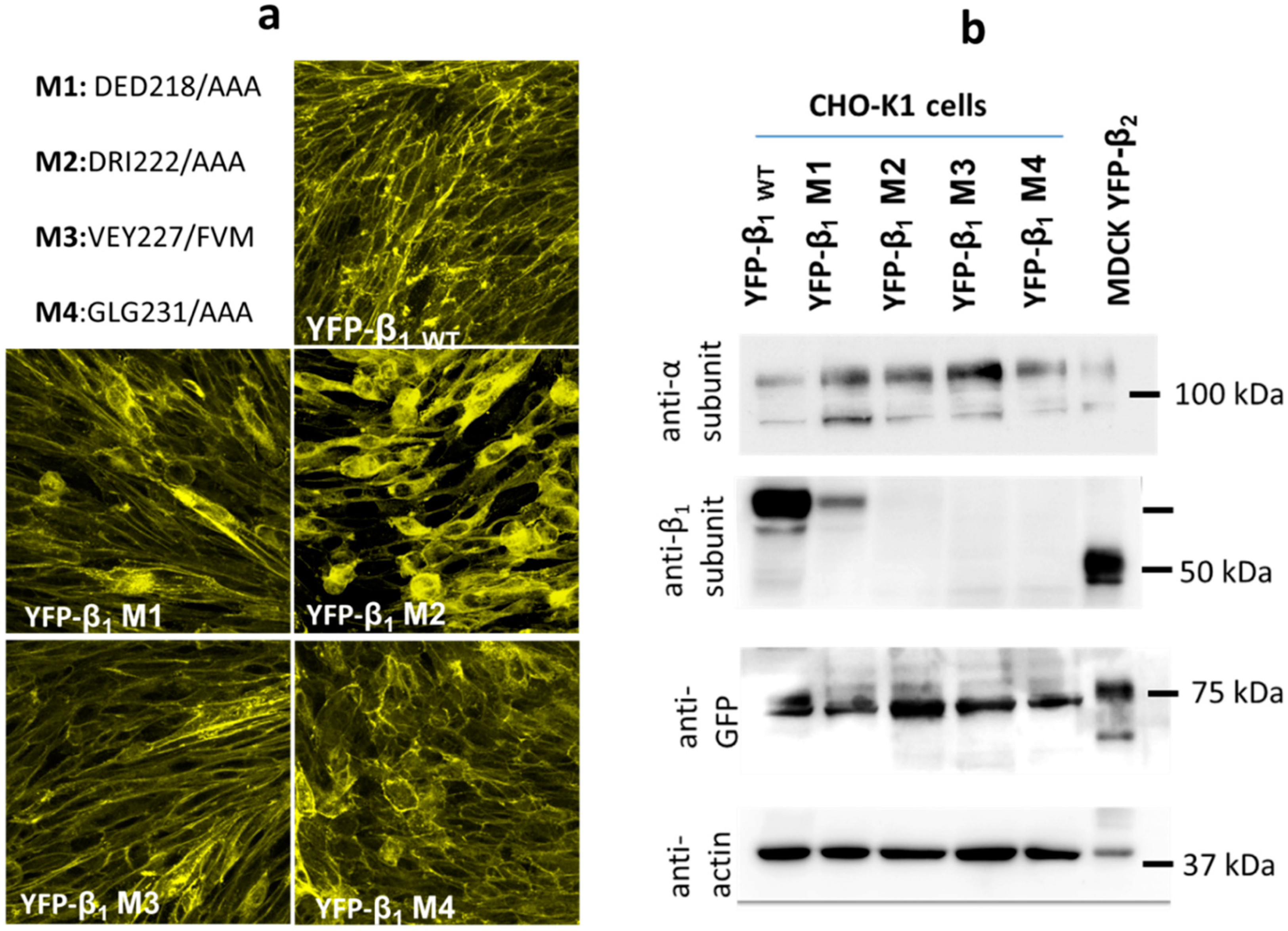
2.4. Expression of YFP-β1 Mutants in CHO Fibroblasts
2.5. Cho Cells Expressing Yfp-β1 m2 and m3 Show Decreased Na+,K+-Atpase β1 Subunit- Mediated Intercellular Adhesion In Vivo
2.6. Cho Cells Expressing Yfp-Β1 M2, M3 And M4 Show Decreased Na+,K+-Atpase Β1-Β1 Interaction In Vitro
3. Discussion
4. Materials and Methods
4.1. Na+, K+-Atpase Β1-Subunit Modeling
4.2. Molecular Dynamics Simulations
4.3. Protein-Protein Docking
4.4. Interface Analysis and Molecular Visualization
4.5. Site-Directed Mutagenesis
- 5′-CAGTGCACTGGCAAGCGAGCTGCTGCCAAGGATAGAATTGGGAAC-3′,
- 5′-CAAGCGAGACGAAGACAAGGCTGCAGCTGGGAACGTGGAGTATTTTG-3′,
- 5′-CAAGGATAGAATTGGGAACTTCGTGATGTTTGGCCTGGGCGGCTACCC-3′
- 5′-GGAACGTGGAGTATTTTGCTGCTGCTGGCTACCCGGGCTTTCCTC-3′.
4.6. Cell Culture, Transfection, and Imaging
4.7. Western Blot
4.8. Dispase Adhesion Assay
4.9. Pull-Down Assay
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cereijido, M.; Shoshani, L.; Contreras, R.G. The Polarized Distribution of Na+, K+-ATPase and Active Transport across Epithelia. J. Membr. Biol. 2001, 184, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Lobato Álvarez, J.A.; López Murillo, T.D.C.; Vilchis Nestor, C.A.; Roldán Gutierrez, M.L.; Páez Gómez, O.; Shoshani, L. Epithelial Na+,K+-ATPase—A Sticky Pump. In Cell Biology-New Insights; InTechOpen: London, UK, 2016. [Google Scholar] [CrossRef]
- Sweadner, K.J. Isozymes of the Na+/K+-ATPase. Biohim. Biophys. Acta 1989, 2, 185–220. [Google Scholar] [CrossRef]
- Geering, K. Functional Roles of Na,K-ATPase Subunits. In Current Opinion in Nephrology and Hypertension; Springer: New York, NY, USA, 2008; pp. 526–532. [Google Scholar] [CrossRef]
- Cereijido, M.; Larre, I.; Paez, O.; Shoshani, L.; Ponce, A. Na+/K+-ATPase Drives Most Asymmetric Transports and Modulates the Phenotype of Epithelial Cells. In Ion Channels and Transporters of Epithelia in Health and Disease; Springer: New York, NY, USA, 2015; pp. 351–374. [Google Scholar] [CrossRef]
- Campbell, H.K.; Maiers, J.L.; DeMali, K.A. Interplay between Tight Junctions & Adherens Junctions. In Experimental Cell Research; Elsevier: Amsterdam, The Netherlands, 2017; pp. 39–44. [Google Scholar] [CrossRef]
- Padilla-Benavides, T.; Roldan, M.L.; Larre, I.; Flores-Benitez, D.; Nicolas, V.-S.; Contreras, R.G.; Marcelino, C.; Liora, S. The Polarized Distribution of Na+,K+-ATPase: Role of the Interaction between Beta Subunits. Mol. Biol. Cell 2010, 21, 4325–4337. [Google Scholar] [CrossRef] [PubMed]
- Tokhtaeva, E.; Sachs, G.; Souda, P.; Bassilian, S.; Whitelegge, J.P.; Shoshani, L.; Vagin, O. Epithelial Junctions Depend on Intercellular Trans-Interactions between the Na,K-ATPase β1subunits. J. Biol. Chem. 2011, 286, 25801–25812. [Google Scholar] [CrossRef] [PubMed]
- Cereijido, M.; Contreras, R.G.; Shoshani, L.; Larre, I. The Na+-K+-ATPase as Self-Adhesion Molecule and Hormone Receptor. AJP Cell Physiol. 2012, 302, C473–C481. [Google Scholar] [CrossRef] [PubMed]
- Shoshani, L.; Contreras, R.G.; Roldán, M.L.; Moreno, J.; Lázaro, A.; Balda, M.S.; Matter, K.; Cereijido, M. The Polarized Expression of Na+,K+-ATPase in Epithelia Depends on the Association between Beta-Subunits Located in Neighboring Cells. Mol. Biol. Cell 2005, 16, 1071–1081. [Google Scholar] [CrossRef] [PubMed]
- Gloor, S.; Antonicek, H.; Sweadner, K.J.; Pagliusi, S.; Frank, R.; Moos, M.; Schachner, M. The Adhesion Molecule on Glia (AMOG) Is a Homologue of the β Subunit of the Na, K-ATPase. J. Cell Biol. 1990, 110, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Antonicek, H.; Schachner, M. The Adhesion Molecule on Glia (AMOG) Incorporated into Lipid Vesicles Binds to Subpopulations of Neurons. J. Neurosci. 2018, 8, 2961–2966. [Google Scholar] [CrossRef]
- Vagin, O.; Tokhtaeva, E.; Sachs, G. The Role of the β1 Subunit of the Na,K-ATPase and Its Glycosylation in Cell-Cell Adhesion. J. Biol. Chem. 2006, 281, 39573–39587. [Google Scholar] [CrossRef]
- Espineda, C.E. Repression of Na, K-ATPase 1-Subunit by the Transcription Factor Snail in Carcinoma. Mol. Biol. Cell 2003, 15, 1364–1373. [Google Scholar] [CrossRef]
- Inge, L.J.; Rajasekaran, S.A.; Yoshimoto, K.; Mischel, P.S.; McBride, W.; Landaw, E.; Rajasekaran, A.K. Evidence for a Potential Tumor Suppressor Role for the Na,K-ATPase beta1-Subunit. Histol. Histopathol. 2008, 23, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Rajasekaran, S.A.; Palmer, L.G.; Quan, K.; Harper, J.F.; Ball, W.J.; Bander, N.H.; Peralta Soler, A.; Rajasekaran, A.K. Na,K-ATPase Beta-Subunit Is Required for Epithelial Polarization, Suppression of Invasion, and Cell Motility. Mol. Biol. Cell 2001, 12, 279–295. [Google Scholar] [CrossRef] [PubMed]
- Lubarski-Gotliv, I.; Dey, K.; Kuznetsov, Y.; Kalchenco, V.; Asher, C.; Garty, H. FXYD5 (Dysadherin) May Mediate Metastatic Progression through Regulation of the β-Na + -K + -ATPase Subunit in the 4T1 Mouse Breast Cancer Model. Am. J. Physiol. Physiol. 2017, 313, C108–C117. [Google Scholar] [CrossRef] [PubMed]
- Madan, P.; Rose, K.; Watson, A.J. Na/K-ATPase β1 Subunit Expression Is Required for Blastocyst Formation and Normal Assembly of Trophectoderm Tight Junction-Associated Proteins. J. Biol. Chem. 2007, 282, 12127–12134. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.M. The Na+/K+ ATPase Is Required for Septate Junction Function and Epithelial Tube-Size Control in the Drosophila Tracheal System. Development 2003, 130, 4963–4974. [Google Scholar] [CrossRef] [PubMed]
- Treuheit, M.J.; Costello, C.E.; Kirley, T.L. Structures of the Complex Glycans Found on the beta-Subunit of (Na,K)-ATPase. J. Biol. Chem. 1993, 268, 13914–13919. [Google Scholar] [PubMed]
- Oda, H.; Takeichi, M. Structural and Functional Diversity of Cadherin at the Adherens Junction. J. Cell Biol. 2011, 193, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Aricescu, A.R.; Jones, E.Y. Immunoglobulin Superfamily Cell Adhesion Molecules: Zippers and Signals. Curr. Opin. Cell Biol. 2007, 19, 543–550. [Google Scholar] [CrossRef]
- Shimono, Y.; Rikitake, Y.; Mandai, K.; Mori, M.; Takai, Y. Immunoglobulin Superfamily Receptors and Adherens Junctions. Subcell. Biochem. 2012, 60, 137–170. [Google Scholar] [CrossRef]
- Chen, J.; Wu, Y. Understanding the Functional Roles of Multiple Extracellular Domains in Cell Adhesion Molecules with a Coarse-Grained Model. J. Mol. Biol. 2017, 429, 1081–1095. [Google Scholar] [CrossRef]
- Tokhtaeva, E.; Sachs, G.; Sun, H.; Dada, L.A.; Sznajder, J.I.; Vagin, O. Identification of the Amino Acid Region Involved in the Intercellular Interaction between the 1 Subunits of Na+/K+-ATPase. J. Cell Sci. 2012, 125, 1605–1616. [Google Scholar] [CrossRef] [PubMed]
- Tokhtaeva, E.; Sun, H.; Deiss-Yehiely, N.; Wen, Y.; Soni, P.N.; Gabrielli, N.M.; Marcus, E.A.; Ridge, K.M.; Sachs, G.; Vazquez-Levin, M.; et al. The O-Glycosylated Ectodomain of FXYD5 Impairs Adhesion by Disrupting Cell–cell Trans-Dimerization of Na,K-ATPase β 1 Subunits. J. Cell Sci. 2016, 129, 2394–2406. [Google Scholar] [CrossRef] [PubMed]
- Harrison, O.J.; Vendome, J.; Brasch, J.; Jin, X.; Hong, S.; Katsamba, P.S.; Ahlsen, G.; Troyanovsky, R.B.; Troyanovsky, S.M.; Honig, B.; et al. Nectin Ectodomain Structures Reveal a Canonical Adhesive Interface. Nat. Struct. Mol. Biol. 2012, 19, 906–915. [Google Scholar] [CrossRef] [PubMed]
- Brasch, J.; Katsamba, P.S.; Harrison, O.J.; Ahlsén, G.; Troyanovsky, R.B.; Indra, I.; Kaczynska, A.; Kaeser, B.; Troyanovsky, S.; Honig, B.; et al. Homophilic and Heterophilic Interactions of Type II Cadherins Identify Specificity Groups Underlying Cell-Adhesive Behavior. Cell Rep. 2018, 23, 1840–1852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janin, J.; Bahadur, R.P.; Chakrabarti, P. Protein-Protein Interaction and Quaternary Structure. Q. Rev. Biophys. 2008, 41, 133–180. [Google Scholar] [CrossRef] [PubMed]
- Kanai, R.; Ogawa, H.; Vilsen, B.; Cornelius, F.; Toyoshima, C. Crystal Structure of a Na+-Bound Na+,K+-ATPase Preceding the E1P State. Nature 2013, 502, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Salmaso, V.; Moro, S. Bridging Molecular Docking to Molecular Dynamics in Exploring Ligand-Protein Recognition Process: An Overview. Front. Pharmacol. 2018, 9. [Google Scholar] [CrossRef]
- Maximova, T.; Moffatt, R.; Ma, B.; Nussinov, R.; Shehu, A. Principles and Overview of Sampling Methods for Modeling Macromolecular Structure and Dynamics. PLoS Comput. Biol. 2016, 12. [Google Scholar] [CrossRef]
- Vajda, S.; Yueh, C.; Beglov, D.; Bohnuud, T.; Mottarella, S.E.; Xia, B.; Hall, D.R.; Kozakov, D. New Additions to the ClusPro Server Motivated by CAPRI. Proteins Struct. Funct. Bioinform. 2017, 85, 435–444. [Google Scholar] [CrossRef]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro Web Server for Protein-Protein Docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef]
- Vilchis-Nestor, C.A.; Roldán, M.L.; Leonardi, A.; Navea, J.G.; Padilla-Benavides, T.; Shoshani, L. Ouabain Enhances Cell-Cell Adhesion Mediated by β1 Subunits of the Na+,K+-ATPase in CHO Fibroblasts. Int. J. Mol. Sci. 2019, 20, 2111. [Google Scholar] [CrossRef] [PubMed]
- Castillo, J.P.; Rui, H.; Basilio, D.; Das, A.; Roux, B.; Latorre, R.; Bezanilla, F.; Holmgren, M. Mechanism of Potassium Ion Uptake by the Na+/K+-ATPase. Nat. Commun. 2015, 6, 7622. [Google Scholar] [CrossRef] [PubMed]
- Hilbers, F.; Kopec, W.; Isaksen, T.J.; Holm, T.H.; Lykke-Hartmann, K.; Nissen, P.; Khandelia, H.; Poulsen, H. Tuning of the Na,K-ATPase by the Beta Subunit. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Bab-Dinitz, E.; Albeck, S.; Peleg, Y.; Brumfeld, V.; Gottschalk, K.E.; Karlish, S.J.D. A C-Terminal Lobe of the beta Subunit of Na,K-ATPase and H,K-ATPase Resembles Cell Adhesion Molecules. Biochemistry 2009, 48, 8684–8691. [Google Scholar] [CrossRef] [PubMed]
- Shinoda, T.; Ogawa, H.; Cornelius, F.; Toyoshima, C. Crystal Structure of the Sodium–potassium Pump at 2.4 Å Resolution. Nature 2009, 459, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, J.P.G.L.M.; Melquiond, A.S.J.; Karaca, E.; Trellet, M.; Van Dijk, M.; Van Zundert, G.C.P.; Schmitz, C.; De Vries, S.J.; Bordogna, A.; Bonati, L.; et al. Defining the Limits of Homology Modeling in Information-Driven Protein Docking. Proteins Struct. Funct. Bioinform. 2013, 81, 2119–2128. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.R.; Sternberg, M.J.E.; Bates, P.A. The Relationship between the Flexibility of Proteins and Their Conformational States on Forming Protein-Protein Complexes with an Application to Protein-Protein Docking. J. Mol. Biol. 2005, 347, 1077–1101. [Google Scholar] [CrossRef]
- Dempski, R.E.; Friedrich, T.; Bamberg, E. The β Subunit of the Na+/K+-ATPase Follows the Conformational State of the Holoenzyme. J. Gen. Physiol. 2005, 125, 505–520. [Google Scholar] [CrossRef]
- Razavi, A.M.; Delemotte, L.; Berlin, J.R.; Carnevale, V.; Voelz, V.A. Molecular Simulations and Free-Energy Calculations Suggest Conformation-Dependent Anion Binding to a Cytoplasmic Site as a Mechanism for Na+/K+-ATPase Ion Selectivity. J. Biol. Chem. 2017, 292, 12412–12423. [Google Scholar] [CrossRef]
- Čechová, P.; Berka, K.; Kubala, M. Ion Pathways in the Na+/K+-ATPase. J. Chem. Inf. Model. 2016, 56, 2434–2444. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology Modelling of Protein Structures and Complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- Scott, W.R.P.; Hünenberger, P.H.; Tironi, I.G.; Mark, A.E.; Billeter, S.R.; Fennen, J.; Torda, A.E.; Huber, T.; Krüger, P.; Van Gunsteren, W.F. The GROMOS Biomolecular Simulation Program Package. J. Phys. Chem. A 1999, 103, 3596–3607. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, Flexible, and Free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Páez, O.; Martínez-Archundia, M.; Villegas-Sepúlveda, N.; Roldan, M.L.; Correa-Basurto, J.; Shoshani, L. A Model for the Homotypic Interaction between Na+,K+-ATPase β1 Subunits Reveals the Role of Extracellular Residues 221–229 in Its Ig-Like Domain. Int. J. Mol. Sci. 2019, 20, 4538. https://doi.org/10.3390/ijms20184538
Páez O, Martínez-Archundia M, Villegas-Sepúlveda N, Roldan ML, Correa-Basurto J, Shoshani L. A Model for the Homotypic Interaction between Na+,K+-ATPase β1 Subunits Reveals the Role of Extracellular Residues 221–229 in Its Ig-Like Domain. International Journal of Molecular Sciences. 2019; 20(18):4538. https://doi.org/10.3390/ijms20184538
Chicago/Turabian StylePáez, Omar, Marlet Martínez-Archundia, Nicolás Villegas-Sepúlveda, María Luisa Roldan, José Correa-Basurto, and Liora Shoshani. 2019. "A Model for the Homotypic Interaction between Na+,K+-ATPase β1 Subunits Reveals the Role of Extracellular Residues 221–229 in Its Ig-Like Domain" International Journal of Molecular Sciences 20, no. 18: 4538. https://doi.org/10.3390/ijms20184538
APA StylePáez, O., Martínez-Archundia, M., Villegas-Sepúlveda, N., Roldan, M. L., Correa-Basurto, J., & Shoshani, L. (2019). A Model for the Homotypic Interaction between Na+,K+-ATPase β1 Subunits Reveals the Role of Extracellular Residues 221–229 in Its Ig-Like Domain. International Journal of Molecular Sciences, 20(18), 4538. https://doi.org/10.3390/ijms20184538