Mapping the Interactions of HBV cccDNA with Host Factors


1
Department of Microbiology and Immunology, Yong Loo Lin School of Medicine, National University Health System (NUHS), National University of Singapore, Singapore 117545, Singapore
2
Department of Medicine, Yong Loo Lin School of Medicine, National University Health System (NUHS), National University of Singapore, Singapore 119228, Singapore
3
Institute of Molecular and Cell Biology, A*STAR (Agency for Science, Technology and Research), Singapore 138673, Singapore
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2019, 20(17), 4276; https://doi.org/10.3390/ijms20174276
Submission received: 6 August 2019
/
Revised: 26 August 2019
/
Accepted: 27 August 2019
/
Published: 1 September 2019
(This article belongs to the Special Issue Molecular Research in Emerging Viruses 2019)
Abstract
:Hepatitis B virus (HBV) infection is a major health problem affecting about 300 million people globally. Although successful administration of a prophylactic vaccine has reduced new infections, a cure for chronic hepatitis B (CHB) is still unavailable. Current anti-HBV therapies slow down disease progression but are not curative as they cannot eliminate or permanently silence HBV covalently closed circular DNA (cccDNA). The cccDNA minichromosome persists in the nuclei of infected hepatocytes where it forms the template for all viral transcription. Interactions between host factors and cccDNA are crucial for its formation, stability, and transcriptional activity. Here, we summarize the reported interactions between HBV cccDNA and various host factors and their implications on HBV replication. While the virus hijacks certain cellular processes to complete its life cycle, there are also host factors that restrict HBV infection. Therefore, we review both positive and negative regulation of HBV cccDNA by host factors and the use of small molecule drugs or sequence-specific nucleases to target these interactions or cccDNA directly. We also discuss several reporter-based surrogate systems that mimic cccDNA biology which can be used for drug library screening of cccDNA-targeting compounds as well as identification of cccDNA-related targets.
1. Introduction
Viral hepatitis is a major public health concern, accounting for more than 1.3 million deaths annually [1]. Life-threatening complications such as liver cirrhosis and hepatocellular carcinoma (HCC) arise when chronic viral infections are left undiagnosed or untreated. Chronic hepatitis B virus (HBV) infection, a major contributor to viral hepatitis, is estimated to afflict nearly 300 million people globally, of which only 10% are diagnosed and an even smaller proportion are receiving treatment [2]. The availability of a prophylactic vaccine has led to a significant reduction in new HBV infections among children aged below five who are most vulnerable to developing persistent infection [1]. However, a reliable cure for individuals already living with chronic hepatitis B (CHB) is still elusive.
Currently, there are two therapeutic strategies approved for the management of chronic HBV infection, namely nucleoside/nucleotide analogs (NAs) and interferon alpha (IFN-α)/pegylated interferon (PEG-IFN) [3]. NAs suppress HBV replication and promote virological clearance by directly inhibiting viral reverse transcription. Compared to first-generation NAs lamivudine (LAM) and adefovir (ADV), newer drugs such as entecavir (ETV), tenofovir disoproximal fumarate (TDF) and tenofovir alafenamide (TAF) are more potent and have high barrier to resistance [4,5,6]. Continuous administration of these drugs can reverse cirrhosis and reduce the risk of developing end-stage liver disease and HCC, thereby improving survival in CHB patients [7,8]. NAs are generally well tolerated and ease of oral administration promotes compliance to treatment. However, the required length of treatment and the safety of these drugs in the long run are still unclear. Immune modulation and limited direct antiviral action by IFN-α/PEG-IFN over a defined treatment period resulted in higher rates of hepatitis B surface antigen (HBsAg) loss and/or seroconversion [9,10]. Following treatment with NAs for between 2 to 5 years, HBsAg loss was observed in 2–10% of hepatitis B e antigen-positive (HBeAg +ve) patients and 0–5% of HBe antigen-negative (HBeAg −ve) patients while HBsAg clearance was around 11% in both HBeAg +ve and HBeAg –ve patients 4 years after PEG-IFN treatment [11,12,13,14,15]. However, response to interferon treatment varied considerably and it is usually poorly tolerated due to adverse side effects [16,17]. Combining NA and IFN treatments either simultaneously or sequentially have shown some promise but more long-term studies are needed to determine the optimal combination for patients with different disease backgrounds or stages [18,19]. Furthermore, both these strategies are not curative as they do not primarily target HBV covalently closed circular DNA (cccDNA), which remains in infected hepatocytes and contributes to viral rebound after stopping treatment.
Targeting cccDNA is thus the next crucial step in the development of anti-HBV therapies and many reviews have comprehensively discussed different aspects of this approach [20,21,22,23,24]. Adding to that, this review will mainly focus on the regulation of HBV cccDNA activity through its interactions with host factors, as well as, recently available surrogate reporter-based systems that can be used to screen for novel cccDNA inhibitors.
2. HBV Covalently Closed Circular DNA
The HBV life cycle includes multiple steps which are highly dependent on various host cell machinery and factors (Figure 1). Upon virus entry, the HBV genome, a partially double-stranded relaxed circular DNA (rcDNA), is transported to the nucleus of host hepatocytes. Subsequently, rcDNA is converted into cccDNA through deproteinization, removal of RNA oligomer linkage and DNA repair [25]. The cccDNA molecule serves as a template for transcription of all viral RNAs, including the pregenomic RNA (pgRNA) essential for making new copies of rcDNA to be packaged into progeny virions [26]. HBV cccDNA stably exists in the nuclei of infected cells as episomal DNA packaged into a minichromosome through interactions with cellular histone and nucleosomal proteins [27]. Although cccDNA cannot self-replicate, its levels are replenished through re-infection, de novo secondary infection and, to a lesser extent, intracellular recycling of newly synthesized rcDNA to the nucleus [28,29]. A recent study using a cell culture model of HBV infection estimates the cccDNA half-life to be around 40 days [29]. This is consistent with earlier in vivo studies that determined the half-lives of cccDNA in hepadnavirus-infected woodchucks (33 to 50 days) and ducks (35 to 57 days) [30,31]. Although the longevity of an individual cccDNA molecule has not been clearly defined, evidence suggests that cccDNA may persist for the life span of the cell [32]. In addition to its long half-life, quantification of the cccDNA level in liver biopsies of CHB patients revealed that a low copy number (median 1.5 copies per cell) is sufficient to maintain chronic infection [33]. Furthermore, reactivation of latent cccDNA may occur post-treatment leading to HBV recurrence [34,35]. Hence, cccDNA has to be eliminated from infected cells or, at least, permanently silenced in order to achieve a functional cure.
2.1. Blocking Formation of New cccDNA Molecules
Due to cell division and natural turnover of hepatocytes, cccDNA level in an infected liver is expected to decline over time, provided new copies are not being synthesized [36]. Formation of new cccDNA molecules through infection can be blocked at several steps including virus attachment, uptake and conversion of rcDNA into cccDNA. Identification of sodium taurocholate cotransporting polypeptide (NTCP) as a high-affinity HBV entry receptor has enabled development of and screenings for small molecule inhibitors that compete with the virus for receptor binding [37,38]. Treatment with entry inhibitor Myrcludex B, a synthetic N-myristoylated lipopeptide designed based on the HBV preS1 motif, blocked cccDNA amplification in infected human hepatocytes [39]. This drug has shown clinical efficacy against CHB and chronic hepatitis D in Phase 2 trial, although its long-term benefits and adverse effects remain to be seen [40]. Aside from its anti-HBV activity, Myrcludex B also disrupts the bile acid transporter function of NTCP as evidenced by an asymptomatic increase in bile acid levels in some patients who received the treatment [40,41]. Using a cell culture infection system, derivatives of cyclosporin A (CsA) that interact with NTCP and inhibit HBV entry without interfering with NTCP transporter function were identified [42]. This finding suggests that the HBV receptor function of NTCP may be selectively targeted to avoid potential side effects associated with inhibition of a host function.
Following virus attachment, uptake of HBV via endocytosis has been shown to be caveolin-dependent in HepaRG cells while clathrin-dependent in immortalized primary human hepatocytes (PHH) and HepG2 cells [43,44,45]. Inhibitors of clathrin-mediated endocytosis such as silibinin and chlorpromazine are capable of blocking HBV uptake [44,46]. Synthesis of new cccDNA copies via both de novo infection and intracellular recycling routes require the conversion of rcDNA into cccDNA. This step involves numerous host enzymes including DNA repair enzyme TDP2 or TDP-related proteins, flap structure-specific endonuclease 1 (FEN1), DNA polymerase alpha (Pol α) and kappa (POLK), DNA topoisomerase I (TOP1) and II (TOP2) and DNA ligases [47,48,49,50,51,52]. Chemical inhibition of these enzymes and siRNA or CRISPR/Cas9-mediated knockdown of their expression were shown to suppress cccDNA amplification. In a screen for compounds that directly target cccDNA, two disubstituted sulfonamides (DSS), CCC-0975 and CCC-0346, were identified as specific inhibitors of rcDNA conversion into cccDNA [53]. Using a similar screening strategy, hydrolyzable tannins were found to inhibit cccDNA formation as well as promote its degradation [54]. However, the mechanism and targets of inhibition by DSS and hydrolyzable tannins are still unknown. Another class of drugs that interferes with cccDNA biosynthesis is capsid assembly modulators (CAMs). These molecules target the HBV core (HBc) protein to either induce premature disassembly of nucleus-bound capsids or prevent encapsidation of newly synthesized pgRNA [55,56]. These events inhibit the generation and delivery of rcDNA to the nucleus and subsequent conversion to cccDNA.
2.2. Eliminating cccDNA from the Infected Liver
To eradicate HBV infection, it will not be sufficient to inhibit synthesis of new cccDNA molecules as clearance of the pre-existing cccDNA will also be required. Clearance of cccDNA can be achieved by killing infected hepatocytes or noncytolytically through the actions of pro-inflammatory cytokines and DNA-targeting enzymes. Host innate and adaptive immune responses can reduce cccDNA levels in the liver through the elimination of infected hepatocytes by natural killer cells and cytotoxic T lymphocytes [57]. Two reports suggest that certain cytokines, namely IFN-α, IFN-γ and tumor necrosis factor-α (TNF-α), and activation of lymphotoxin-β receptor (LT-βR) are capable of inducing cccDNA degradation without causing cell death through the activation of nuclear deaminases APOBEC3A and 3B [58,59]. However, due to the technical limitations of the assays employed in these studies, the claim that cytokines can induce cccDNA loss noncytopathically is slightly controversial and requires independent verification since this may have important implications for HBV cure [60]. HBV-specific immunity is usually lacking or defective in CHB patients and immune modulatory therapies are needed to mount an efficient antiviral response [61].
The use of sequence-specific nuclease technology has shown promise in inactivating or eliminating cccDNA from infected cells. Multiple studies done using zing finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs) and clustered regularly interspaced short palindromic repeats associated nuclease 9 (CRISPR/Cas9) in cell culture and animal models showed reductions in cccDNA copy number (see a recent review by Bloom et al.) [23]. Although they were designed to specifically target and disrupt HBV DNA sequences, off-target DNA cleavage events and possible increased genomic instability are still a concern [62]. Delivery of these nucleases to target cells also needs to be optimized. Nevertheless, this gene editing approach continues to be an attractive possibility for anti-HBV therapy with two CRISPR/Cas9 candidates, EBT106 and HBV, currently in preclinical testing [63].
2.3. Targeting cccDNA Transcriptional Activity
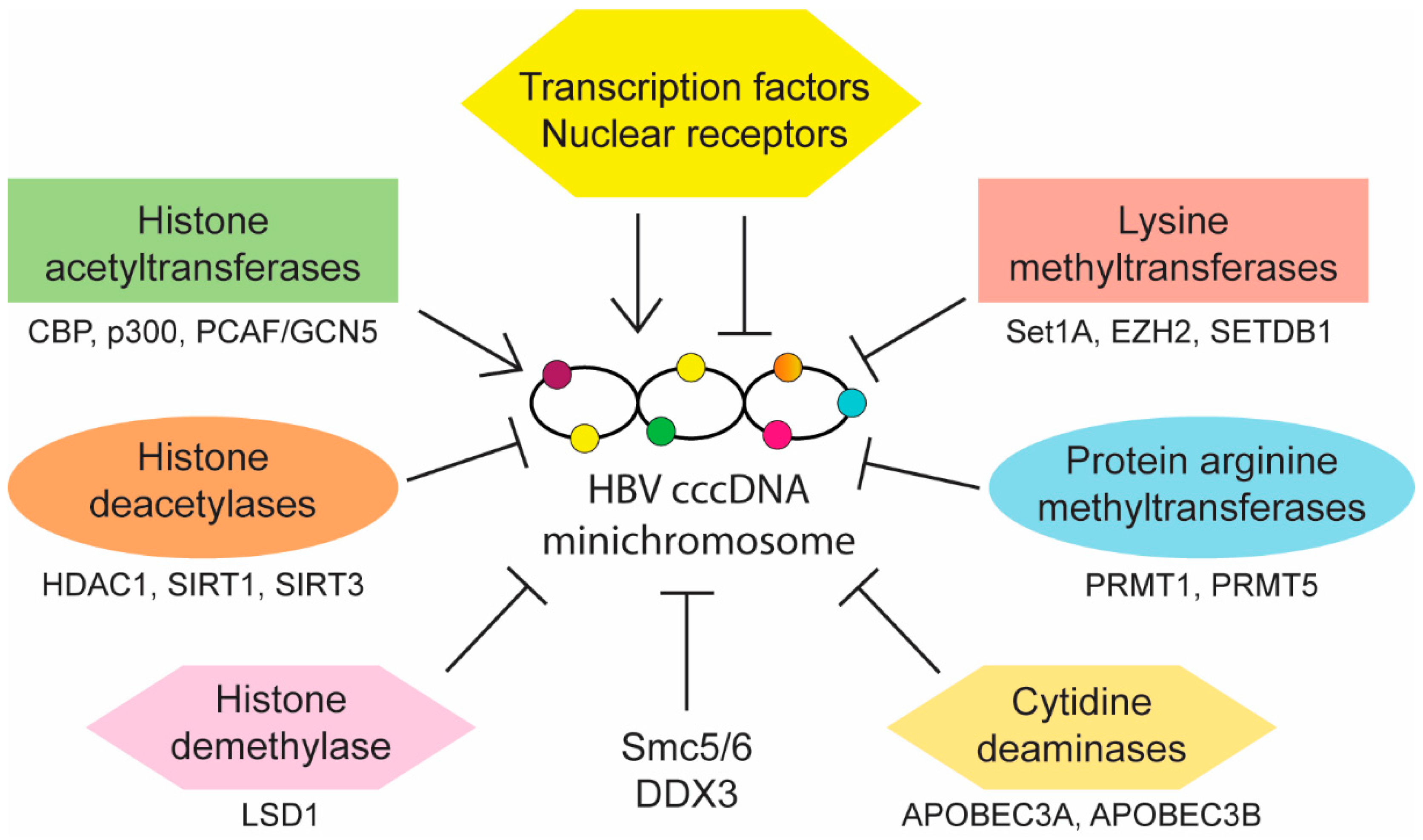
Aside from reducing cccDNA copy number, control of HBV infection may also be achieved through the regulation of cccDNA activity. Transcription of viral pgRNA and mRNAs is regulated by four distinct promoters (preS1, preS2, core, and X) and two known enhancers (EnhI and EnhII). Multiple host transcription factors and nuclear receptors are known to bind and modulate these elements. HBV cccDNA also contains three predicted CpG islands (islands I, II, and III), of which methylation of island II has been shown to be associated with reduced cccDNA transcription in vitro and low level of circulating HBV DNA in CHB patients [64]. In addition, epigenetic modifications of cccDNA-associated histones also affect cccDNA activity. Interactions between cccDNA and host nuclear factors can either promote or inhibit its transcriptional activity (summarized in Figure 2) and understanding these interactions may provide opportunities for targeted therapy development.
2.3.1. Host Factors That Positively Regulate cccDNA Activity
The cccDNA promoter and enhancer elements contain binding sites for liver-enriched as well as ubiquitous transcription factors and nuclear receptors. Those that activate viral transcription include hepatocyte nuclear factors 1, 3 and 4 (HNF1, HNF3/FoxA, HNF4), CCAAT-enhancer-binding protein (C/EBP), retinoid X receptor alpha/peroxisome proliferator-activated receptor alpha (RXRα/PPARα), Farnesoid X receptor (FXR), nuclear factor (NF1), specificity protein 1 (SP1), activator protein 1 (AP-1), TATA-binding protein (TBP), cAMP response element binding protein (CREB), octamer transcription factor 1 (Oct1), and nuclear respiratory factor 1 (NRF1) (references listed in Table 1). Involvement of transcriptional coactivators such as CREB-regulated transcriptional coactivator 1 (CRTC1) has also been demonstrated [65].
Various histone modifying enzymes including acetyltransferases (HATs), deacetylases (HDACs), lysine methyltransferases (KMTs) and protein arginine methyltransferases (PRMTs) as well as DNA methyltransferases (DNMTs) alter the methylation and acetylation status of cccDNA and its associated histones 3 and 4 (H3 and H4). Hypoacetylation of H3 and H4 correlates with low HBV replication in vitro and in vivo. Conversely, acetylation of cccDNA-bound H4 is associated with increased HBV replication and HDAC inhibitors allow maintenance of the active acetylated cccDNA minichromosome [107]. In line with this, HBx-dependent recruitment of HATs such as CREB-binding protein (CBP), p300 and p300/CBP-associated factor/general control nonderepressible 5 (PCAF/GCN5) onto cccDNA promotes histone acetylation and active transcription [92]. Besides histone acetylation, the presence of activating and repressive methylation marks on cccDNA-bound histones also determines cccDNA activity. Mapping of histone modifications in HBV-infected cells revealed high levels of activating marks such as trimethylation of lysine 4 on H3 (H3K4me3) and a corresponding absence of repressive marks such as di-or trimethylation of lysine 9 on H3 (H3K9me2 and H3K9me3) on HBV DNA, resulting in active viral transcription [108]. While a similar pattern of histone marks was observed in a subsequent study involving CHB patients, interpatient variability was evident depending on disease stage and the presence of repressive marks did not necessarily correlate with reduced viral transcription, suggesting a more complex epigenetic interplay in vivo [109]. In contrast to the earlier in vitro infection study, cccDNA was observed to be in a transcriptionally repressed state with hypoacetylated and lysine 9-methylated H3 on viral promoters 24 hours after HBV genome transfection [93]. The viral regulatory protein HBx overcomes this repression by recruiting histone demethylase lysine-specific demethylase-1 (LSD1) and KMT SET domain containing 1A (Set1A) to viral promoters. LSD1 and Set1A activate viral transcription through demethylation of repressive H3K9me2 and accumulation of activating H3K4me3, respectively [93].
2.3.2. Host Restriction Factors That Directly Repress cccDNA Activity
While HBV makes use of multiple cellular proteins to complete different steps in its life cycle, it is worthwhile to note that there are also host factors that negatively regulate HBV infection, possibly as a host defense mechanism. Several transcription factors and epigenetic modifiers such as nuclear factor-kappa B (NF-κB), signal transducer and activator of transcription 1 and 2 (STAT1 and STAT2), prospero homeobox protein 1 (PROX1), enhancer of zeste homolog 2 (EZH2), Yin Yang 1 (YY1), sirtuins 1 and 3 (SIRT1 and SIRT3), PRMT1, PRMT5, HDAC1, SET domain bifurcated 1 (SETDB1), structural maintenance of chromosome 5/6 complex (Smc5/6), zinc finger and homeoboxes 2 (ZHX2), DEAD-box polypeptide 3 (DDX3) and apolipoprotein B mRNA editing catalytic polypeptide-like 3A and 3B (APOBEC3A and APOBEC3B) have been identified as host restriction factors of HBV infection (references listed in Table 1). Some of these factors directly repress the activity of cccDNA leading to reduction in viral replication and Smc5/6 is a well-characterized restriction factor (see a recent review by Livingston) [110]. Smc5/6 binds cccDNA and blocks its transcription [103]. Importantly, HBx binds to DNA-damage binding protein 1 (DDB1) which then bind to Cul4-ROC1 E3 ligase to ubiquitylate Smc5/6 for degradation so as to achieve HBV gene expression and replication [103,111]. Interestingly, a recent study found that nitazoxanide inhibits the interaction between HBx and DDB1 which restores Smc5/6 expression, leading to a reduction in viral transcription [112]. Nitazoxanide, which has been approved by the FDA for treating multiple virus and parasite infections, may be repurposed as a new therapeutic drug for HBV. As the ubiquitin-dependent degradation of Smc5/6 also requires the NEDD8 protein, pevonedistat (MLN 4924) is also able to block the degradation of Smc5/6 because it inhibits the NEDD8-activating enzyme E1 [113]. As the current HBV therapies are not able to target cccDNA, the successful use of nitazoxanide and pevonedistat to block cccDNA transcription suggests that it is feasible to develop new classes of therapies to target the activity of cccDNA.
Besides Smc5/6, ZHX2 has also been shown to restrict HBV replication [104]. Based on immunohistochemical staining of adjacent non-tumor sections from 80 HCC patients, nuclear expression of ZHX2 was found to be lower in patients who are HBV active (defined as being HBeAg positive or HBV DNA > 1000 copies/mL in serum) than those who are HBV inactive. Furthermore, overexpression of ZHX2 in Huh7-NTCP cells, which have low endogenous ZHX2 level, reduced the production of pgRNA, HBsAg and HBeAg as well as secretion of HBV core particle DNA. Conversely, knockdown of ZHX2 in HepG2-NTCP cells, which have high endogenous ZHX2 level, enhanced viral replication. It seems that ZHX2 transcriptionally represses HBV promoters as well as regulates post-translational modification (PTMs) of cccDNA-bound histones. By using cccDNA-ChIP, the authors showed that ZHX2 down-regulated PTMs associated with transcriptional activation, leading to inhibition of cccDNA transcription. Similarly, DDX3, which is a member of the DEAD-box RNA helicase family, is a host restriction factor that inhibits HBV replication at the transcriptional level [105]. The overexpression of DDX3 resulted in decrease viral RNA transcripts and the ATPase activity, but not helicase activity, of DDX3 is required for this inhibition. Interestingly, another member DDX5 did not show an inhibitory effect. By using a HBV promoter-driven reporter assay, overexpression of DDX3 inhibited the core, S1 and S2 promoter activities. Consistently, the knockdown of DDX3 in HepG2-NTCP cells increased viral replication without increasing the level of cccDNA, indicating that DDX3 represses the transcription of cccDNA.
By testing the expression of SIRT1 to 7, which are class III HDACs, SIRT3 was found to be down-regulated transcriptionally in HBV-infected HepG2-NTCP as well as HepAD38 cell line [99]. Consistently, the inhibitory effect of SIRT3 on cccDNA transcriptional activity was confirmed using ectopic SIRT3 expression as well as short hairpin RNA targeting. Although SIRT3 is localized to both mitochondrial and nucleus, it was shown that the nuclear form of SIRT3 functions as a nuclear NAD+-dependent HDAC and causes deacetylation of cccDNA-bound H3. Besides causing the hypoacetylation of H3K9 on cccDNA, which inhibits HBV transcription, overexpression of SIRT3 increased the recruitment of KMT SUV39H1 but decreased the recruitment of another KMT SETD1A to cccDNA in HBV-infected HepG2-NTCP cells. Consequently, the overexpression of SIRT3 caused an increase in repressive H3K9me3 and a decrease in activating H3K4me3 marks on cccDNA, resulting in an inactive chromatin structure and transcriptional silencing. Importantly, the levels of SIRT3 mRNA and SIRT3 protein on cccDNA were decreased in the liver of CHB patients when compared with inactive carriers, indicating SIRT3 acts as a HBV restriction factor in vivo. However, HBV counteracts this SIRT3-mediated repression because overexpression of HBx reduced the mRNA and protein levels of SIRT3 in Huh-7 and HepG2 cells. In HBV-infected PHH, both HBx and SIRT3 were found to bind to cccDNA and the recruitment of SIRT3 on cccDNA negatively correlated with recruitment of HBx.
PRMT5 is another host restriction factor that represses cccDNA transcription through epigenetic regulation [101]. Overexpressed PRMT5 was found to bind to cccDNA in HBV-infected dHepaRG cells and this interaction was also observed for a mutant PRMT5 lacking the methyltransferase domain. However, only wild-type but not mutant PRMT5 increased the symmetric dimethylation of cccDNA-bound H4R3, leading to decreased cccDNA transcription. Conversely, knockdown of PRMT5 decreased symmetric dimethylation of cccDNA-bound H4R3 and increased cccDNA transcription. The action of PRMT5 seems to be related to its regulation of the interaction between cccDNA and Brg-1-based human SWI/SNF chromatin remodeler as well as RNA Pol II. In a separate study, PRMT1 was identified as a binding partner of HBx [100]. While PRMT5 increased the symmetric dimethylation of cccDNA-bound H4R3, PRMT1 catalysed asymmetric dimethylation instead. Nevertheless, PRMT1 is also recruited to cccDNA in HBV infected PHH. Knockdown of PRMT1 in HepG2 cells transfected with HBV vector increased HBV transcription, indicating that PRMT1 is also a HBV restriction factor. Conversely, the overexpression of PRMT1 decreased HBV transcription and this effect is dependent on PRMT1′s methyltransferase activity because it was not observed with enzymatically-inactive PRMT1 mutant. It seems that both asymmetric and symmetric dimethylation of cccDNA-bound H4R3, which can be catalyzed by PRMT1 and PRMT5 respectively, lead to a reduction in HBV transcription. However, their relative importance in regulating cccDNA activity during HBV infection have not been determined and further studies using mouse model of infection will be required. Interestingly, HBx may counteract the repressive activity of PRMT1 because HBx is able to inhibit PRMT1′s methyltransferase activity. On the other hand, PRMT5 interacts with HBc and regulates its post-translational modifications [101,114]. However, PRMT5-HBc interaction regulates pregenomic RNA encapsidation but may not be relevant for PRMT5′s repressive activity on cccDNA.
While most of the above restriction factors regulate cccDNA epigenetically, APOBEC3A and APOBEC3B edit cccDNA and cause its degradation [58,59]. Several members of the APOBEC3 family of cytidine deaminases have previously been shown to inhibit HBV replication through deaminase-dependent and -independent mechanisms [115,116,117,118,119,120]. The exact HBV DNA species targeted by these enzymes is still unclear although a recent study suggests HBV rcDNA to be the primary substrate for deamination [121]. APOBEC3A and APOBEC3B were found to be up-regulated in differentiated HepaRG (dHepaRG) cells treated with IFN-α and LT-βR agonists respectively. Consequently, these treatments led to a reduction in the levels of cccDNA in infected dHepaRG cells as well as PHH. Both APOBEC3A and APOBEC3B colocalize with HBc in the nucleus of infected cells where they deaminate and degrade cccDNA. Furthermore, HBV core interacts with APOBEC3A and the central region of HBc (amino acids 77 to 149) is required for this interaction. In another study, APOBEC3B was also shown to interact with HBc and the presence of RNA is required for this interaction. APOBEC3B has two cytidine deaminase domains but the domain at the C-terminal is the main contributor towards inhibition of HBV replication. In addition to editing cccDNA, APOBEC3B also edits the HBV minus- and plus-strand DNAs found in the cytoplasm during reverse transcription but does not edit pgRNA. Similar to Smc5/6, an E3 ubiquitin ligase, MSL complex subunit 2 (MSL2), was found to interact with and promote ubiquitylation of APOBEC3B, resulting in its degradation and an increase in cccDNA stability [106]. Consequently, knockdown of MSL2 resulted in the up-regulation of APOBEC3B on the protein level but not at the mRNA level. In contrast, APOBEC3A expression was not affected by MSL2. Furthermore, HBx up-regulates MSL2 transcriptionally, thus counteracting the restriction of APOBEC3B on HBV replication.
HBx is a multi-functional protein known to interact with many cellular proteins and has a central role in viral replication (see a recent review by Slagle and Bouchard) [122]. As described above, HBx also regulates several host restriction factors of HBV directly or indirectly. As HBx is being expressed in infected cells, it regulates several restriction factors like Smc5/6, SIRT3, PRMT1, and APOBEC3B and counteracts their repressive actions on cccDNA, thus allowing efficient viral replication. As illustrated by the ability of nitazoxanide to inhibit viral replication by blocking the interaction between HBx and DDB1 [112], future therapies could be developed to target HBx’s ability to counteract host restriction factors of HBV.
3. Surrogate Systems for Screening of Anti-HBV cccDNA Drugs
Given the importance of cccDNA in HBV life cycle and its persistence in CHB patients, efficient and accurate cccDNA quantification methods are required. However, studying HBV cccDNA biology has been challenging, primarily due to the low copy number of cccDNA in infected PHH, hepatic stem cells, and NTCP-expressing hepatoma cell lines [37,123]. Currently, Southern blotting and real-time quantitative polymerase chain reaction (qPCR) are the commonly employed methods for quantifying cccDNA [124,125,126]. Detection and quantification of cccDNA by classical Southern blotting is still considered the gold standard despite its limitations. Due to high cost and laborious workflow, application of Southern blotting in high throughput screening and clinical setting is restricted. These limitations have been circumvented with the emergence of real-time qPCR approach. Compared to Southern blotting, real-time qPCR is less laborious, inexpensive, and can easily be upscaled for high throughput screening [127]. In some cases, a single copy of cccDNA can be detected using advanced PCR approaches such as Droplet Digital PCR (ddPCR) [128]. Although more sensitive, real-time qPCR is more susceptible to false positive errors due to the presence of other viral DNA intermediates, such as rcDNA and single-stranded DNA (ssDNA), which are in high abundance [129,130,131,132]. Though several strategies have been developed recently to reduce false positives, the uptake of these approaches by researchers is slow [132,133]. Given the restrictions of methods mentioned here, several surrogate systems based on recombinant cccDNA (rcccDNA) have been developed and used for screening of anti-HBV cccDNA drugs.
In an approach developed by Cai et al., an inducible reporter-based system was developed to quantify cccDNA formation through measurement of HBeAg secretion [53]. HBeAg was used as a surrogate marker for cccDNA formation in this system as the HBeAg open reading frame (ORF) and its 5′UTR were engineered to localize to opposite ends of the linearized HBV transgene cassette [53]. After formation of cccDNA, the HBeAg transcriptional cassette will be complete, allowing expression and secretion of HBeAg [53]. By quantifying the level of secreted HBeAg using ELISA, the efficiency of cccDNA formation can be determined [53]. This approach was further improved by fusing the HBeAg ORF to a human influenza hemagglutinin (HA) epitope tag [134]. This improvement enabled detection using HA tag antibody instead of the less specific HBeAg antibody that also recognizes secreted HBcAg, which is not indicative of cccDNA formation in this system [134]. Indeed, compounds that inhibit HBV cccDNA formation were identified using this system, highlighting the potential of rcccDNA technology in facilitating identification of novel cccDNA-targeting drugs [53].
In another rcccDNA-based system, the formation of rcccDNA was facilitated through Cre/loxP-mediated DNA recombination [135,136]. In this approach, the linearized cccDNA monomer was engineered to be flanked with loxP sites before introduction into HepG2 cell genome through transposon-mediated integration, forming HepG2-HBV/loxP cell lines [136]. When Cre was expressed in HepG2-HBV/loxP cells via adenoviral transduction, the rcccDNAs will be produced through Cre/loxP-mediated DNA recombination, with the loxP sites forming part of the chimeric intron. The loxP sites will be removed from the viral transcript by RNA splicing, thus leaving the function of pgRNA and viral transcripts intact [136]. In the linearized form of cccDNA, HBsAg was truncated and formation of the rcccDNA is required for HBsAg expression. Therefore, the dynamics of cccDNA can be observed through quantification of HBsAg, which now functions as a surrogate marker of cccDNA. Similarly, this system can be used to address the questions on how cccDNA is regulated and maintained in infected cells and is useful for screening cccDNA-targeting therapeutic compounds.
Despite the ability to generate cccDNA, the copy number of cccDNA formed from these approaches remained low, probably due to low recombination efficiency, thus limiting its application [135]. Using DNA minicircle technology, a new rcccDNA, termed HBVcircle, was generated. Instead of in vivo formation of cccDNA in hepatocytes, the HBVcircle DNA was formed and extracted in E. coli via integrase-mediated intramolecular recombination [137,138,139,140]. With this approach, the cccDNA generated does not contain the bacterial backbone except for a short attR sequence at the recombination site, which has no effect on viral replication [128,139]. This allows the rcccDNA to closely mimic the unmodified HBV cccDNA following transfection into cells. This is beneficial as the transcription of cccDNA from HBV plasmids containing bacterial backbone was demonstrated to be rapidly suppressed in cells [137]. In addition, when coupled with a Gaussia luciferase reporter, the expression of the rcccDNA can be conveniently monitored through luminescence, thus providing an attractive alternative for the screening of therapeutic agents against cccDNA [141].
Reporter genes such as luciferase are useful for the development of rapid, high throughput assays. However, their application in HBV studies is limited by the intolerance of insertions into the compact viral genome. To overcome this problem, Nishitsuji and colleagues produced infectious reporter HBV by co-transfecting recombinant HBV plasmid bearing the NanoLuc (NL) gene with a helper plasmid that codes for missing viral proteins [142]. Although it was first developed to monitor early steps in the HBV replication cycle such as viral entry, a subsequent study showed that when propagated with the wildtype virus, the HBV/NL reporter virus is able to mimic the entire life cycle of HBV [143]. NL activity is repressed by ETV treatment and enhanced by ectopic HBx expression, suggesting a correlation between NL activity and cccDNA level and/or transcription. However, since this system recapitulates many steps in the virus life cycle, anti-HBV agents identified using this engineered reporter HBV may not be acting directly on cccDNA and further studies would be needed to determine their mode of action.
4. Conclusions
The dynamic nature and complexity of cccDNA minichromosomal structure pose great challenges to the development of strategies to eliminate cccDNA and viral persistence in CHB patients. As summarized here, small molecule drugs and sequence-specific nuclease technologies have been used to target cccDNA synthesis and/or stability. In addition, multiple host factors regulating cccDNA activities have also been identified (Table 1). While some of these host factors are required for efficient cccDNA transcription, there are also many host restriction factors that directly repress the activity of cccDNA and HBV replication. More research is required to understand the interplay between HBV and different host factors. As such, several highly sensitive methods have been developed to detect and quantify cccDNA, whose level in HBV infected cells is intrinsically low. However, such assays are not suitable for high throughput screening of drugs targeting cccDNA. To overcome this, various surrogate systems that mimic cccDNA biology and have simple readouts have been developed. Some of the latest developments in this area are summarized here and these assays have been shown to be useful for drug library screening as well as identification of drug targets related to cccDNA regulation.
Author Contributions
All authors contributed to this paper with conception and design of the study, literature review and analysis, drafting, critical revision and editing, and approval of the final version.
Funding
Supported by “Eradication of HBV TCR Program: NMRC/TCR/014-NUHS/2015”.
Conflicts of Interest
The authors declare no conflict of interest.
References
- WHO. Global Hepatitis Report 2017. Available online: https://www.who.int/hepatitis/publications/global-hepatitis-report2017/en/ (accessed on 3 April 2019).
- Polaris Observatory Collaborators. Global prevalence, treatment, and prevention of hepatitis B virus infection in 2016: A modelling study. Lancet Gastroenterol. Hepatol. 2018, 3, 383–403. [Google Scholar]
- Ghany, M.G. Current treatment guidelines of chronic hepatitis B: The role of nucleos(t)ide analogues and peginterferon. Best Pract. Res. Clin. Gastroenterol. 2017, 31, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Ghany, M.G.; Doo, E.C. Antiviral resistance and hepatitis B therapy. Hepatology 2009, 49, S174–S184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ke, W.; Liu, L.; Zhang, C.; Ye, X.; Gao, Y.; Zhou, S.; Yang, Y. Comparison of efficacy and safety of tenofovir and entecavir in chronic hepatitis B virus infection: A systematic review and meta-analysis. PLoS ONE 2014, 9, e98865. [Google Scholar] [CrossRef] [PubMed]
- Abdul Basit, S.; Dawood, A.; Ryan, J.; Gish, R. Tenofovir alafenamide for the treatment of chronic hepatitis B virus infection. Expert Rev. Clin. Pharmacol. 2017, 10, 707–716. [Google Scholar] [CrossRef]
- Marcellin, P.; Gane, E.; Buti, M.; Afdhal, N.; Sievert, W.; Jacobson, I.M.; Washington, M.K.; Germanidis, G.; Flaherty, J.F.; Aguilar Schall, R.; et al. Regression of cirrhosis during treatment with tenofovir disoproxil fumarate for chronic hepatitis B: A 5-year open-label follow-up study. Lancet 2013, 381, 468–475. [Google Scholar] [CrossRef]
- Chang, T.T.; Liaw, Y.F.; Wu, S.S.; Schiff, E.; Han, K.H.; Lai, C.L.; Safadi, R.; Lee, S.S.; Halota, W.; Goodman, Z.; et al. Long-term entecavir therapy results in the reversal of fibrosis/cirrhosis and continued histological improvement in patients with chronic hepatitis B. Hepatology 2010, 52, 886–893. [Google Scholar] [CrossRef] [PubMed]
- Marcellin, P.; Bonino, F.; Yurdaydin, C.; Hadziyannis, S.; Moucari, R.; Kapprell, H.P.; Rothe, V.; Popescu, M.; Brunetto, M.R. Hepatitis B surface antigen levels: Association with 5-year response to peginterferon alfa-2a in hepatitis B e-antigen-negative patients. Hepatol. Int. 2013, 7, 88–97. [Google Scholar] [CrossRef]
- Xia, Y.; Protzer, U. Control of hepatitis B virus by cytokines. Viruses 2017, 9, 18. [Google Scholar] [CrossRef]
- Lau, G.K.; Piratvisuth, T.; Luo, K.X.; Marcellin, P.; Thongsawat, S.; Cooksley, G.; Gane, E.; Fried, M.W.; Chow, W.C.; Paik, S.W.; et al. Peginterferon alfa-2a, lamivudine, and the combination for hbeag-positive chronic hepatitis B. N. Engl. J. Med. 2005, 352, 2682–2695. [Google Scholar] [CrossRef]
- Marcellin, P.; Lau, G.K.; Bonino, F.; Farci, P.; Hadziyannis, S.; Jin, R.; Lu, Z.M.; Piratvisuth, T.; Germanidis, G.; Yurdaydin, C.; et al. Peginterferon alfa-2a alone, lamivudine alone, and the two in combination in patients with hbeag-negative chronic hepatitis B. N. Engl. J. Med. 2004, 351, 1206–1217. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.T.; Lai, C.L.; Kew Yoon, S.; Lee, S.S.; Coelho, H.S.; Carrilho, F.J.; Poordad, F.; Halota, W.; Horsmans, Y.; Tsai, N.; et al. Entecavir treatment for up to 5 years in patients with hepatitis B e antigen-positive chronic hepatitis B. Hepatology 2010, 51, 422–430. [Google Scholar] [CrossRef] [PubMed]
- Wursthorn, K.; Jung, M.; Riva, A.; Goodman, Z.D.; Lopez, P.; Bao, W.; Manns, M.P.; Wedemeyer, H.; Naoumov, N.V. Kinetics of hepatitis b surface antigen decline during 3 years of telbivudine treatment in hepatitis b e antigen-positive patients. Hepatology 2010, 52, 1611–1620. [Google Scholar] [CrossRef] [PubMed]
- Buti, M.; Tsai, N.; Petersen, J.; Flisiak, R.; Gurel, S.; Krastev, Z.; Aguilar Schall, R.; Flaherty, J.F.; Martins, E.B.; Charuworn, P.; et al. Seven-year efficacy and safety of treatment with tenofovir disoproxil fumarate for chronic hepatitis B virus infection. Dig. Dis. Sci. 2015, 60, 1457–1464. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.C.; Yang, S.S.; Su, C.W.; Wang, Y.J.; Lee, K.C.; Huo, T.I.; Lin, H.C.; Huang, Y.H. Predictors of response to pegylated interferon in chronic hepatitis b: A real-world hospital-based analysis. Sci. Rep. 2016, 6, 29605. [Google Scholar] [CrossRef] [PubMed]
- Van Zonneveld, M.; Flink, H.J.; Verhey, E.; Senturk, H.; Zeuzem, S.; Akarca, U.S.; Cakaloglu, Y.; Simon, C.; So, T.M.; Gerken, G.; et al. The safety of pegylated interferon alpha-2b in the treatment of chronic hepatitis b: Predictive factors for dose reduction and treatment discontinuation. Aliment. Pharmacol. Ther. 2005, 21, 1163–1171. [Google Scholar] [CrossRef]
- Su, T.H.; Liu, C.J. Combination therapy for chronic hepatitis B: Current updates and perspectives. Gut Liver 2017, 11, 590–603. [Google Scholar] [CrossRef]
- Wu, D.; Ning, Q. Toward a cure for hepatitis B virus infection: Combination therapy involving viral suppression and immune modulation and long-term outcome. J. Infect. Dis. 2017, 216, S771–S777. [Google Scholar] [CrossRef]
- Lucifora, J.; Protzer, U. Attacking hepatitis B virus cccdna—The holy grail to hepatitis B cure. J. Hepatol. 2016, 64, S41–S48. [Google Scholar] [CrossRef]
- Anikhindi, S.A.; Kumar, A.; Sharma, P.; Singla, V.; Bansal, N.; Arora, A. Ideal cure for hepatitis B infection: The target is in sight. J. Clin. Exp. Hepatol. 2018, 8, 188–194. [Google Scholar] [CrossRef]
- Dong, J.; Ying, J.; Qiu, X.; Lu, Y.; Zhang, M. Advanced strategies for eliminating the cccDNA of HBV. Dig. Dis. Sci. 2018, 63, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Bloom, K.; Maepa, M.B.; Ely, A.; Arbuthnot, P. Gene therapy for chronic HBV-can we eliminate cccdna. Genes 2018, 9, 207. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.G.; Testoni, B.; Zoulim, F. Biological basis for functional cure of chronic hepatitis B. J. Viral. Hepat. 2019. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Jiang, D.; Zhou, T.; Cuconati, A.; Block, T.M.; Guo, J.T. Characterization of the intracellular deproteinized relaxed circular DNA of hepatitis B virus: An intermediate of covalently closed circular DNA formation. J. Virol. 2007, 81, 12472–12484. [Google Scholar] [CrossRef] [PubMed]
- Summers, J.; Mason, W.S. Replication of the genome of a hepatitis B--like virus by reverse transcription of an rna intermediate. Cell 1982, 29, 403–415. [Google Scholar] [CrossRef]
- Bock, C.T.; Schranz, P.; Schroder, C.H.; Zentgraf, H. Hepatitis B virus genome is organized into nucleosomes in the nucleus of the infected cell. Virus Genes 1994, 8, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Kock, J.; Rosler, C.; Zhang, J.J.; Blum, H.E.; Nassal, M.; Thoma, C. Generation of covalently closed circular DNA of hepatitis B viruses via intracellular recycling is regulated in a virus specific manner. PLoS Pathog. 2010, 6, e1001082. [Google Scholar] [CrossRef]
- Ko, C.; Chakraborty, A.; Chou, W.M.; Hasreiter, J.; Wettengel, J.M.; Stadler, D.; Bester, R.; Asen, T.; Zhang, K.; Wisskirchen, K.; et al. Hepatitis B virus genome recycling and de novo secondary infection events maintain stable cccDNA levels. J. Hepatol. 2018, 69, 1231–1241. [Google Scholar] [CrossRef]
- Zhu, Y.; Yamamoto, T.; Cullen, J.; Saputelli, J.; Aldrich, C.E.; Miller, D.S.; Litwin, S.; Furman, P.A.; Jilbert, A.R.; Mason, W.S. Kinetics of hepadnavirus loss from the liver during inhibition of viral DNA synthesis. J. Virol. 2001, 75, 311–322. [Google Scholar] [CrossRef]
- Addison, W.R.; Walters, K.A.; Wong, W.W.; Wilson, J.S.; Madej, D.; Jewell, L.D.; Tyrrell, D.L. Half-life of the duck hepatitis B virus covalently closed circular DNA pool in vivo following inhibition of viral replication. J. Virol. 2002, 76, 6356–6363. [Google Scholar] [CrossRef]
- Moraleda, G.; Saputelli, J.; Aldrich, C.E.; Averett, D.; Condreay, L.; Mason, W.S. Lack of effect of antiviral therapy in nondividing hepatocyte cultures on the closed circular DNA of woodchuck hepatitis virus. J. Virol. 1997, 71, 9392–9399. [Google Scholar] [PubMed]
- Laras, A.; Koskinas, J.; Dimou, E.; Kostamena, A.; Hadziyannis, S.J. Intrahepatic levels and replicative activity of covalently closed circular hepatitis B virus DNA in chronically infected patients. Hepatology 2006, 44, 694–702. [Google Scholar] [CrossRef] [PubMed]
- Werle-Lapostolle, B.; Bowden, S.; Locarnini, S.; Wursthorn, K.; Petersen, J.; Lau, G.; Trepo, C.; Marcellin, P.; Goodman, Z.; Delaney, W.E.t.; et al. Persistence of cccDNA during the natural history of chronic hepatitis B and decline during adefovir dipivoxil therapy. Gastroenterology 2004, 126, 1750–1758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nassal, M. HBV cccDNA: Viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut 2015, 64, 1972–1984. [Google Scholar] [CrossRef] [PubMed]
- Allweiss, L.; Dandri, M. The role of cccDNA in HBV maintenance. Viruses 2017, 9, 156. [Google Scholar] [CrossRef]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and d virus. Elife 2012, 1, e00049. [Google Scholar] [CrossRef] [PubMed]
- Saso, W.; Tsukuda, S.; Ohashi, H.; Fukano, K.; Morishita, R.; Matsunaga, S.; Ohki, M.; Ryo, A.; Park, S.Y.; Suzuki, R.; et al. A new strategy to identify hepatitis B virus entry inhibitors by alphascreen technology targeting the envelope-receptor interaction. Biochem. Biophys. Res. Commun. 2018, 501, 374–379. [Google Scholar] [CrossRef] [PubMed]
- Volz, T.; Allweiss, L.; Ben, M.M.; Warlich, M.; Lohse, A.W.; Pollok, J.M.; Alexandrov, A.; Urban, S.; Petersen, J.; Lutgehetmann, M.; et al. The entry inhibitor myrcludex-b efficiently blocks intrahepatic virus spreading in humanized mice previously infected with hepatitis B virus. J. Hepatol. 2013, 58, 861–867. [Google Scholar] [CrossRef]
- Bogomolov, P.; Alexandrov, A.; Voronkova, N.; Macievich, M.; Kokina, K.; Petrachenkova, M.; Lehr, T.; Lempp, F.A.; Wedemeyer, H.; Haag, M.; et al. Treatment of chronic hepatitis d with the entry inhibitor myrcludex b: First results of a phase ib/iia study. J. Hepatol. 2016, 65, 490–498. [Google Scholar] [CrossRef]
- Blank, A.; Markert, C.; Hohmann, N.; Carls, A.; Mikus, G.; Lehr, T.; Alexandrov, A.; Haag, M.; Schwab, M.; Urban, S.; et al. First-in-human application of the novel hepatitis B and hepatitis d virus entry inhibitor myrcludex b. J. Hepatol. 2016, 65, 483–489. [Google Scholar] [CrossRef]
- Shimura, S.; Watashi, K.; Fukano, K.; Peel, M.; Sluder, A.; Kawai, F.; Iwamoto, M.; Tsukuda, S.; Takeuchi, J.S.; Miyake, T.; et al. Cyclosporin derivatives inhibit hepatitis B virus entry without interfering with ntcp transporter activity. J. Hepatol. 2017, 66, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Macovei, A.; Radulescu, C.; Lazar, C.; Petrescu, S.; Durantel, D.; Dwek, R.A.; Zitzmann, N.; Nichita, N.B. Hepatitis B virus requires intact caveolin-1 function for productive infection in heparg cells. J. Virol. 2010, 84, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.C.; Chen, C.C.; Chang, W.C.; Tao, M.H.; Huang, C. Entry of hepatitis B virus into immortalized human primary hepatocytes by clathrin-dependent endocytosis. J. Virol. 2012, 86, 9443–9453. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, M.; Watashi, K.; Tsukuda, S.; Aly, H.H.; Fukasawa, M.; Fujimoto, A.; Suzuki, R.; Aizaki, H.; Ito, T.; Koiwai, O.; et al. Evaluation and identification of hepatitis B virus entry inhibitors using hepg2 cells overexpressing a membrane transporter ntcp. Biochem. Biophys. Res. Commun. 2014, 443, 808–813. [Google Scholar] [CrossRef] [PubMed]
- Umetsu, T.; Inoue, J.; Kogure, T.; Kakazu, E.; Ninomiya, M.; Iwata, T.; Takai, S.; Nakamura, T.; Sano, A.; Shimosegawa, T. Inhibitory effect of silibinin on hepatitis B virus entry. Biochem. Biophys. Rep. 2018, 14, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; McAllister, R.; Boregowda, R.; Sohn, J.A.; Cortes Ledesma, F.; Caldecott, K.W.; Seeger, C.; Hu, J. Does tyrosyl DNA phosphodiesterase-2 play a role in hepatitis B virus genome repair. PLoS ONE 2015, 10, e0128401. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, K.; Que, L.; Shimadu, M.; Koura, M.; Ishihara, Y.; Wakae, K.; Nakamura, T.; Watashi, K.; Wakita, T.; Muramatsu, M. Flap endonuclease 1 is involved in cccDNA formation in the hepatitis B virus. PLoS Pathog. 2018, 14, e1007124. [Google Scholar] [CrossRef]
- Tang, L.; Sheraz, M.; McGrane, M.; Chang, J.; Guo, J.T. DNA polymerase alpha is essential for intracellular amplification of hepatitis B virus covalently closed circular DNA. PLoS Pathog. 2019, 15, e1007742. [Google Scholar] [CrossRef]
- Qi, Y.; Gao, Z.; Xu, G.; Peng, B.; Liu, C.; Yan, H.; Yao, Q.; Sun, G.; Liu, Y.; Tang, D.; et al. DNA polymerase kappa is a key cellular factor for the formation of covalently closed circular DNA of hepatitis B virus. PLoS Pathog. 2016, 12, e1005893. [Google Scholar] [CrossRef]
- Sheraz, M.; Cheng, J.; Tang, L.; Chang, J.; Guo, J.T. Cellular DNA topoisomerases are required for the synthesis of hepatitis B virus covalently closed circular DNA. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Long, Q.; Yan, R.; Hu, J.; Cai, D.; Mitra, B.; Kim, E.S.; Marchetti, A.; Zhang, H.; Wang, S.; Liu, Y.; et al. The role of host DNA ligases in hepadnavirus covalently closed circular DNA formation. PLoS Pathog. 2017, 13, e1006784. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Mills, C.; Yu, W.; Yan, R.; Aldrich, C.E.; Saputelli, J.R.; Mason, W.S.; Xu, X.; Guo, J.T.; Block, T.M.; et al. Identification of disubstituted sulfonamide compounds as specific inhibitors of hepatitis B virus covalently closed circular DNA formation. Antimicrob. Agents Chemother. 2012, 56, 4277–4288. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Cai, D.; Zhang, L.; Tang, W.; Yan, R.; Guo, H.; Chen, X. Identification of hydrolyzable tannins (punicalagin, punicalin and geraniin) as novel inhibitors of hepatitis B virus covalently closed circular DNA. Antiviral Res. 2016, 134, 97–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, F.; Zhao, Q.; Sheraz, M.; Cheng, J.; Qi, Y.; Su, Q.; Cuconati, A.; Wei, L.; Du, Y.; Li, W.; et al. HBV core protein allosteric modulators differentially alter cccDNA biosynthesis from de novo infection and intracellular amplification pathways. PLoS Pathog. 2017, 13, e1006658. [Google Scholar] [CrossRef] [PubMed]
- Berke, J.M.; Dehertogh, P.; Vergauwen, K.; Van Damme, E.; Mostmans, W.; Vandyck, K.; Pauwels, F. Capsid assembly modulators have a dual mechanism of action in primary human hepatocytes infected with hepatitis B virus. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [PubMed]
- Schuch, A.; Hoh, A.; Thimme, R. The role of natural killer cells and cd8(+) t cells in hepatitis B virus infection. Front. Immunol. 2014, 5, 258. [Google Scholar] [CrossRef]
- Lucifora, J.; Xia, Y.; Reisinger, F.; Zhang, K.; Stadler, D.; Cheng, X.; Sprinzl, M.F.; Koppensteiner, H.; Makowska, Z.; Volz, T.; et al. Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA. Science 2014, 343, 1221–1228. [Google Scholar] [CrossRef]
- Xia, Y.; Stadler, D.; Lucifora, J.; Reisinger, F.; Webb, D.; Hosel, M.; Michler, T.; Wisskirchen, K.; Cheng, X.; Zhang, K.; et al. Interferon-gamma and tumor necrosis factor-alpha produced by t cells reduce the HBV persistence form, cccDNA, without cytolysis. Gastroenterology 2016, 150, 194–205. [Google Scholar] [CrossRef]
- Chisari, F.V.; Mason, W.S.; Seeger, C. Virology. Comment on “specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA”. Science 2014, 344, 1237. [Google Scholar] [CrossRef]
- Bertoletti, A.; Le Bert, N. Immunotherapy for chronic hepatitis B virus infection. Gut Liver 2018, 12, 497–507. [Google Scholar] [CrossRef]
- Anderson, K.R.; Haeussler, M.; Watanabe, C.; Janakiraman, V.; Lund, J.; Modrusan, Z.; Stinson, J.; Bei, Q.; Buechler, A.; Yu, C.; et al. Crispr off-target analysis in genetically engineered rats and mice. Nat. Methods 2018, 15, 512–514. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Liang, T.J. Development of direct-acting antiviral and host-targeting agents for treatment of hepatitis B virus infection. Gastroenterology 2019, 156, 311–324. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Mao, R.; Yan, R.; Cai, D.; Zhang, Y.; Zhu, H.; Kang, Y.; Liu, H.; Wang, J.; Qin, Y.; et al. Transcription of hepatitis B virus covalently closed circular DNA is regulated by cpg methylation during chronic infection. PLoS ONE 2014, 9, e110442. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.M.; Gao, W.W.; Chan, C.P.; Cheng, Y.; Chaudhary, V.; Deng, J.J.; Yuen, K.S.; Wong, C.M.; Ng, I.O.; Kok, K.H.; et al. Requirement of crtc1 coactivator for hepatitis B virus transcription. Nucleic Acids Res. 2014, 42, 12455–12468. [Google Scholar] [CrossRef] [PubMed]
- Courtois, G.; Baumhueter, S.; Crabtree, G.R. Purified hepatocyte nuclear factor 1 interacts with a family of hepatocyte-specific promoters. Proc. Natl. Acad. Sci. USA 1988, 85, 7937–7941. [Google Scholar] [CrossRef]
- Zhou, D.X.; Yen, T.S. The ubiquitous transcription factor oct-1 and the liver-specific factor hnf-1 are both required to activate transcription of a hepatitis B virus promoter. Mol. Cell Biol. 1991, 11, 1353–1359. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Li, J.; Ou, J.H. Regulation of hepatitis B virus core promoter by transcription factors hnf1 and hnf4 and the viral x protein. J. Virol. 2004, 78, 6908–6914. [Google Scholar] [CrossRef]
- Ori, A.; Shaul, Y. Hepatitis B virus enhancer binds and is activated by the hepatocyte nuclear factor 3. Virology 1995, 207, 98–106. [Google Scholar] [CrossRef]
- Chen, M.; Hieng, S.; Qian, X.; Costa, R.; Ou, J.H. Regulation of hepatitis B virus eni enhancer activity by hepatocyte-enriched transcription factor HNF3. Virology 1994, 205, 127–132. [Google Scholar] [CrossRef]
- Raney, A.K.; Zhang, P.; McLachlan, A. Regulation of transcription from the hepatitis B virus large surface antigen promoter by hepatocyte nuclear factor 3. J. Virol. 1995, 69, 3265–3272. [Google Scholar] [Green Version]
- Li, M.; Xie, Y.; Wu, X.; Kong, Y.; Wang, Y. HNF3 binds and activates the second enhancer, enii, of hepatitis B virus. Virology 1995, 214, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Raney, A.K.; McLachlan, A. Characterization of the hepatitis B virus major surface antigen promoter hepatocyte nuclear factor 3 binding site. J. Gen. Virol. 1997, 78(Pt. 11), 3029–3038. [Google Scholar] [CrossRef]
- He, F.; Chen, E.Q.; Liu, L.; Zhou, T.Y.; Liu, C.; Cheng, X.; Liu, F.J.; Tang, H. Inhibition of hepatitis B virus replication by hepatocyte nuclear factor 4-alpha specific short hairpin rna. Liver Int. 2012, 32, 742–751. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Cabrera, M.; Letovsky, J.; Hu, K.Q.; Siddiqui, A. Multiple liver-specific factors bind to the hepatitis B virus core/pregenomic promoter: Trans-activation and repression by ccaat/enhancer binding protein. Proc. Natl. Acad. Sci. USA 1990, 87, 5069–5073. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Cabrera, M.; Letovsky, J.; Hu, K.Q.; Siddiqui, A. Transcriptional factor c/ebp binds to and transactivates the enhancer element ii of the hepatitis B virus. Virology 1991, 183, 825–829. [Google Scholar] [CrossRef]
- Huan, B.; Kosovsky, M.J.; Siddiqui, A. Retinoid x receptor alpha transactivates the hepatitis B virus enhancer 1 element by forming a heterodimeric complex with the peroxisome proliferator-activated receptor. J. Virol. 1995, 69, 547–551. [Google Scholar] [PubMed]
- Raney, A.K.; Kline, E.F.; Tang, H.; McLachlan, A. Transcription and replication of a natural hepatitis B virus nucleocapsid promoter variant is regulated in vivo by peroxisome proliferators. Virology 2001, 289, 239–251. [Google Scholar] [CrossRef]
- Ramiere, C.; Scholtes, C.; Diaz, O.; Icard, V.; Perrin-Cocon, L.; Trabaud, M.A.; Lotteau, V.; Andre, P. Transactivation of the hepatitis B virus core promoter by the nuclear receptor fxralpha. J. Virol. 2008, 82, 10832–10840. [Google Scholar] [CrossRef]
- Shaul, Y.; Ben-Levy, R.; De-Medina, T. High affinity binding site for nuclear factor i next to the hepatitis B virus s gene promoter. EMBO J. 1986, 5, 1967–1971. [Google Scholar] [CrossRef]
- Ori, A.; Atzmony, D.; Haviv, I.; Shaul, Y. An nf1 motif plays a central role in hepatitis B virus enhancer. Virology 1994, 204, 600–608. [Google Scholar] [CrossRef]
- Raney, A.K.; Le, H.B.; McLachlan, A. Regulation of transcription from the hepatitis B virus major surface antigen promoter by the Sp1 transcription factor. J. Virol. 1992, 66, 6912–6921. [Google Scholar] [PubMed]
- Raney, A.K.; McLachlan, A. Characterization of the hepatitis B virus large surface antigen promoter Sp1 binding site. Virology 1995, 208, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ou, J.H. Differential regulation of hepatitis B virus gene expression by the Sp1 transcription factor. J. Virol. 2001, 75, 8400–8406. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.H.; Tao, Y.; Zhang, Z.Z.; Chen, W.X.; Cai, X.F.; Chen, K.; Ko, B.C.; Song, C.L.; Ran, L.K.; Li, W.Y.; et al. Sirtuin 1 regulates hepatitis B virus transcription and replication by targeting transcription factor ap-1. J. Virol. 2014, 88, 2442–2451. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.H.; Park, C.J.; Rho, H.M. Insulin activates the hepatitis B virus x gene through the activating protein-1 binding site in hepg2 cells. DNA Cell Biol. 1998, 17, 951–956. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.H.; Huang, C.J.; Ting, L.P. Overlapping initiator and tata box functions in the basal core promoter of hepatitis B virus. J. Virol. 1995, 69, 3647–3657. [Google Scholar] [PubMed]
- Bogomolski-Yahalom, V.; Klein, A.; Greenblat, I.; Haviv, Y.; Tur-Kaspa, R. The tata-less promoter of hepatitis B virus s gene contains a tbp binding site and an active initiator. Virus Res. 1997, 49, 1–7. [Google Scholar] [CrossRef]
- Kim, B.K.; Lim, S.O.; Park, Y.G. Requirement of the cyclic adenosine monophosphate response element-binding protein for hepatitis B virus replication. Hepatology 2008, 48, 361–373. [Google Scholar] [CrossRef] [PubMed]
- Song, C.L.; Ren, J.H.; Ran, L.K.; Li, Y.G.; Li, X.S.; Chen, X.; Li, W.Y.; Huang, A.L.; Chen, J. Cyclin d2 plays a regulatory role in HBV replication. Virology 2014, 462–463, 149–157. [Google Scholar] [CrossRef]
- Tokusumi, Y.; Zhou, S.; Takada, S. Nuclear respiratory factor 1 plays an essential role in transcriptional initiation from the hepatitis B virus x gene promoter. J. Virol. 2004, 78, 10856–10864. [Google Scholar] [CrossRef] [PubMed]
- Belloni, L.; Pollicino, T.; De Nicola, F.; Guerrieri, F.; Raffa, G.; Fanciulli, M.; Raimondo, G.; Levrero, M. Nuclear HBx binds the HBV minichromosome and modifies the epigenetic regulation of cccDNA function. Proc. Natl. Acad. Sci. USA 2009, 106, 19975–19979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alarcon, V.; Hernandez, S.; Rubio, L.; Alvarez, F.; Flores, Y.; Varas-Godoy, M.; De Ferrari, G.V.; Kann, M.; Villanueva, R.A.; Loyola, A. The enzymes lsd1 and set1a cooperate with the viral protein hbx to establish an active hepatitis B viral chromatin state. Sci. Rep. 2016, 6, 25901. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.C.; Hsu, E.C.; Ting, L.P. Repression of hepatitis B viral gene expression by transcription factor nuclear factor-kappab. Cell Microbiol. 2009, 11, 645–660. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Zhai, J.; Hong, R.; Shan, S.; Kong, Y.; Wen, Y.; Wang, Y.; Liu, J.; Xie, Y. Prospero-related homeobox protein (Prox1) inhibits hepatitis B virus replication through repressing multiple cis regulatory elements. J. Gen. Virol. 2009, 90, 1246–1255. [Google Scholar] [CrossRef] [PubMed]
- Robek, M.D.; Boyd, B.S.; Wieland, S.F.; Chisari, F.V. Signal transduction pathways that inhibit hepatitis B virus replication. Proc. Natl. Acad. Sci. USA 2004, 101, 1743–1747. [Google Scholar] [CrossRef] [Green Version]
- Belloni, L.; Allweiss, L.; Guerrieri, F.; Pediconi, N.; Volz, T.; Pollicino, T.; Petersen, J.; Raimondo, G.; Dandri, M.; Levrero, M. Ifn-alpha inhibits HBV transcription and replication in cell culture and in humanized mice by targeting the epigenetic regulation of the nuclear cccDNA minichromosome. J. Clin. Investig. 2012, 122, 529–537. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi-Matsui, M.; Hayashi, Y.; Kitamura, Y.; Koike, K. Integrated hepatitis B virus DNA preserves the binding sequence of transcription factor yin and yang 1 at the virus-cell junction. J. Virol. 2000, 74, 5562–5568. [Google Scholar] [CrossRef]
- Ren, J.H.; Hu, J.L.; Cheng, S.T.; Yu, H.B.; Wong, V.K.W.; Law, B.Y.K.; Yang, Y.F.; Huang, Y.; Liu, Y.; Chen, W.X.; et al. Sirt3 restricts hepatitis B virus transcription and replication through epigenetic regulation of covalently closed circular DNA involving suppressor of variegation 3-9 homolog 1 and set domain containing 1a histone methyltransferases. Hepatology 2018, 68, 1260–1276. [Google Scholar] [CrossRef]
- Benhenda, S.; Ducroux, A.; Riviere, L.; Sobhian, B.; Ward, M.D.; Dion, S.; Hantz, O.; Protzer, U.; Michel, M.L.; Benkirane, M.; et al. Methyltransferase prmt1 is a binding partner of hbx and a negative regulator of hepatitis B virus transcription. J. Virol. 2013, 87, 4360–4371. [Google Scholar] [CrossRef]
- Zhang, W.; Chen, J.; Wu, M.; Zhang, X.; Zhang, M.; Yue, L.; Li, Y.; Liu, J.; Li, B.; Shen, F.; et al. Prmt5 restricts hepatitis B virus replication through epigenetic repression of covalently closed circular DNA transcription and interference with pregenomic rna encapsidation. Hepatology 2017, 66, 398–415. [Google Scholar] [CrossRef]
- Riviere, L.; Gerossier, L.; Ducroux, A.; Dion, S.; Deng, Q.; Michel, M.L.; Buendia, M.A.; Hantz, O.; Neuveut, C. HBx relieves chromatin-mediated transcriptional repression of hepatitis B viral cccDNA involving setdb1 histone methyltransferase. J. Hepatol. 2015, 63, 1093–1102. [Google Scholar] [CrossRef] [PubMed]
- Decorsiere, A.; Mueller, H.; van Breugel, P.C.; Abdul, F.; Gerossier, L.; Beran, R.K.; Livingston, C.M.; Niu, C.; Fletcher, S.P.; Hantz, O.; et al. Hepatitis B virus x protein identifies the smc5/6 complex as a host restriction factor. Nature 2016, 531, 386–389. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Wu, Z.; Tan, S.; Wang, Z.; Lin, Q.; Li, X.; Song, X.; Liu, Y.; Song, Y.; Zhang, J.; et al. Tumor suppressor zhx2 restricts hepatitis B virus replication via epigenetic and non-epigenetic manners. Antiviral Res. 2018, 153, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Ko, C.; Lee, S.; Windisch, M.P.; Ryu, W.S. Ddx3 dead-box rna helicase is a host factor that restricts hepatitis B virus replication at the transcriptional level. J. Virol. 2014, 88, 13689–13698. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Feng, J.; Yang, G.; Zhang, S.; Liu, Y.; Bu, Y.; Sun, M.; Zhao, M.; Chen, F.; Zhang, W.; et al. Hepatitis B virus x protein-elevated MSL2 modulates hepatitis B virus covalently closed circular DNA by inducing degradation of APOBEC3B to enhance hepatocarcinogenesis. Hepatology 2017, 66, 1413–1429. [Google Scholar] [CrossRef] [PubMed]
- Pollicino, T.; Belloni, L.; Raffa, G.; Pediconi, N.; Squadrito, G.; Raimondo, G.; Levrero, M. Hepatitis B virus replication is regulated by the acetylation status of hepatitis B virus cccDNA-bound h3 and h4 histones. Gastroenterology 2006, 130, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Tropberger, P.; Mercier, A.; Robinson, M.; Zhong, W.; Ganem, D.E.; Holdorf, M. Mapping of histone modifications in episomal HBV cccDNA uncovers an unusual chromatin organization amenable to epigenetic manipulation. Proc. Natl. Acad. Sci. USA 2015, 112, E5715–E5724. [Google Scholar] [CrossRef] [Green Version]
- Flecken, T.; Meier, M.A.; Skewes-Cox, P.; Barkan, D.T.; Heim, M.H.; Wieland, S.F.; Holdorf, M.M. Mapping the heterogeneity of histone modifications on hepatitis B virus DNA using liver needle biopsies obtained from chronically infected patients. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Livingston, C.M.; Ramakrishnan, D.; Strubin, M.; Fletcher, S.P.; Beran, R.K. Identifying and characterizing interplay between hepatitis B virus x protein and smc5/6. Viruses 2017, 9, 69. [Google Scholar] [CrossRef]
- Murphy, C.M.; Xu, Y.; Li, F.; Nio, K.; Reszka-Blanco, N.; Li, X.; Wu, Y.; Yu, Y.; Xiong, Y.; Su, L. Hepatitis B virus x protein promotes degradation of smc5/6 to enhance HBV replication. Cell Rep. 2016, 16, 2846–2854. [Google Scholar] [CrossRef]
- Sekiba, K.; Otsuka, M.; Ohno, M.; Yamagami, M.; Kishikawa, T.; Suzuki, T.; Ishibashi, R.; Seimiya, T.; Tanaka, E.; Koike, K. Inhibition of HBV transcription from cccDNA with nitazoxanide by targeting the hbx-ddb1 interaction. Cell Mol. Gastroenterol. Hepatol. 2019, 7, 297–312. [Google Scholar] [CrossRef] [PubMed]
- Sekiba, K.; Otsuka, M.; Ohno, M.; Yamagami, M.; Kishikawa, T.; Seimiya, T.; Suzuki, T.; Tanaka, E.; Ishibashi, R.; Funato, K.; et al. Pevonedistat, a neuronal precursor cell-expressed developmentally down-regulated protein 8-activating enzyme inhibitor, is a potent inhibitor of hepatitis B virus. Hepatology 2019, 69, 1903–1915. [Google Scholar] [CrossRef] [PubMed]
- Lubyova, B.; Hodek, J.; Zabransky, A.; Prouzova, H.; Hubalek, M.; Hirsch, I.; Weber, J. Prmt5: A novel regulator of hepatitis B virus replication and an arginine methylase of HBV core. PLoS ONE 2017, 12, e0186982. [Google Scholar] [CrossRef] [PubMed]
- Turelli, P.; Mangeat, B.; Jost, S.; Vianin, S.; Trono, D. Inhibition of hepatitis B virus replication by APOBEC3G. Science 2004, 303, 1829. [Google Scholar] [CrossRef] [PubMed]
- Seppen, J. Unedited inhibition of HBV replication by APOBEC3G. J. Hepatol. 2004, 41, 1068–1069. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.H.; Gummuluru, S.; Hu, J. Deamination-independent inhibition of hepatitis B virus reverse transcription by APOBEC3G. J. Virol. 2007, 81, 4465–4472. [Google Scholar] [CrossRef]
- Bonvin, M.; Achermann, F.; Greeve, I.; Stroka, D.; Keogh, A.; Inderbitzin, D.; Candinas, D.; Sommer, P.; Wain-Hobson, S.; Vartanian, J.P.; et al. Interferon-inducible expression of APOBEC3 editing enzymes in human hepatocytes and inhibition of hepatitis B virus replication. Hepatology 2006, 43, 1364–1374. [Google Scholar] [CrossRef]
- Chen, Y.; Hu, J.; Cai, X.; Huang, Y.; Zhou, X.; Tu, Z.; Hu, J.; Tavis, J.E.; Tang, N.; Huang, A.; et al. APOBEC3B edits HBV DNA and inhibits HBV replication during reverse transcription. Antiviral Res. 2018, 149, 16–25. [Google Scholar] [CrossRef]
- Baumert, T.F.; Rosler, C.; Malim, M.H.; von Weizsacker, F. Hepatitis B virus DNA is subject to extensive editing by the human deaminase APOBEC3C. Hepatology 2007, 46, 682–689. [Google Scholar] [CrossRef]
- Nair, S.; Zlotnick, A. Asymmetric modification of hepatitis B virus (HBV) genomes by an endogenous cytidine deaminase inside HBV cores informs a model of reverse transcription. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Slagle, B.L.; Bouchard, M.J. Role of HBx in hepatitis B virus persistence and its therapeutic implications. Curr. Opin. Virol. 2018, 30, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Hantz, O.; Parent, R.; Durantel, D.; Gripon, P.; Guguen-Guillouzo, C.; Zoulim, F. Persistence of the hepatitis B virus covalently closed circular DNA in heparg human hepatocyte-like cells. J. Gen. Virol. 2009, 90, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Nie, H.; Yan, R.; Guo, J.T.; Block, T.M.; Guo, H. A southern blot assay for detection of hepatitis B virus covalently closed circular DNA from cell cultures. Methods Mol. Biol. 2013, 1030, 151–161. [Google Scholar] [PubMed]
- Suzuki, F.; Miyakoshi, H.; Kobayashi, M.; Kumada, H. Correlation between serum hepatitis B virus core-related antigen and intrahepatic covalently closed circular DNA in chronic hepatitis B patients. J. Med. Virol. 2009, 81, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Han, J.; Zou, Z.; Liu, S.; Tang, B.; Ren, X.; Li, X.; Zhao, Y.; Liu, Y.; Zhou, D.; et al. Quantitation of HBV covalently closed circular DNA in micro formalin fixed paraffin-embedded liver tissue using rolling circle amplification in combination with real-time pcr. Clin. Chim. Acta 2011, 412, 1905–1911. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhao, J.; Yuan, Q.; Xia, N. Detection of HBV covalently closed circular DNA. Viruses 2017, 9, 139. [Google Scholar] [CrossRef] [PubMed]
- Mu, D.; Yan, L.; Tang, H.; Liao, Y. A sensitive and accurate quantification method for the detection of hepatitis B virus covalently closed circular DNA by the application of a droplet digital polymerase chain reaction amplification system. Biotechnol. Lett. 2015, 37, 2063–2073. [Google Scholar] [CrossRef]
- Levrero, M.; Pollicino, T.; Petersen, J.; Belloni, L.; Raimondo, G.; Dandri, M. Control of cccDNA function in hepatitis B virus infection. J. Hepatol. 2009, 51, 581–592. [Google Scholar] [CrossRef] [Green Version]
- Singh, M.; Dicaire, A.; Wakil, A.E.; Luscombe, C.; Sacks, S.L. Quantitation of hepatitis B virus (HBV) covalently closed circular DNA (cccDNA) in the liver of HBV-infected patients by lightcycler real-time pcr. J. Virol. Methods 2004, 118, 159–167. [Google Scholar] [CrossRef]
- Jiang, P.X.; Mao, R.C.; Dong, M.H.; Yu, X.P.; Xun, Q.; Wang, J.Y.; Jing, L.; Qiang, D.; Zhang, J.M. Exonuclease I and III improve the detection efficacy of hepatitis B virus covalently closed circular DNA. Hepatobiliary Pancreat Dis. Int. 2018. [Google Scholar] [CrossRef]
- Luo, J.; Cui, X.; Gao, L.; Hu, J. Identification of an intermediate in hepatitis B virus covalently closed circular (ccc) DNA formation and sensitive and selective ccc DNA detection. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Y.; Zhang, B.H.; Theele, D.; Litwin, S.; Toll, E.; Summers, J. Single-cell analysis of covalently closed circular DNA copy numbers in a hepadnavirus-infected liver. Proc. Natl. Acad. Sci. USA 2003, 100, 12372–12377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, D.; Wang, X.; Yan, R.; Mao, R.; Liu, Y.; Ji, C.; Cuconati, A.; Guo, H. Establishment of an inducible HBV stable cell line that expresses cccDNA-dependent epitope-tagged hbeag for screening of cccDNA modulators. Antiviral Res. 2016, 132, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Qi, Z.; Li, G.; Hu, H.; Yang, C.; Zhang, X.; Leng, Q.; Xie, Y.; Yu, D.; Zhang, X.; Gao, Y.; et al. Recombinant covalently closed circular hepatitis B virus DNA induces prolonged viral persistence in immunocompetent mice. J. Virol. 2014, 88, 8045–8056. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Li, J.; Yue, L.; Bai, L.; Li, Y.; Chen, J.; Zhang, X.; Yuan, Z. Establishment of cre-mediated HBV recombinant cccDNA (rcccDNA) cell line for cccDNA biology and antiviral screening assays. Antiviral Res. 2018, 152, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.Y.; He, C.Y.; Ehrhardt, A.; Kay, M.A. Minicircle DNA vectors devoid of bacterial DNA result in persistent and high-level transgene expression in vivo. Mol. Ther. 2003, 8, 495–500. [Google Scholar] [CrossRef]
- Kay, M.A.; He, C.Y.; Chen, Z.Y. A robust system for production of minicircle DNA vectors. Nat. Biotechnol. 2010, 28, 1287–1289. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Chen, P.; Hou, X.; Xu, W.; Wang, D.; Wang, T.Y.; Zhang, L.; Zheng, G.; Gao, Z.L.; He, C.Y.; et al. The recombined cccDNA produced using minicircle technology mimicked HBV genome in structure and function closely. Sci. Rep. 2016, 6, 25552. [Google Scholar] [CrossRef] [Green Version]
- Yan, Z.; Zeng, J.; Yu, Y.; Xiang, K.; Hu, H.; Zhou, X.; Gu, L.; Wang, L.; Zhao, J.; Young, J.A.T.; et al. HBV CIRCLE: A novel tool to investigate hepatitis B virus covalently closed circular DNA. J. Hepatol. 2017, 66, 1149–1157. [Google Scholar] [CrossRef]
- Li, F.; Cheng, L.; Murphy, C.M.; Reszka-Blanco, N.J.; Wu, Y.; Chi, L.; Hu, J.; Su, L. Minicircle HBV cccDNA with a gaussia luciferase reporter for investigating HBV cccDNA biology and developing cccDNA-targeting drugs. Sci. Rep. 2016, 6, 36483. [Google Scholar] [CrossRef]
- Nishitsuji, H.; Ujino, S.; Shimizu, Y.; Harada, K.; Zhang, J.; Sugiyama, M.; Mizokami, M.; Shimotohno, K. Novel reporter system to monitor early stages of the hepatitis B virus life cycle. Cancer Sci. 2015, 106, 1616–1624. [Google Scholar] [CrossRef] [PubMed]
- Nishitsuji, H.; Harada, K.; Ujino, S.; Zhang, J.; Kohara, M.; Sugiyama, M.; Mizokami, M.; Shimotohno, K. Investigating the hepatitis B virus life cycle using engineered reporter hepatitis B viruses. Cancer Sci. 2018, 109, 241–249. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic representation of the hepatitis B virus (HBV) life cycle.

Figure 2.
Summary of main classes of host factors that promote or inhibit covalently closed circular DNA (cccDNA) transcription.
Figure 2.
Summary of main classes of host factors that promote or inhibit covalently closed circular DNA (cccDNA) transcription.


{kind=link}
{kind=link}
Table 1.
Host factors that promote or repress HBV cccDNA activity.
| Name | Function | Effect on cccDNA | References |
|---|---|---|---|
| HNF1 | Transcription factor | Activation | [66,67,68] |
| HNF3/FoxA | Transcription factor | Activation | [69,70,71,72,73] |
| HNF4 | Nuclear receptor | Activation | [68,74] |
| C/EBP | Transcription factor | Activation | [75,76] |
| RXRα/PPARα | Nuclear receptor | Activation | [77,78] |
| FXR | Nuclear receptor | Activation | [79] |
| NF1 | Transcription factor | Activation | [80,81] |
| SP1 | Transcription factor | Activation | [82,83,84] |
| AP-1 | Transcription factor | Activation | [85,86] |
| TBP | Transcription factor | Activation | [87,88] |
| CREB | Transcription factor | Activation | [89,90] |
| Oct1 | Transcription factor | Activation | [67] |
| NRF1 | Transcription factor | Activation | [91] |
| CRTC1 | Transcriptional coactivator | Activation | [65] |
| CBP | HAT | Activation | [92] |
| p300 | HAT | Activation | [92] |
| PCAF/GCN5 | HAT | Activation | [92] |
| LSD1 | Histone demethylase | Activation | [93] |
| Set1A | KMT | Activation | [93] |
| NF-κB | Transcription factor | Inhibition | [94] |
| PROX1 | Transcription factor | Inhibition | [95] |
| STAT1/2 | Transcription factor | Inhibition | [96,97] |
| EZH2 | KMT | Inhibition | [97] |
| YY1 | Transcription factor | Inhibition | [97,98] |
| SIRT1 | Class III HDAC | Inhibition | [85,97] |
| SIRT3 | Class III HDAC | Inhibition | [99] |
| PRMT1 | PRMT | Inhibition | [100] |
| PRMT5 | PRMT | Inhibition | [101] |
| HDAC1 | Class I HDAC | Inhibition | [97] |
| SETDB1 | KMT | Inhibition | [102] |
| Smc5/6 | Structural maintenance of chromosomes | Inhibition | [103] |
| ZHX2 | Transcription factor | Inhibition | [104] |
| DDX3 | DEAD-box RNA helicase | Inhibition | [105] |
| APOBEC3A | Cytidine deaminase | Inhibition | [58] |
| APOBEC3B | Cytidine deaminase | Inhibition | [58,106] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Mohd-Ismail, N.K.; Lim, Z.; Gunaratne, J.; Tan, Y.-J. Mapping the Interactions of HBV cccDNA with Host Factors. Int. J. Mol. Sci. 2019, 20, 4276. https://doi.org/10.3390/ijms20174276
AMA Style
Mohd-Ismail NK, Lim Z, Gunaratne J, Tan Y-J. Mapping the Interactions of HBV cccDNA with Host Factors. International Journal of Molecular Sciences. 2019; 20(17):4276. https://doi.org/10.3390/ijms20174276
Chicago/Turabian StyleMohd-Ismail, Nur K., Zijie Lim, Jayantha Gunaratne, and Yee-Joo Tan. 2019. "Mapping the Interactions of HBV cccDNA with Host Factors" International Journal of Molecular Sciences 20, no. 17: 4276. https://doi.org/10.3390/ijms20174276
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.