Neurodegeneration and Neuro-Regeneration—Alzheimer’s Disease and Stem Cell Therapy

1
Institute for Microscopic Anatomy and Neurobiology, University Medical Center of the Johannes Gutenberg University, 55131 Mainz, Germany
2
Institute of Anatomy, Medical Faculty Carl Gustav Carus, Technische Universität Dresden School of Medicine, 01069 Dresden, Germany
*
Author to whom correspondence should be addressed.
†
Verica Vasic and Kathrin Barth are both co-first author.
Int. J. Mol. Sci. 2019, 20(17), 4272; https://doi.org/10.3390/ijms20174272
Submission received: 23 July 2019
/
Revised: 27 August 2019
/
Accepted: 28 August 2019
/
Published: 31 August 2019
(This article belongs to the Special Issue Feature Annual Reviews in Molecular Sciences 2019)
Abstract
:Aging causes many changes in the human body, and is a high risk for various diseases. Dementia, a common age-related disease, is a clinical disorder triggered by neurodegeneration. Brain damage caused by neuronal death leads to cognitive decline, memory loss, learning inabilities and mood changes. Numerous disease conditions may cause dementia; however, the most common one is Alzheimer’s disease (AD), a futile and yet untreatable illness. Adult neurogenesis carries the potential of brain self-repair by an endogenous formation of newly-born neurons in the adult brain; however it also declines with age. Strategies to improve the symptoms of aging and age-related diseases have included different means to stimulate neurogenesis, both pharmacologically and naturally. Finally, the regulatory mechanisms of stem cells neurogenesis or a functional integration of newborn neurons have been explored to provide the basis for grafted stem cell therapy. This review aims to provide an overview of AD pathology of different neural and glial cell types and summarizes current strategies of experimental stem cell treatments and their putative future use in clinical settings.
1. Introduction
According to the World Alzheimer Report 2018 there are about 50 million people suffering from dementia in the world. It is expected that this number will increase to about 82 million in 2030, and to about 152 million in 2050. There are over 200 subtypes of dementia, 50%–60% of all cases caused by Alzheimer’s disease (AD). This type of dementia was originally described by Alois Alzheimer in 1907 in Frankfurt am Main, and has become designated later as the most widespread neurodegenerative disease. However, its etiology is still so far unknown, despite many facts about its pathology having been uncovered during the last decades. It is well known that dementia is mainly a sporadic, age-related disease, and only less than 5% of all cases are caused by inherited mutations. The risk of AD development increases 14-fold between the age of 65–85, and affects almost 47% of people over the age of 85 [1]. This disease is characterized by the progressive deterioration of cognitive functions. Clinically, these patients are characterized by an impairment of short-term memory interfering and complicating the activities of daily life, later followed by impairment in the other cognitive fields, e.g., language, logical understanding, orientation, executive function, judgment, behavior and, finally, motor difficulties. This progress is linked to a significant reduction in the volume of the brain in these patients [2]. This atrophy results from the death of neurons and the degeneration of synapses, in particular in the hippocampus formation [3], which is the region responsible for memory and spatial orientation. One of the hallmarks of AD is the occurrence of amyloid plaques in extracellular space of AD brains.
They consist of the agglutinated peptide amyloid-β (mainly Aβ1–40/42) containing β-sheet structures and which forms fibrils [4]. The accumulation of amyloid-β peptide is caused by a disturbance of the homeostasis of amyloid-β peptides arising from proteolytic degradation of the amyloid precursor protein (APP). Furthermore, AD brains are characterized by the presence of neurofibrillary tangles, composed of hyperphosphorylated tau protein [5], loss of synapses, dystrophic neurites, and a prominent gliosis.
The pathogenesis of this progressive brain abnormality is multi-factorally caused. The main reasons are age, environmental factors, like chronic stress, traumatic brain injury, internal processes like chronic pain, oxidative stress or inflammation, as well as several genetic factors, like mutations in the genes encoding the amyloid precursor protein (APP), preselinin-1 and -2 or the apolipo-protein E (ApoE) protein.
2. Cellular Systems Related to AD
The nervous system is formed mainly by various cell types, neurons and glial cells. Microglia, as well as glial cells, namely oligodendrocytes, astrocytes, and oligodendrocyte progenitor cells (polydendrocytes, NG2 glia), and neurons, may play an important role in the pathogenesis of AD, caused by their roles in neuroprotection, maintenance of CNS homeostasis (concentration of ions, neurotransmitter, etc.), and the brain immune system.
2.1. Microglia
Recently, extensive reviews on the role of microglia in healthy brains and the pathogenesis of neurological disorders including AD have been published recently [6,7,8]. Here we will focus upon the main findings, only. Microglia are the brain’s resident macrophages arising from the mesenchyme [9], or are blood-derived (review, see [10]). Microglia display a ramified morphology with numerous branching processes, which enable them to get a survey of the brain environment [11]. Microglial cells have an important function for the preservation of a healthy brain by their ability to attack and remove potential pathogens and detritus, as well as by the secretion of tissue rebuilding factors [12,13]. Furthermore, several data implicate a potential role of microglia for synaptic remodeling [14]. Microglial cells sense neuronal activity, and thereby regulate synaptic plasticity as well as learning and memory mechanisms. Therefore they are a main component in the determination of cognitive function [15,16].
Microglia are linked to neuro-inflammation and are involved in the phagocytic clearance of Aβ peptide. An imbalance in the homeostasis of Aβ peptides as observed in neurological disorders like AD leads to the accumulation and aggregation of Aβ peptide, which is accompanied by a prominent gliosis. This involves changes in the morphology and function of microglia in the Aβ peptide fibrils surrounding parenchyma as an inflammatory response (reviews see [17,18,19,20,21]. The inflammation process is clearly associated with the pathogenesis of the sporadic forms of AD [22]. Microglial cells release pro-inflammatory neurotoxins and cytokine/chemokines like TNF-α, interferon (IFN)−γ or interleukins IL-1β and IL-6.
The human genome-wide association studies (GWAS) have shown that the microglial triggering receptor expressed in myeloid cells 2 (TREM2) plays an important role in the AD-related immune response [23]. It has been shown that TREM2 as a lipid and lipoprotein sensor supports reactive microgliosis [24,25,26] and triggers the transcriptional activation of microglial cells by interacting with the apolipoprotein E4 (APOE4), one of the major risk factors for AD [27].
A further gene which is involved in the etiology of sporadic AD is the ATP-binding cassette subfamily A member 7 (ABCA7) gene, encoding a protein involved in the regulation of lipid homeostasis, and probably also in Aβ homeostasis [28]. This protein indirectly effects the phagocytosis of apoptotic cells and Aβ peptides by mediating the formation of phagocytic cups [29,30,31].
During progress of the neuroinflammatory process in AD pathogenesis, ATP is released into the extracellular space and induces an increased expression of the P2X7 receptor (P2X7R) in microglial cells. This results in the promotion and activation of microglia. P2X7R is an ATP-gated, non-selective cation channel localized in cellular membrane and responsible for the influx of Ca2+ or Na2+ and efflux of K+ [32]. Besides the promotion and activation of microglial cells, an increased expression of this receptor in microglial cells results also in a decrease of their phagocytic activity [33,34,35] and a stimulation of production of neurotoxic molecules like TNF-a, COX-2, IL-6, MMP-9 and reactive oxygen species (ROS) [36,37,38,39]. Hey et al. [40] have shown that P2X7R, modulator of neuroinflammation, plays an important role in mediating microglial cell death and cytokine release, which may be coupled to AKT and ERK pathways. Furthermore, it has been demonstrated that the P2X7/NLRP3 inflammasome pathway leads to the release of the pro-inflammatory cytokines IL-1β, IL-18 and IL-33 [41,42,43]. Therefore, it was suggested that the overactivation of P2X7R in microglial cells mediated by extracellular ATP is most important for the generation of neuroinflammation-induced AD [44].
2.2. Glial Cells
2.2.1. Astrocytes
Astrocytes interact with neurons by releasing and recycling glio-transmitter, controlling ion homeostasis, energy metabolism, synaptic remodeling and the modulation of oxidative stress [8]. This cooperative interplay controls neurotransmission as well as synaptic plasticity in different brain regions [45,46,47], and modulates cognitive functions [46,48]. Astrocytes respond quickly to pathological changes inside the brain, react chemotactically to compounds of Aβ plaques, express receptors that bind Aβ, and therefore accumulate at sites of aggregated Aβ depositions, where they take up and degrade Aβ [49,50,51,52]. Extracellular degradation of Aβ may also occur by a secretion of several enzymes like insulin degrading enzyme and metalloproteases [49,53,54,55]. Aggregated Aβ induces an increased Ca2+ uptake in astrocytes by an enhanced activation of P2Y1 receptors [56], transient receptor potential channel 4 [57], nicotinic acid receptor [58,59] and the glutamate metabotropic receptor mGluR5 [60,61]. Furthermore, uptake of glutamate is decreased by Aβ in astrocytes by changing the activity of adenosine A2A receptors [62].
Most importantly, the presence of Aβ induces the release of cytokines and chemokines by its secretion of Aβ peptides by astrocytes [63]. Apolipoprotein E (ApoE), which plays an important role in AD pathogenesis, is also expressed in astrocytes. Furthermore, hyperphosphorylated tau-forming intracellular tangles, which besides Aβ are the second hallmark of AD, can also be produced in astrocytes (see Section 2.3). Taken together, all these data support the strong connection of astrocytes with the pathogenesis of AD.
2.2.2. Oligodendrocytes
Oligodendrocytes together with myelin lipid layers form the envelope of the neuronal axons required for the fast action potential propagation. They are well known targets for immune reactions in neuronal disorders and exhibit specific morphological changes during AD progression. However, their special function remains still unclear, despite the fact that various experimental data have been published (review see [8]). Oligodendrocytes exhibit specific morphological changes during AD progression. Deterioration in myelin integrity and axonal destruction have been observed [64,65,66,67,68,69]. However, studies on mouse models and hAD samples have shown that there is no reduction of the overall amount of myelin [70,71,72]. Furthermore, it has been demonstrated that Aβ has a cytotoxic effect on oligodendrocytes [73].
2.2.3. NG2-Glia
NG2-glia, also called polydendrocytes or oligodendrocyte precursor cells (OPCs), are the newest discovered glia cell type [74], which play an important role in the pathogenesis of AD. It has been shown that in APP23 mice, Aβ activates GSK3β, resulting in the increased phosphorylation of β-catenin followed by β-catenin degradation accompanied with the inhibition of the Wingles/integrated (Wnt) signaling pathway [75]. This results in an inhibition of the differentiation of NG2-glia [76]. Further studies are required to uncover the specific role of NG-2 glia in the pathogenesis of AD.
2.3. Neurons
Neurons express a large number of molecules, protecting them against inflammatory attacks and inducing neurological disorders. Some of these cells have been detected to be injured and to be defective in AD. The most prominent pathological process in neurons is the formation of intracellular neurofibrillary tangles by hyperphosphorylated tau protein, which is one of the pathological hallmarks of AD. The tau protein normally binds to microtubule, and is involved in the axonal transport of mitochondria in the cell. It contains more than 45 phosphorylation sites [77]. The controlled phosphorylation of these sites affects the capacity of tau to bind to microtubules [78]. During AD progress, tau is hyperphosphorylated, dissociates from microtubules and aggregates intracellularly into neurofibrillary tangles [5,79,80,81]. This results in impaired axonal transport of mitochondria between cell body and synapsis, leading to energy dysfunction, generation of reactive oxygen species (ROS) and nitrogen species [82,83].
3. Organelles and Organelle Related Processes
3.1. Mitochondria
Constantly, mitochondria undergo a balanced fission and fusion process [84] which promotes mitochondrial distribution along micro-tubulare axons into synapses [85]. This process enables mitochondria to react to high energy demands and to facilitate neuroprotective effects, eliminating defective mitochondria constituents and protecting against reactive oxygen species (ROS) damage during ageing [86,87,88].
As mentioned above (see Section 2.3), mitochondria dysfunction has a fundamental role in the pathogenesis of AD [89]. Dysfunctional mitochondria exhibit abnormal morphology-decreased ATP synthesis, impaired antioxidant enzymes and defective oxidative phosphorylation complexes. Mitochondria in the neuronal cells of AD patients show a perinuclear mis-localization resulting in ATP depletion, oxidative stress and synaptic dysfunction [90,91]. The main reason for these disturbances seems to be the aggregation of hyperphosphorylated tau in neurofibrillary tangles (see Section 2.2). However, it needs to be taken into account that this is a very complex process in which also Aβ interactions, ROS production by free metal ions (review see [91], defective autophagy, in particular dysfunctional mitophagy, as well as other processes, may be involved.
3.2. Autophagy (Mitophagy)
Autophagy is an organelle-, protein- and lipid-degrading pathway mediated by membranes, vesicles and lysosomes, and is essential for protein, lipid and organelle homeostasis to ensure cell health. Autophagy is the process responsible for the turn-over of mitochondria (called mitophagy) to adapt mitochondria to different energy demands and to eliminate dysfunctional mitochondria. These are actively transported into lysosomes and degraded there. Trafficking may occur via three different pathways:
- Macroautophagy—engulfment of cytoplasmic components by autophagy vesicles and fusion with lysosomes regulated by autophagy-related proteins and specific autophagy receptors.
- Microautophagy—direct engulfment of cytoplasmic components by lysosomes [92].
Autophagy is essential for synaptic plasticity, anti-inflammatory function in glial cells, oligodendrocyte development, and the myelination process [97,98]. Recently, it has been demonstrated that autophagy plays a major role in the etiology of AD [99,100,101]. Genetic studies have also demonstrated a link between the expression of several autophagy genes and AD [102,103]. Furthermore it has been shown that impaired mitophagy results in reduced cellular energy levels, increased ROS, and impaired neuroplasticity [104,105]. Recent transcriptome analysis of two AD models (3xTG-AD and Apo3/Apo4-targeted replacement mice) supports the previous data that autophagy, including mitophagy, is clearly dysregulated in AD [106].
3.3. Endocytic Processes
Cells use different pathways to internalize materials from the cellular surface into the cell. Most cargos enter cells via clathrin-mediated endocytosis (CME) [107]. Besides these there exist two clathrin-independent endocytotic pathways (CIE), one is using flotillin-1 and -2, and the other one is going via caveolae [108,109]. The cargo including endosomes deliver their material to a variety of intracellular compartments like the endosomes and the trans-Golgi network, or recycle the material back to the plasma membrane [110]. Flotillin-1 and -2 are proteins associated with membrane lipid-rafts, regions enriched with cholesterol, glycosphingolipids and sphingomyelin essential for synapse development, maintenance and stabilization [111,112]. They form micro-domains, and are highly expressed in neurons [113,114]. Caveolae are invaginations in ordered lipid raft domains of the plasma membrane [115], and may contain caveolin-1, -2 or -3 [110,116], depending on cell type. However, all three isoforms are found in the nervous system of mammals [117]. Among them caveolin-1 (Cav-1) is a cholesterol binding protein which organizes and targets synaptic components of the neurotransmitter and neurotropic receptor signaling pathway to the lipid rafts [36,118,119,120,121]. Lipid rafts, cholesterol and Cav-1 may be involved in protection processes against progressing APP and Aβ toxicity. APP is enriched in lipid rafts physically associated with Cav-1 [122]. Overexpression of Cav-1 was associated with α-secretase-mediated proteolysis of APP [117,122]. Cav-2 and Cav-1 interact together and form a heteromeric complex [123]. Contrary to Cav-1, Cav-2 increases with ageing, suggesting a possible increase in the endocytosis of APP [124]. Cav-3 is predominantly expressed in astroglial cells, and is reduced in older cells [124]. Both flotillins are also localized to lipid rafts, and participate in the generation of Aβ [125]. Flotillin-1 is unchanged by ageing, contrary to flotillin-2, which is significantly more highly expressed in aged brains [124].
There are only a few studies on changes in endocytic processes in AD brains, however, remarkable changes in endocytosis have been observed already [108,124,126]. Early endocytic changes like increased volume of total endosomes [127] and increased levels of several CME proteins [128] were detected. Down-regulation of Cav-1 increased the accumulation of APP in AD [122,129]. This is supported by studies of Cav-1 KO mice which develop CNS pathology similar to AD [120,126,130,131,132]. An accumulation of flotillin-1 has been detected in the endosomes of neurons in AD in comparison with healthy patients [125].
An overview representing the involvement of specific cell types and the role of organelles and cell processes in the development of AD is presented in Table 1.
4. Stem Cell Therapies
4.1. Endogenous Regeneration
Adult hippocampal neurogenesis, the generation of adult-born neurons in the dentate gyrus (DG) from residing stem cells, was broadly analyzed in the context of AD. Firstly, the hippocampus and its neurogenic DG are one of the brain regions that have a key role in learning and memory [133,134,135,136,137]. The hippocampus is one of the first regions of the brain to suffer from damage, observed by the symptoms such as short-term memory loss and disorientation. Secondly, severe neuronal loss in hippocampal regions is another hallmark of AD. Approximately, 1 million neurons are lost in the DG, and about 5 million in the CA1 [138]. On the contrary, the rate of hippocampal neurogenesis was estimated to be about 700 adult-born neurons a day in the adult brain, but is moderately declining with age [139]. Therefore, hippocampal neurogenesis was suspected as the brain’s endogenous regenerative process that may be used for treatment purposes.
Despite the idea that studies of adult neurogenesis in animal models of AD provided a great variability in results, which was dependent on experimental conditions, the age or promoter studied, and though neurogenesis was decreased and increased in its dependence of study conditions (reviewed in [140,141]), most strategies focused on the induction of neurogenesis. Adult neurogenesis can be stimulated through extrinsic administration of chemical agents and growth factors in the context of AD, such as: Erythropoietin [142,143,144], fluoxetine [145], granulocyte colony stimulating factor (G-CSF) and AMD3100 [146], brain-derived neurotrophic factor (BDNF) [147,148,149], insulin growth factor-1 (IGF-1) [150], nerve growth factor (NGF) [151,152], vascular endothelial growth factor (VEGF) [153], growth, and transforming growth factor β (TGF-β) [154]. In addition, neurogenesis becomes stimulated by voluntary running and physical exercise, as well as an enriched environment [155,156,157,158]. Long-term physical activity was even found to diminish neuronal loss in the CA1 region in the AD transgenic mouse model [159], which is of particular interest.
The replacement of CA1 neurons is still unclear, as this is one of the major destruction sites. Therefore, further and additional approaches are required.
4.2. Engrafted Regeneration
In general, there are several major groups of stem cells that are used for therapeutic purposes. Mostly, embryonic stem cells (ESCs), neural stem cells (NSCs), induced pluripotent stem cells (iPSCs) and mesenchymal (MSCs) stem cells are applied in this context. Different strategies regarding their application in the context of stem cells therapies in AD is overviewed in Figure 1. Other types of stem cells, such as olfactory ensheathing cells and hematopoietic stem cells, may as well be applied for treatment [160], however they are not discussed in this review.
4.1.1. ESCs
ESCs possess enormous potential due to their pluripotency, the ability to generate cell types from the ectodermal, mesodermal and endodermal germ layers, and due to their unlimited self-renewal capacities [161]. This makes them a very attractive candidate as a therapeutic means. They are derived from the inner cell mass of a developing blastocyst at embryonic day 5 to 6. For research purposes however, most ESCs are derived from embryos that develop from eggs that have been fertilized in vitro and then donated for research purposes (with the informed consent of the donors). Nevertheless, the use of ESCs for research purposes naturally remains a controversy, and is differentially regulated from country to country. For example, in Germany the use of ESCs is prohibited, and yet in Austria it is allowed. In the USA, on the federal level, there is no official ban, however particular states employ their own restrictions.
Several reports have explored the role of ESCs in AD rodent models. Pluripotency, one the biggest advantages of ESCs, represents one of their main drawbacks, as their differentiation can occur towards any direction, and lead to neoplasia or teratoms [162,163]. Therefore, research strategies focus on establishing pre-differentiation protocols. Mouse ESCs (mESCs) were successfully used to generate basal forebrain cholinergic neurons (BFCNs), which are severely affected in AD patients, and this process was applied for driving ESC-derived, as well as primed NPC differentiation, upon the transplantation into the AD rat model [164]. In addition, these rats displayed significant behavioral improvement in memory deficits. Human ESCs (hESCs) were also able to produce cholinergic neurons in vitro and upon engraftment into cultured entorhinal-hippocampal mouse slices, they connected to the existing neuronal network [165]. Similarly, both mESCs and hESCs were directed into mature BFCNs, and following transplantation into AD mice, improvement in learning and memory performance was observed [166]. Another approach was to differentiate hESCs into MGE (medial ganglionic eminence)-like progenitors cells, as MGE is the place of origin of basal forebrain neurons, including BFCNs and GABA interneurons, during development. Transplantation of these MGE-like progenitors into the mouse hippocampus yielded comparable results as the studies mentioned above [167].
4.1.2. NSCs
Adult NSCs reside in the sub-granular zone (SGZ) of hippocampal DG and in the subventricular zone (SVZ) of the lateral walls of the ventricles. They are self-renewing, multipotent cells that can give rise to neurons, oligodendrocytes or astrocytes [168]. Discovery of the NSCs’ existence in the adult brain a few decades ago opened a whole new field of treatment possibilities for incurable brain diseases. Aside from strategies for their endogenous repair applications, they are also used in exogenous repair strategies. Critical points of NSCs’ transplantation include their poor survival outcomes [169,170], therefore, aside from substituting damaged cells via the differentiation and autocrine production of neurotrophic and neuroprotective factors, therapeutic approaches have focused upon a combination of grafting and the application of beneficial factors and therapeutic genes.
hNSCs from fetal telencephalon were transplanted into the lateral ventricles of an AD mouse brain where they migrated, engrafted and differentiated into neuronal and glial cells. The results of this process included improved spatial memory, decreased tau phosphorylation and Aβ42 levels, reduced microgliosis and astrogliosis [171], enhanced endogenous synaptogenesis [172] and increased neuronal, synaptic and nerve fiber density [173]. Interestingly, these effects were achieved via multiple mechanisms, including the modulation of signaling pathways, metabolic activity, secretion of anti-inflammatory factors and cell-to-cell contacts. BDNF, an important NSC-derived neuroprotective factor, was found to be crucial for improving the cognition of AD rodents with transplanted NSCs (obtained from the brain or the hippocampus), by improving hippocampal synaptic density [147] and enhancing the number of cholinergic neurons [174,175]. Transplantation of an hNSC line that over-expresses choline acetyltransferase, an enzyme that synthesizes acetylcholine (ACh) into the aged ICR mice, led to the improved cognitive function and physical activity of aging mice [176]. This was achieved by producing ACh directly and restoring cholinergic neuronal integrity, probably mediated by increased levels of BDNF and NGF neurotrophins. In addition, hNSCs were genetically modified to express NGF, and were transplanted in mice with induced cognitive dysfunction where they improved learning and memory [152]. Other neurotrophic effects were accomplished by a combination of NSC grafting and the supply of neurotrophic drugs like (+)-phenserine (amyloid modulator) [177] or Cerebrolysin (mixture of neurotrophic peptides) [178], to support the survival of NSCs. Finally, NSCs’ transplantation reduced neuroinflammation by reducing glial and toll-like receptor 4 (TLR4) activation and its downstream signaling pathways [179].
4.1.3. iPSCs
Using defined reprogramming factors to reprogram fully differentiated somatic cells into iPSCs has become a novel strategy to produce pluripotent cells derived from patients that enable autologous transplantation [180]. Although many ethical issues regarding immune rejection can be circumvented by the use of iPSCs, their application, however, bares limitations in terms of AD-related pathological phenotypes of generated neurons (such as abnormal Aβ levels and increased tau phosphorylation) [181,182]. Currently, there is a limited amount of studies using transplanted iPSCs, due to its novelty and mentioned issues, however their application in vitro might be the best tool for studying AD pathology and screening for therapeutic drugs.
Under in vitro conditions, hiPSCs derived from skin fibroblasts of patients (with APOE alterations) were used to produce neurons that expressed apolipoprotein E4 (ApoE4) variant, which is a major risk factor for AD. The study identified ApoE4 and its altered conformation (and not ApoE3, which showed rescue properties) as the cause of the pathogenic phenotype of AD, and provided the possibility of treatment by applying a corrector of the pathogenic conformation of ApoE4 [183]. Transplantation of iPSCs in vivo was achieved with iPSCs derived from mouse skin fibroblasts by treating protein extracts of embryonic stem cells. These cells differentiated into glial cells and decreased plaque depositions in the 5XFAD transgenic AD mouse model and moreover, improved cognitive dysfunction [184]. Separately, neuronal precursors developed from human iPSCs were transplanted into the hippocampus of transgenic mice with severe amyloid β deposition and progressive spatial memory dysfunction, where they differentiated into cholinergic neurons and significantly improved memory impairments [185]. In addition, iPSCs transplantation was successfully used in various Parkinson’s disease models [186,187,188] and stroke models [189].
4.1.4. MSCs
Mesenchymal stem cells are multipotent stromal cells that can be isolated from various organs and tissues, such as bone marrow (BM-MSCs), umbilical cord (UCB-MSCs), adipose tissue (AD-MSCs), peripheral blood, amniotic fluid, muscle and lung [190]. MSCs can differentiate into a variety of cells types, are highly proliferative and are easily accessed and easily handled. Therefore, these cells have found a very broad application in research. However, the rates of neuronal differentiation tend to be rather low, and glial cell generation can be preferred [191]. Nevertheless, MSCs can also be applied intravenously, as these cells are able to cross the blood-brain barrier and travel to the injury site [192,193], which offers enormous benefits for patient treatments.
Similarly to other types of stem cell transplantation effects, transplantation of MSCs reduced Aβ deposits and tau-phosphorylation, increased neurogenesis and provided support with secreted factors and improved learning and memory deficits [194,195,196,197,198]. Immune modulating and anti-inflammatory effects have also been observed, through the upregulation of neuroprotective and downregulation of pro-inflammatory cytokines. Another important pathway by which MSCs participate in tissue repair is by the secretion of extracellular vesicles (EVs), particularly exosomes and micro-vesicles (MV), and this approach is extensively explored. MSCs can be genetically altered to release EVs supplemented with therapeutic agents, including siRNAs and enzymes, that target Aβ deposits [199,200]. Alternatively, MSCs can be manipulated to overexpress cytokines like VEGF that exhibit regenerative effects in AD models [201].
4.3. Translation
There is an increasing number of clinical trials that employ stem cell therapies in order to treat Alzheimer’s disease, and most of them are still ongoing. The treatment concept includes the capability of transplanted stem cells to differentiate into neuronal and glial cells that suffered the damage, to act in a paracrine manner by secreting neurotrophic and neuroprotective agents and to stimulate endogenous repair mechanisms. The most-used cell type for this purpose is the MSC, due to easy harvest, the possibility for intravenous transplantation, lack of immune reaction and ethical issues. The advantages and disadvantages of distinct stem cell types are presented in Table 2.
In one of the first studies (ClinicalTrials.gov Identifier: NCT01297218) allogenic hUBC-MSCs were injected intracranially into bilateral hippocampi of AD patients in phase I mostly to test safety and cells dosage, followed by effect evaluation phase (ClinicalTrials.gov Identifier: NCT01696591) where no Aβ decrease or cognitive improvement was observed, but limitations of the study need to be considered [202]. Another study in a phase I clinical trial focused on employing autologous fibroblasts transduced to express nerve growth factor (NGF) and could show positive evidence of a response to NGF through improved function of cholinergic neurons followed by improved cognitive decline after 22 months [203]. In a larger clinical trial (25 subjects) BM-MSCs from healthy donors are used intravenously subjects will be followed up at 2,4,13, 26, 39 and 52 week post study product infusion with expected end on March 2020 (ClinicalTrials.gov Identifier: NCT02600130). Similarly, BM-MSCs are used intravenously but in combination with near infrared light which should provide additive support (ClinicalTrials.gov Identifier: NCT03724136) on 100 participants with an estimated completion date on October 2021. In another study autologous AdMSC will be applied (ClinicalTrials.gov Identifier: NCT03117738) where intravenous administration will be repeated 9 times at a 2-week interval.
5. Conclusions
Since Aloysius Alzheimer’s first publication on a patient exhibiting a wide-spread case of dementia, it became more and more evident that the etiology of this disease is a multifactorial-caused process characterized by a high neuropathological heterogeneity. In spite of an explosion of studies on the neurophysiology of dementia, we are still far from a complete understanding of the etiology of AD, and a more global view has to be developed. Nevertheless, some new data may be a basis for developing new concepts for the successful treatment of AD.
Stem cell therapy bares enormous potential for the treatment of AD. Pre-clinical analyses yielded successful results and paved way for clinical trials; however, it is evident that translation from rodent models to AD patients is not straightforward. Issues with participant enrollment, choosing the right time for transplantation, possible issues regarding gender differences and long duration of monitoring are all factors that affect the outcomes of clinical trials. Therefore, a comprehensive evaluation of the key factors, including the cell type and source, delivery systems, long-term safety and efficacy, the reaction of the implanted cell to the AD environment and mechanisms of action in the AD model, is needed before prior to the start of clinical trials. Altogether, it takes rather a long time before conclusions can be drawn and the results of currently ongoing studies will provide answers on the efficiency of stem cell therapy in AD patients. AD is a progressive disease which is initiated usually years before the onset of first symptoms. To this day, early diagnosis remains one of the most crucial points in AD treatment. In this context, additional research is needed that can provide reliable diagnostic tools. Moreover, it is possible that a synergy of methods needs to be employed in treatment strategies which would involve exogenous neuroreplacement, endogenous neurogenesis, genetic manipulations and pharmacological agents.
Author Contributions
V.V. and K.B. wrote and edited the manuscript and M.H.H.S. edited the manuscript.
Funding
This research was funded by German Research Foundation (DFG) via the collaborative research center 1080, project A3 as well as the individual grant SCHM2159 4-1 to MHHS. Open Access Funding by the Publication Funds of the TU Dresden.
Acknowledgments
We thank members of the Schmidt lab for all their support.
Conflicts of Interest
The authors declare that they have no competing interests.
Abbreviations
| AD | Alzheimer’s disease |
| APP | Amyloid Precursor Protein |
| APOE | Apolipoprotein E |
| CIE | Clathrin-independent endocytosis |
| CME | Clathrin-mediated endocytosis |
| HC | Hippocampus |
| SGZ | Sub-granular zone |
| SVZ | Subventricular zone |
| ESCs | Embryonic stem cells |
| NSCs | Neural stem cells |
| iPSCs | Induced Pluripotent stem cells |
| MSCs | Mesenchymal stem cells |
References
- Yankner, B.A.; Lu, T. Amyloid beta-protein toxicity and the pathogenesis of alzheimer disease. J. Biol. Chem. 2009, 284, 4755–4759. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P. Pathways towards and away from alzheimer’s disease. Nature 2004, 430, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Giraldo, E.; Lloret, A.; Fuchsberger, T.; Vina, J. Abeta and tau toxicities in Alzheimer’s are linked via oxidative stress-induced p38 activation: Protective role of vitamin e. Redox Biol. 2014, 2, 873–877. [Google Scholar] [CrossRef] [PubMed]
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease and down’s syndrome: Sharing of a unique cerebrovascular amyloid fibril protein. Biochem. Biophys. Res. Commun. 1984, 122, 1131–1135. [Google Scholar] [CrossRef]
- Grundke-Iqbal, I.; Iqbal, K.; Tung, Y.C.; Quinlan, M.; Wisniewski, H.M.; Binder, L.I. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. USA 1986, 83, 4913–4917. [Google Scholar] [CrossRef] [PubMed]
- Bisht, K.; Sharma, K.; Tremblay, M.E. Chronic stress as a risk factor for alzheimer’s disease: Roles of microglia-mediated synaptic remodeling, inflammation, and oxidative stress. Neurobiol. Stress 2018, 9, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Katsumoto, A.; Takeuchi, H.; Takahashi, K.; Tanaka, F. Microglia in alzheimer’s disease: Risk factors and inflammation. Front. Neurol. 2018, 9, 978. [Google Scholar] [CrossRef] [PubMed]
- Wyss-Coray, T.; Rogers, J. Inflammation in alzheimer disease-a brief review of the basic science and clinical literature. Cold Spring Harb. Perspect. Med. 2012, 2, a006346. [Google Scholar] [CrossRef]
- Rezaie, P.; Male, D. Mesoglia & microglia--a historical review of the concept of mononuclear phagocytes within the central nervous system. J. Hist. Neurosci. 2002, 11, 325–374. [Google Scholar]
- Chan, W.Y.; Kohsaka, S.; Rezaie, P. The origin and cell lineage of microglia: New concepts. Brain Res. Rev. 2007, 53, 344–354. [Google Scholar] [CrossRef]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [PubMed]
- Wyss-Coray, T. Tgf-beta pathway as a potential target in neurodegeneration and alzheimer’s. Curr. Alzheimer Res. 2006, 3, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J.; Xue, Q.S. Life and death of microglia. J. Neuroimmune. Pharm. 2009, 4, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Wake, H.; Moorhouse, A.J.; Jinno, S.; Kohsaka, S.; Nabekura, J. Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J. Neurosci. 2009, 29, 3974–3980. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.P.; Clark, I.A.; Zinn, R.; Vissel, B. Microglia: A new frontier for synaptic plasticity, learning and memory, and neurodegenerative disease research. Neurobiol. Learn. Mem. 2013, 105, 40–53. [Google Scholar] [CrossRef] [PubMed]
- Sipe, G.O.; Lowery, R.L.; Tremblay, M.E.; Kelly, E.A.; Lamantia, C.E.; Majewska, A.K. Microglial p2y12 is necessary for synaptic plasticity in mouse visual cortex. Nat. Commun. 2016, 7, 10905. [Google Scholar] [CrossRef] [PubMed]
- Cuello, A.C. Early and late cns inflammation in alzheimer’s disease: Two extremes of a continuum? Trends Pharm. Sci. 2017, 38, 956–966. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef]
- Calsolaro, V.; Edison, P. Neuroinflammation in alzheimer’s disease: Current evidence and future directions. Alzheimers Dement. 2016, 12, 719–732. [Google Scholar] [CrossRef]
- Heppner, F.L.; Ransohoff, R.M.; Becher, B. Immune attack: The role of inflammation in alzheimer disease. Nat. Rev. Neurosci. 2015, 16, 358–372. [Google Scholar] [CrossRef]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. Nlrp3 is activated in alzheimer’s disease and contributes to pathology in app/ps1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Sarlus, H.; Heneka, M.T. Microglia in alzheimer’s disease. J. Clin. Invest. 2017, 127, 3240–3249. [Google Scholar] [CrossRef] [PubMed]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in alzheimer’s disease. J. Cell. Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Yeh, F.L.; Wang, Y.; Tom, I.; Gonzalez, L.C.; Sheng, M. Trem2 binds to apolipoproteins, including apoe and clu/apoj, and thereby facilitates uptake of amyloid-beta by microglia. Neuron 2016, 91, 328–340. [Google Scholar] [CrossRef] [PubMed]
- Yeh, F.L.; Hansen, D.V.; Sheng, M. Trem2, microglia, and neurodegenerative diseases. Trends Mol. Med. 2017, 23, 512–533. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cella, M.; Mallinson, K.; Ulrich, J.D.; Young, K.L.; Robinette, M.L.; Gilfillan, S.; Krishnan, G.M.; Sudhakar, S.; Zinselmeyer, B.H.; et al. Trem2 lipid sensing sustains the microglial response in an alzheimer’s disease model. Cell 2015, 160, 1061–1071. [Google Scholar] [CrossRef] [PubMed]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The trem2-apoe pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 2017, 47, 566–581 e569. [Google Scholar] [CrossRef] [PubMed]
- Cuyvers, E.; De Roeck, A.; Van den Bossche, T.; Van Cauwenberghe, C.; Bettens, K.; Vermeulen, S.; Mattheijssens, M.; Peeters, K.; Engelborghs, S.; Vandenbulcke, M.; et al. Mutations in abca7 in a belgian cohort of alzheimer’s disease patients: A targeted resequencing study. Lancet Neurol. 2015, 14, 814–822. [Google Scholar] [CrossRef]
- Kim, W.S.; Li, H.; Ruberu, K.; Chan, S.; Elliott, D.A.; Low, J.K.; Cheng, D.; Karl, T.; Garner, B. Deletion of abca7 increases cerebral amyloid-beta accumulation in the j20 mouse model of alzheimer’s disease. J. Neurosci. 2013, 33, 4387–4394. [Google Scholar] [CrossRef]
- Jehle, A.W.; Gardai, S.J.; Li, S.; Linsel-Nitschke, P.; Morimoto, K.; Janssen, W.J.; Vandivier, R.W.; Wang, N.; Greenberg, S.; Dale, B.M.; et al. Atp-binding cassette transporter a7 enhances phagocytosis of apoptotic cells and associated erk signaling in macrophages. J. Cell. Biol. 2006, 174, 547–556. [Google Scholar] [CrossRef]
- Abe-Dohmae, S.; Ikeda, Y.; Matsuo, M.; Hayashi, M.; Okuhira, K.; Ueda, K.; Yokoyama, S. Human abca7 supports apolipoprotein-mediated release of cellular cholesterol and phospholipid to generate high density lipoprotein. J. Biol. Chem. 2004, 279, 604–611. [Google Scholar] [CrossRef] [PubMed]
- Volonte, C.; Apolloni, S.; Skaper, S.D.; Burnstock, G. P2x7 receptors: Channels, pores and more. CNS Neurol. Disord. Drug Targets 2012, 11, 705–721. [Google Scholar] [CrossRef] [PubMed]
- Monif, M.; Reid, C.A.; Powell, K.L.; Smart, M.L.; Williams, D.A. The p2x7 receptor drives microglial activation and proliferation: A trophic role for p2x7r pore. J. Neurosci. 2009, 29, 3781–3791. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Wang, P.; Zhang, J.; Chen, W.; Gu, L. Silencing of the p2x(7) receptor enhances amyloid-beta phagocytosis by microglia. Biochem. Biophys. Res. Commun. 2013, 434, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Numakawa, T.; Shimazu, K.; Koshimizu, H.; Hara, T.; Hatanaka, H.; Mei, L.; Lu, B.; Kojima, M. Bdnf-induced recruitment of trkb receptor into neuronal lipid rafts: Roles in synaptic modulation. J. Cell. Biol. 2004, 167, 1205–1215. [Google Scholar] [CrossRef]
- Shieh, C.H.; Heinrich, A.; Serchov, T.; van Calker, D.; Biber, K. P2x7-dependent, but differentially regulated release of il-6, ccl2, and tnf-alpha in cultured mouse microglia. Glia 2014, 62, 592–607. [Google Scholar] [CrossRef]
- Murphy, N.; Lynch, M.A. Activation of the p2x(7) receptor induces migration of glial cells by inducing cathepsin b degradation of tissue inhibitor of metalloproteinase 1. J. Neurochem. 2012, 123, 761–770. [Google Scholar] [CrossRef]
- Bartlett, R.; Yerbury, J.J.; Sluyter, R. P2x7 receptor activation induces reactive oxygen species formation and cell death in murine eoc13 microglia. Mediat. Inflamm. 2013, 2013, 271813. [Google Scholar] [CrossRef]
- He, Y.; Taylor, N.; Fourgeaud, L.; Bhattacharya, A. The role of microglial p2x7: Modulation of cell death and cytokine release. J. Neuroinflamm. 2017, 14, 135. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Jones, D.N.C. Emerging role of the p2x7-nlrp3-il1beta pathway in mood disorders. Psychoneuroendocrinology 2018, 98, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Di Virgilio, F. Liaisons dangereuses: P2x(7) and the inflammasome. Trends Pharm. Sci. 2007, 28, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Thawkar, B.S.; Kaur, G. Inhibitors of nf-kappab and p2x7/nlrp3/caspase 1 pathway in microglia: Novel therapeutic opportunities in neuroinflammation induced early-stage alzheimer’s disease. J. Neuroimmunol. 2019, 326, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Pascual, O.; Casper, K.B.; Kubera, C.; Zhang, J.; Revilla-Sanchez, R.; Sul, J.Y.; Takano, H.; Moss, S.J.; McCarthy, K.; Haydon, P.G. Astrocytic purinergic signaling coordinates synaptic networks. Science 2005, 310, 113–116. [Google Scholar] [CrossRef]
- Fields, R.D.; Araque, A.; Johansen-Berg, H.; Lim, S.S.; Lynch, G.; Nave, K.A.; Nedergaard, M.; Perez, R.; Sejnowski, T.; Wake, H. Glial biology in learning and cognition. Neuroscientist 2014, 20, 426–431. [Google Scholar] [CrossRef]
- Jourdain, P.; Becq, F.; Lengacher, S.; Boinot, C.; Magistretti, P.J.; Marquet, P. The human cftr protein expressed in cho cells activates aquaporin-3 in a camp-dependent pathway: Study by digital holographic microscopy. J. Cell. Sci. 2014, 127, 546–556. [Google Scholar] [CrossRef]
- Perea, G.; Navarrete, M.; Araque, A. Tripartite synapses: Astrocytes process and control synaptic information. Trends Neurosci. 2009, 32, 421–431. [Google Scholar] [CrossRef]
- Wyss-Coray, T.; Loike, J.D.; Brionne, T.C.; Lu, E.; Anankov, R.; Yan, F.; Silverstein, S.C.; Husemann, J. Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat. Med. 2003, 9, 453–457. [Google Scholar] [CrossRef]
- Koistinaho, M.; Lin, S.; Wu, X.; Esterman, M.; Koger, D.; Hanson, J.; Higgs, R.; Liu, F.; Malkani, S.; Bales, K.R.; et al. Apolipoprotein e promotes astrocyte colocalization and degradation of deposited amyloid-beta peptides. Nat. Med. 2004, 10, 719–726. [Google Scholar] [CrossRef]
- Mohamed, A.; Posse de Chaves, E. Abeta internalization by neurons and glia. Int. J. Alzheimers Dis. 2011, 2011, 127984. [Google Scholar] [PubMed]
- Nagele, R.G.; D’Andrea, M.R.; Lee, H.; Venkataraman, V.; Wang, H.Y. Astrocytes accumulate a beta 42 and give rise to astrocytic amyloid plaques in alzheimer disease brains. Brain Res. 2003, 971, 197–209. [Google Scholar] [CrossRef]
- Leal, M.C.; Dorfman, V.B.; Gamba, A.F.; Frangione, B.; Wisniewski, T.; Castano, E.M.; Sigurdsson, E.M.; Morelli, L. Plaque-associated overexpression of insulin-degrading enzyme in the cerebral cortex of aged transgenic tg2576 mice with alzheimer pathology. J. Neuropathol. Exp. Neurol. 2006, 65, 976–987. [Google Scholar] [CrossRef] [PubMed]
- Mulder, S.D.; Veerhuis, R.; Blankenstein, M.A.; Nielsen, H.M. The effect of amyloid associated proteins on the expression of genes involved in amyloid-beta clearance by adult human astrocytes. Exp. Neurol. 2012, 233, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Pihlaja, R.; Koistinaho, J.; Kauppinen, R.; Sandholm, J.; Tanila, H.; Koistinaho, M. Multiple cellular and molecular mechanisms are involved in human abeta clearance by transplanted adult astrocytes. Glia 2011, 59, 1643–1657. [Google Scholar] [CrossRef]
- Delekate, A.; Fuchtemeier, M.; Schumacher, T.; Ulbrich, C.; Foddis, M.; Petzold, G.C. Metabotropic p2y1 receptor signalling mediates astrocytic hyperactivity in vivo in an alzheimer’s disease mouse model. Nat. Commun. 2014, 5, 5422. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.Z.; Lipski, J. Involvement of trpv4 channels in abeta(40)-induced hippocampal cell death and astrocytic ca(2+) signalling. Neurotoxicology 2014, 41, 64–72. [Google Scholar] [CrossRef]
- Xiu, J.; Nordberg, A.; Zhang, J.T.; Guan, Z.Z. Expression of nicotinic receptors on primary cultures of rat astrocytes and up-regulation of the alpha7, alpha4 and beta2 subunits in response to nanomolar concentrations of the beta-amyloid peptide(1-42). Neurochem. Int. 2005, 47, 281–290. [Google Scholar] [CrossRef]
- Lee, L.; Kosuri, P.; Arancio, O. Picomolar amyloid-beta peptides enhance spontaneous astrocyte calcium transients. J. Alzheimers Dis. 2014, 38, 49–62. [Google Scholar] [CrossRef]
- Grolla, A.A.; Sim, J.A.; Lim, D.; Rodriguez, J.J.; Genazzani, A.A.; Verkhratsky, A. Amyloid-beta and alzheimer’s disease type pathology differentially affects the calcium signalling toolkit in astrocytes from different brain regions. Cell. Death Dis. 2013, 4, e623. [Google Scholar] [CrossRef]
- Ronco, V.; Grolla, A.A.; Glasnov, T.N.; Canonico, P.L.; Verkhratsky, A.; Genazzani, A.A.; Lim, D. Differential deregulation of astrocytic calcium signalling by amyloid-beta, tnfalpha, Il-1beta and LPS. Cell Calcium 2014, 55, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Matos, M.; Augusto, E.; Machado, N.J.; dos Santos-Rodrigues, A.; Cunha, R.A.; Agostinho, P. Astrocytic adenosine a2a receptors control the amyloid-beta peptide-induced decrease of glutamate uptake. J. Alzheimers Dis. 2012, 31, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Blasko, I.; Veerhuis, R.; Stampfer-Kountchev, M.; Saurwein-Teissl, M.; Eikelenboom, P.; Grubeck-Loebenstein, B. Costimulatory effects of interferon-gamma and interleukin-1beta or tumor necrosis factor alpha on the synthesis of abeta1-40 and abeta1-42 by human astrocytes. Neurobiol. Dis. 2000, 7, 682–689. [Google Scholar] [CrossRef] [PubMed]
- Malone, M.J.; Szoke, M.C. Neurochemical changes in white matter: Aged human brain and alzheimer’s disease. Arch. Neurol. 1985, 42, 1063–1066. [Google Scholar] [CrossRef] [PubMed]
- Barber, R.; Scheltens, P.; Gholkar, A.; Ballard, C.; McKeith, I.; Ince, P.; Perry, R.; O’brien, J. White matter lesions on magnetic resonance imaging in dementia with lewy bodies, alzheimer’s disease, vascular dementia, and normal aging. J. Neurol. Neurosurg. Psychiatry 1999, 67, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Bartzokis, G.; Cummings, J.L.; Sultzer, D.; Henderson, V.W.; Nuechterlein, K.H.; Mintz, J. White matter structural integrity in healthy aging adults and patients with alzheimer disease: A magnetic resonance imaging study. Arch. Neurol. 2003, 60, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Rose, S.E.; Chen, F.; Chalk, J.B.; Zelaya, F.O.; Strugnell, W.E.; Benson, M.; Semple, J.; Doddrell, D.M. Loss of connectivity in alzheimer’s disease: An evaluation of white matter tract integrity with colour coded mr diffusion tensor imaging. J. Neurol. Neurosurg. Psychiatry 2000, 69, 528–530. [Google Scholar] [CrossRef] [PubMed]
- Song, S.-K.; Kim, J.H.; Lin, S.-J.; Brendza, R.P.; Holtzman, D.M. Diffusion tensor imaging detects age-dependent white matter changes in a transgenic mouse model with amyloid deposition. Neurobiol. Dis. 2004, 15, 640–647. [Google Scholar] [CrossRef]
- Desai, M.K.; Sudol, K.L.; Janelsins, M.C.; Mastrangelo, M.A.; Frazer, M.E.; Bowers, W.J. Triple-transgenic alzheimer’s disease mice exhibit region-specific abnormalities in brain myelination patterns prior to appearance of amyloid and tau pathology. Glia 2009, 57, 54–65. [Google Scholar] [CrossRef]
- Desai, M.K.; Mastrangelo, M.A.; Ryan, D.A.; Sudol, K.L.; Narrow, W.C.; Bowers, W.J. Early oligodendrocyte/myelin pathology in alzheimer’s disease mice constitutes a novel therapeutic target. Am. J. Pathol. 2010, 177, 1422–1435. [Google Scholar] [CrossRef]
- Kobayashi, K.; Hayashi, M.; Nakano, H.; Fukutani, Y.; Sasaki, K.; Shimazaki, M.; Koshino, Y. Apoptosis of astrocytes with enhanced lysosomal activity and oligodendrocytes in white matter lesions in alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2002, 28, 238–251. [Google Scholar] [CrossRef] [PubMed]
- Sjöbeck, M.; Englund, E. Glial levels determine severity of white matter disease in alzheimer’s disease: A neuropathological study of glial changes. Neuropathol. Appl. Neurobiol. 2003, 29, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Chen, S.; Ahmed, S.H.; Chen, H.; Ku, G.; Goldberg, M.P.; Hsu, C.Y. Amyloid-β peptides are cytotoxic to oligodendrocytes. J. Neurosci. 2001, 21, RC118. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, A.; Komitova, M.; Suzuki, R.; Zhu, X. Polydendrocytes (ng2 cells): Multifunctional cells with lineage plasticity. Nat. Rev. Neurosci. 2009, 10, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Salins, P.; Shawesh, S.; He, Y.; Dibrov, A.; Kashour, T.; Arthur, G.; Amara, F. Lovastatin protects human neurons against abeta-induced toxicity and causes activation of beta-catenin-tcf/lef signaling. Neurosci. Lett. 2007, 412, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.P.; Zhao, J.; Li, S. Roles of ng2 glial cells in diseases of the central nervous system. Neurosci. Bull. 2011, 27, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Hanger, D.P.; Anderton, B.H.; Noble, W. Tau phosphorylation: The therapeutic challenge for neurodegenerative disease. Trends Mol. Med. 2009, 15, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Li, B.; Tung, E.J.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X. Site-specific effects of tau phosphorylation on its microtubule assembly activity and self-aggregation. Eur. J. Neurosci. 2007, 26, 3429–3436. [Google Scholar] [CrossRef] [Green Version]
- Nukina, N.; Ihara, Y. One of the antigenic determinants of paired helical filaments is related to tau protein. J. Biochem. 1986, 99, 1541–1544. [Google Scholar] [CrossRef]
- Mondragon-Rodriguez, S.; Basurto-Islas, G.; Santa-Maria, I.; Mena, R.; Binder, L.I.; Avila, J.; Smith, M.A.; Perry, G.; Garcia-Sierra, F. Cleavage and conformational changes of tau protein follow phosphorylation during alzheimer’s disease. Int. J. Exp. Pathol. 2008, 89, 81–90. [Google Scholar] [CrossRef]
- Mondragon-Rodriguez, S.; Basurto-Islas, G.; Lee, H.G.; Perry, G.; Zhu, X.; Castellani, R.J.; Smith, M.A. Causes versus effects: The increasing complexities of alzheimer’s disease pathogenesis. Expert. Rev. Neurother. 2010, 10, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Kopeikina, K.J.; Carlson, G.A.; Pitstick, R.; Ludvigson, A.E.; Peters, A.; Luebke, J.I.; Koffie, R.M.; Frosch, M.P.; Hyman, B.T.; Spires-Jones, T.L. Tau accumulation causes mitochondrial distribution deficits in neurons in a mouse model of tauopathy and in human alzheimer’s disease brain. Am. J. Pathol. 2011, 179, 2071–2082. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, S.I. Coupled reductions in brain oxidative phosphorylation and synaptic function can be quantified and staged in the course of alzheimer disease. Neurotox. Res. 2003, 5, 385–398. [Google Scholar] [CrossRef] [PubMed]
- Westermann, B. Bioenergetic role of mitochondrial fusion and fission. Biochim. Biophys. Acta 2012, 1817, 1833–1838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; McCaffery, J.M.; Chan, D.C. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell 2007, 130, 548–562. [Google Scholar] [CrossRef] [PubMed]
- Westermann, B. Molecular machinery of mitochondrial fusion and fission. J. Biol. Chem. 2008, 283, 13501–13505. [Google Scholar] [CrossRef]
- Chen, H.; Chan, D.C. Mitochondrial dynamics--fusion, fission, movement, and mitophagy--in neurodegenerative diseases. Hum. Mol. Genet. 2009, 18, R169–R176. [Google Scholar] [CrossRef]
- Santos, R.X.; Correia, S.C.; Wang, X.; Perry, G.; Smith, M.A.; Moreira, P.I.; Zhu, X. Alzheimer’s disease: Diverse aspects of mitochondrial malfunctioning. Int. J. Clin. Exp. Pathol. 2010, 3, 570–581. [Google Scholar]
- Moreira, P.I.; Carvalho, C.; Zhu, X.; Smith, M.A.; Perry, G. Mitochondrial dysfunction is a trigger of alzheimer’s disease pathophysiology. Biochim. Biophys. Acta 2010, 1802, 2–10. [Google Scholar] [CrossRef]
- Chen, Z.; Zhong, C. Oxidative stress in alzheimer’s disease. Neurosci. Bull. 2014, 30, 271–281. [Google Scholar] [CrossRef]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in alzheimer’s disease. Redox. Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Li, W.W.; Li, J.; Bao, J.K. Microautophagy: Lesser-known self-eating. Cell. Mol. Life Sci. 2012, 69, 1125–1136. [Google Scholar] [CrossRef] [PubMed]
- Dice, J.F.; Terlecky, S.R.; Chiang, H.L.; Olson, T.S.; Isenman, L.D.; Short-Russell, S.R.; Freundlieb, S.; Terlecky, L.J. A selective pathway for degradation of cytosolic proteins by lysosomes. Semin. Cell. Biol. 1990, 1, 449–455. [Google Scholar] [PubMed]
- Majeski, A.E.; Dice, J.F. Mechanisms of chaperone-mediated autophagy. Int. J. Biochem. Cell. Biol. 2004, 36, 2435–2444. [Google Scholar] [CrossRef] [PubMed]
- Bejarano, E.; Cuervo, A.M. Chaperone-mediated autophagy. Proc. Am. Thorac. Soc. 2010, 7, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Bandyopadhyay, U.; Sridhar, S.; Kiffin, R.; Martinez-Vicente, M.; Kon, M.; Orenstein, S.J.; Wong, E.; Cuervo, A.M. Chaperone-mediated autophagy at a glance. J. Cell. Sci. 2011, 124, 495–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.A. Neuronal autophagy: A housekeeper or a fighter in neuronal cell survival? Exp. Neurobiol. 2012, 21, 1–8. [Google Scholar] [CrossRef]
- Kesidou, E.; Lagoudaki, R.; Touloumi, O.; Poulatsidou, K.N.; Simeonidou, C. Autophagy and neurodegenerative disorders. Neural. Regen. Res. 2013, 8, 2275–2283. [Google Scholar]
- Frake, R.A.; Ricketts, T.; Menzies, F.M.; Rubinsztein, D.C. Autophagy and neurodegeneration. J. Clin. Invest. 2015, 125, 65–74. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Liu, Y.; Sun, M. Autophagy and alzheimer’s disease. Cell. Mol. Neurobiol. 2017, 37, 377–388. [Google Scholar] [CrossRef]
- Whyte, L.S.; Lau, A.A.; Hemsley, K.M.; Hopwood, J.J.; Sargeant, T.J. Endo-lysosomal and autophagic dysfunction: A driving factor in alzheimer’s disease? J. Neurochem. 2017, 140, 703–717. [Google Scholar] [CrossRef] [PubMed]
- Menzies, F.M.; Fleming, A.; Rubinsztein, D.C. Compromised autophagy and neurodegenerative diseases. Nat. Rev. Neurosci. 2015, 16, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Fullgrabe, J.; Jackson, A.; Jimenez Sanchez, M.; Karabiyik, C.; et al. Autophagy and neurodegeneration: Pathogenic mechanisms and therapeutic opportunities. Neuron 2017, 93, 1015–1034. [Google Scholar] [CrossRef] [PubMed]
- Correia, S.C.; Perry, G.; Moreira, P.I. Mitochondrial traffic jams in alzheimer’s disease—Pinpointing the roadblocks. Biochim. Biophys. Acta 2016, 1862, 1909–1917. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.S.; Adriaanse, B.A.; Greig, N.H.; Mattson, M.P.; Cader, M.Z.; Bohr, V.A.; Fang, E.F. Mitophagy and alzheimer’s disease: Cellular and molecular mechanisms. Trends Neurosci. 2017, 40, 151–166. [Google Scholar] [CrossRef] [PubMed]
- Caberlotto, L.; Nguyen, T.P.; Lauria, M.; Priami, C.; Rimondini, R.; Maioli, S.; Cedazo-Minguez, A.; Sita, G.; Morroni, F.; Corsi, M.; et al. Cross-disease analysis of alzheimer’s disease and type-2 diabetes highlights the role of autophagy in the pathophysiology of two highly comorbid diseases. Sci. Rep. 2019, 9, 3965. [Google Scholar] [CrossRef] [PubMed]
- Bitsikas, V.; Riento, K.; Howe, J.D.; Barry, N.P.; Nichols, B.J. The role of flotillins in regulating abeta production, investigated using flotillin 1-/-, flotillin 2-/- double knockout mice. PLoS ONE 2014, 9, e85217. [Google Scholar] [CrossRef] [PubMed]
- Tate, B.A.; Mathews, P.M. Targeting the role of the endosome in the pathophysiology of alzheimer’s disease: A strategy for treatment. Sci. Aging Knowl. Env. 2006, 2006, re2. [Google Scholar] [CrossRef] [PubMed]
- Gaudreault, S.B.; Chabot, C.; Gratton, J.P.; Poirier, J. The caveolin scaffolding domain modifies 2-amino-3-hydroxy-5-methyl-4-isoxazole propionate receptor binding properties by inhibiting phospholipase a2 activity. J. Biol. Chem. 2004, 279, 356–362. [Google Scholar] [CrossRef]
- Okamoto, C.T. Endocytosis and transcytosis. Adv. Drug Deliv. Rev. 1998, 29, 215–228. [Google Scholar] [CrossRef]
- Mauch, D.H.; Nagler, K.; Schumacher, S.; Goritz, C.; Muller, E.C.; Otto, A.; Pfrieger, F.W. CNS synaptogenesis promoted by glia-derived cholesterol. Science 2001, 294, 1354–1357. [Google Scholar] [CrossRef] [PubMed]
- Willmann, R.; Pun, S.; Stallmach, L.; Sadasivam, G.; Santos, A.F.; Caroni, P.; Fuhrer, C. Cholesterol and lipid microdomains stabilize the postsynapse at the neuromuscular junction. EMBO J. 2006, 25, 4050–4060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakadate, K. Developmental changes in the flotillin-1 expression pattern of the rat visual cortex. Neuroscience 2015, 292, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Santiago, J.M.; Torrado, A.I.; Arocho, L.C.; Rosas, O.R.; Rodriguez, A.E.; Toro, F.K.; Salgado, I.K.; Torres, Y.A.; Silva, W.I.; Miranda, J.D. Expression profile of flotillin-2 and its pathophysiological role after spinal cord injury. J. Mol. Neurosci. 2013, 49, 347–359. [Google Scholar] [CrossRef] [PubMed]
- Harder, T.; Simons, K. Caveolae, digs, and the dynamics of sphingolipid-cholesterol microdomains. Curr. Opin. Cell. Biol. 1997, 9, 534–542. [Google Scholar] [CrossRef]
- Fra, A.M.; Williamson, E.; Simons, K.; Parton, R.G. De novo formation of caveolae in lymphocytes by expression of vip21-caveolin. Proc. Natl. Acad. Sci. USA 1995, 92, 8655–8659. [Google Scholar] [CrossRef] [PubMed]
- Ikezu, T.; Ueda, H.; Trapp, B.D.; Nishiyama, K.; Sha, J.F.; Volonte, D.; Galbiati, F.; Byrd, A.L.; Bassell, G.; Serizawa, H.; et al. Affinity-purification and characterization of caveolins from the brain: Differential expression of caveolin-1, -2, and -3 in brain endothelial and astroglial cell types. Brain Res. 1998, 804, 177–192. [Google Scholar] [CrossRef]
- Bilderback, T.R.; Gazula, V.R.; Lisanti, M.P.; Dobrowsky, R.T. Caveolin interacts with trk a and p75(ntr) and regulates neurotrophin signaling pathways. J. Biol. Chem. 1999, 274, 257–263. [Google Scholar] [CrossRef]
- Hibbert, A.P.; Kramer, B.M.; Miller, F.D.; Kaplan, D.R. The localization, trafficking and retrograde transport of bdnf bound to p75ntr in sympathetic neurons. Mol. Cell. Neurosci. 2006, 32, 387–402. [Google Scholar] [CrossRef] [PubMed]
- Head, B.P.; Patel, H.H.; Tsutsumi, Y.M.; Hu, Y.; Mejia, T.; Mora, R.C.; Insel, P.A.; Roth, D.M.; Drummond, J.C.; Patel, P.M. Caveolin-1 expression is essential for n-methyl-d-aspartate receptor-mediated src and extracellular signal-regulated kinase 1/2 activation and protection of primary neurons from ischemic cell death. FASEB J. 2008, 22, 828–840. [Google Scholar] [CrossRef]
- Gaudreault, S.B.; Blain, J.F.; Gratton, J.P.; Poirier, J. A role for caveolin-1 in post-injury reactive neuronal plasticity. J. Neurochem. 2005, 92, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Ikezu, T.; Trapp, B.D.; Song, K.S.; Schlegel, A.; Lisanti, M.P.; Okamoto, T. Caveolae, plasma membrane microdomains for alpha-secretase-mediated processing of the amyloid precursor protein. J. Biol. Chem. 1998, 273, 10485–10495. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, K.; Trapp, B.D.; Ikezu, T.; Ransohoff, R.M.; Tomita, T.; Iwatsubo, T.; Kanazawa, I.; Hsiao, K.K.; Lisanti, M.P.; Okamoto, T. Caveolin-3 upregulation activates beta-secretase-mediated cleavage of the amyloid precursor protein in alzheimer’s disease. J. Neurosci. 1999, 19, 6538–6548. [Google Scholar] [CrossRef] [PubMed]
- Alsaqati, M.; Thomas, R.S.; Kidd, E.J. Proteins involved in endocytosis are upregulated by ageing in the normal human brain: Implications for the development of alzheimer’s disease. J. Gerontol. A Biol. Sci. Med. Sci. 2018, 73, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Girardot, N.; Allinquant, B.; Langui, D.; Laquerriere, A.; Dubois, B.; Hauw, J.J.; Duyckaerts, C. Accumulation of flotillin-1 in tangle-bearing neurones of alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2003, 29, 451–461. [Google Scholar] [CrossRef]
- Head, B.P.; Peart, J.N.; Panneerselvam, M.; Yokoyama, T.; Pearn, M.L.; Niesman, I.R.; Bonds, J.A.; Schilling, J.M.; Miyanohara, A.; Headrick, J.; et al. Loss of caveolin-1 accelerates neurodegeneration and aging. PLoS ONE 2010, 5, e15697. [Google Scholar] [CrossRef] [PubMed]
- Cataldo, A.M.; Peterhoff, C.M.; Troncoso, J.C.; Gomez-Isla, T.; Hyman, B.T.; Nixon, R.A. Endocytic pathway abnormalities precede amyloid beta deposition in sporadic alzheimer’s disease and down syndrome: Differential effects of apoe genotype and presenilin mutations. Am. J. Pathol. 2000, 157, 277–286. [Google Scholar] [CrossRef]
- Thomas, R.S.; Lelos, M.J.; Good, M.A.; Kidd, E.J. Clathrin-mediated endocytic proteins are upregulated in the cortex of the tg2576 mouse model of alzheimer’s disease-like amyloid pathology. Biochem. Biophys. Res. Commun. 2011, 415, 656–661. [Google Scholar] [CrossRef]
- van Helmond, Z.K.; Miners, J.S.; Bednall, E.; Chalmers, K.A.; Zhang, Y.; Wilcock, G.K.; Love, S.; Kehoe, P.G. Caveolin-1 and -2 and their relationship to cerebral amyloid angiopathy in alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2007, 33, 317–327. [Google Scholar] [CrossRef]
- Trushina, E.; Du Charme, J.; Parisi, J.; McMurray, C.T. Neurological abnormalities in caveolin-1 knock out mice. Behav. Brain Res. 2006, 172, 24–32. [Google Scholar] [CrossRef]
- Gioiosa, L.; Raggi, C.; Ricceri, L.; Jasmin, J.F.; Frank, P.G.; Capozza, F.; Lisanti, M.P.; Alleva, E.; Sargiacomo, M.; Laviola, G. Altered emotionality, spatial memory and cholinergic function in caveolin-1 knock-out mice. Behav. Brain Res. 2008, 188, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Jasmin, J.F.; Malhotra, S.; Singh Dhallu, M.; Mercier, I.; Rosenbaum, D.M.; Lisanti, M.P. Caveolin-1 deficiency increases cerebral ischemic injury. Circ. Res. 2007, 100, 721–729. [Google Scholar] [CrossRef] [PubMed]
- Wais, P.E.; Wixted, J.T.; Hopkins, R.O.; Squire, L.R. The hippocampus supports both the recollection and the familiarity components of recognition memory. Neuron 2006, 49, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Manns, J.R.; Hopkins, R.O.; Reed, J.M.; Kitchener, E.G.; Squire, L.R. Recognition memory and the human hippocampus. Neuron 2003, 37, 171–180. [Google Scholar] [CrossRef]
- Vargha-Khadem, F.; Gadian, D.G.; Watkins, K.E.; Connelly, A.; Van Paesschen, W.; Mishkin, M. Differential effects of early hippocampal pathology on episodic and semantic memory. Science 1997, 277, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Ekstrom, A.D.; Kahana, M.J.; Caplan, J.B.; Fields, T.A.; Isham, E.A.; Newman, E.L.; Fried, I. Cellular networks underlying human spatial navigation. Nature 2003, 425, 184. [Google Scholar] [CrossRef] [PubMed]
- Ono, T.; Nakamura, K.; Fukuda, M.; Tamura, R. Place recognition responses of neurons in monkey hippocampus. Neurosci. Lett. 1991, 121, 194–198. [Google Scholar] [CrossRef]
- West, M.J.; Coleman, P.D.; Flood, D.G.; Troncoso, J.C. Differences in the pattern of hippocampal neuronal loss in normal ageing and alzheimer’s disease. Lancet 1994, 344, 769–772. [Google Scholar] [CrossRef]
- Spalding, K.L.; Bergmann, O.; Alkass, K.; Bernard, S.; Salehpour, M.; Huttner, H.B.; Boström, E.; Westerlund, I.; Vial, C.; Buchholz, B.A. Dynamics of hippocampal neurogenesis in adult humans. Cell 2013, 153, 1219–1227. [Google Scholar] [CrossRef]
- Lazarov, O.; Marr, R.A. Neurogenesis and alzheimer’s disease: At the crossroads. Exp. Neurol. 2010, 223, 267–281. [Google Scholar] [CrossRef]
- Marlatt, M.W.; Lucassen, P.J. Neurogenesis and alzheimer’s disease: Biology and pathophysiology in mice and men. Curr. Alzheimer Res. 2010, 7, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Hassouna, I.; Ott, C.; Wüstefeld, L.; Offen, N.; Neher, R.A.; Mitkovski, M.; Winkler, D.; Sperling, S.; Fries, L.; Goebbels, S. Revisiting adult neurogenesis and the role of erythropoietin for neuronal and oligodendroglial differentiation in the hippocampus. Mol. Psychiatry 2016, 21, 1752. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Fang, X.; Huang, D.; Luo, Q.; Zheng, M.; Wang, K.; Cao, L.; Yin, Z. Erythropoietin signaling increases neurogenesis and oligodendrogenesis of endogenous neural stem cells following spinal cord injury both in vivo and in vitro. Mol. Med. Rep. 2018, 17, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Cevik, B.; Solmaz, V.; Yigitturk, G.; Cavusoğlu, T.; Peker, G.; Erbas, O. Neuroprotective effects of erythropoietin on alzheimer’s dementia model in rats. Adv. Clin. Exp. Med. 2017, 26, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Sachs, B.D.; Caron, M.G. Chronic fluoxetine increases extra-hippocampal neurogenesis in adult mice. Int. J. Neuropsychopharmacol. 2015, 18. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.W.; Lee, J.K.; Lee, J.E.; Min, W.K.; Schuchman, E.H.; Jin, H.K.; Bae, J.s. Combined effects of hematopoietic progenitor cell mobilization from bone marrow by granulocyte colony stimulating factor and amd3100 and chemotaxis into the brain using stromal cell-derived factor-1α in an alzheimer’s disease mouse model. Stem Cells 2011, 29, 1075–1089. [Google Scholar] [CrossRef] [PubMed]
- Blurton-Jones, M.; Kitazawa, M.; Martinez-Coria, H.; Castello, N.A.; Müller, F.-J.; Loring, J.F.; Yamasaki, T.R.; Poon, W.W.; Green, K.N.; LaFerla, F.M. Neural stem cells improve cognition via bdnf in a transgenic model of alzheimer disease. Proc. Natl. Acad. Sci. USA 2009, 106, 13594–13599. [Google Scholar] [CrossRef]
- Choi, S.H.; Bylykbashi, E.; Chatila, Z.K.; Lee, S.W.; Pulli, B.; Clemenson, G.D.; Kim, E.; Rompala, A.; Oram, M.K.; Asselin, C. Combined adult neurogenesis and bdnf mimic exercise effects on cognition in an alzheimer’s mouse model. Science 2018, 361, eaan8821. [Google Scholar] [CrossRef]
- Liu, P.Z.; Nusslock, R. Exercise-mediated neurogenesis in the hippocampus via bdnf. Front. Neurosci. 2018, 12, 52. [Google Scholar] [CrossRef]
- McGinley, L.M.; Sims, E.; Lunn, J.S.; Kashlan, O.N.; Chen, K.S.; Bruno, E.S.; Pacut, C.M.; Hazel, T.; Johe, K.; Sakowski, S.A. Human cortical neural stem cells expressing insulin-like growth factor-i: A novel cellular therapy for alzheimer’s disease. Stem Cells Transl. Med. 2016, 5, 379–391. [Google Scholar] [CrossRef]
- Capsoni, S.; Marinelli, S.; Ceci, M.; Vignone, D.; Amato, G.; Malerba, F.; Paoletti, F.; Meli, G.; Viegi, A.; Pavone, F. Intranasal “painless” human nerve growth factors slows amyloid neurodegeneration and prevents memory deficits in app x ps1 mice. PLoS ONE 2012, 7, e37555. [Google Scholar] [CrossRef]
- Lee, H.J.; Lim, I.J.; Park, S.W.; Kim, Y.B.; Ko, Y.; Kim, S.U. Human neural stem cells genetically modified to express human nerve growth factor (ngf) gene restore cognition in the mouse with ibotenic acid-induced cognitive dysfunction. Cell Transplant. 2012, 21, 2487–2496. [Google Scholar] [CrossRef] [PubMed]
- Herrán, E.; Pérez-González, R.; Igartua, M.; Pedraz, J.L.; Carro, E.; Hernández, R.M. Vegf-releasing biodegradable nanospheres administered by craniotomy: A novel therapeutic approach in the app/ps1 mouse model of alzheimer’s disease. J. Control. Release 2013, 170, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Yousef, H.; Conboy, M.J.; Morgenthaler, A.; Schlesinger, C.; Bugaj, L.; Paliwal, P.; Greer, C.; Conboy, I.M.; Schaffer, D. Systemic attenuation of the tgf-β pathway by a single drug simultaneously rejuvenates hippocampal neurogenesis and myogenesis in the same old mammal. Oncotarget 2015, 6, 11959. [Google Scholar] [CrossRef] [PubMed]
- Speisman, R.B.; Kumar, A.; Rani, A.; Pastoriza, J.M.; Severance, J.E.; Foster, T.C.; Ormerod, B.K. Environmental enrichment restores neurogenesis and rapid acquisition in aged rats. Neurobiol. Aging 2013, 34, 263–274. [Google Scholar] [CrossRef]
- Monteiro, B.M.; Moreira, F.A.; Massensini, A.R.; Moraes, M.F.; Pereira, G.S. Enriched environment increases neurogenesis and improves social memory persistence in socially isolated adult mice. Hippocampus 2014, 24, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Van Praag, H.; Kempermann, G.; Gage, F.H. Running increases cell proliferation and neurogenesis in the adult mouse dentate gyrus. Nat. Neurosci. 1999, 2, 266. [Google Scholar] [CrossRef]
- Van Praag, H.; Shubert, T.; Zhao, C.; Gage, F.H. Exercise enhances learning and hippocampal neurogenesis in aged mice. J. Neurosci. 2005, 25, 8680–8685. [Google Scholar] [CrossRef]
- Hüttenrauch, M.; Brauss, A.; Kurdakova, A.; Borgers, H.; Klinker, F.; Liebetanz, D.; Salinas-Riester, G.; Wiltfang, J.; Klafki, H.; Wirths, O. Physical activity delays hippocampal neurodegeneration and rescues memory deficits in an alzheimer disease mouse model. Transl. Psychiatry 2016, 6, e800. [Google Scholar] [CrossRef]
- Alipour, M.; Nabavi, S.M.; Arab, L.; Vosough, M.; Pakdaman, H.; Ehsani, E.; Shahpasand, K. Stem cell therapy in alzheimer’s disease: Possible benefits and limiting drawbacks. Mol. Biol. Rep. 2019, 46, 1425–1446. [Google Scholar] [CrossRef]
- Pera, M.F.; Trounson, A.O. Human embryonic stem cells: Prospects for development. Development 2004, 131, 5515–5525. [Google Scholar] [CrossRef] [PubMed]
- Fujikawa, T.; Oh, S.-H.; Pi, L.; Hatch, H.M.; Shupe, T.; Petersen, B.E. Teratoma formation leads to failure of treatment for type i diabetes using embryonic stem cell-derived insulin-producing cells. Am. J. Pathol. 2005, 166, 1781–1791. [Google Scholar] [CrossRef]
- Richards, M.; Fong, C.-Y.; Chan, W.-K.; Wong, P.-C.; Bongso, A. Human feeders support prolonged undifferentiated growth of human inner cell masses and embryonic stem cells. Nat. Biotechnol. 2002, 20, 933. [Google Scholar] [CrossRef] [PubMed]
- Moghadam, F.H.; Alaie, H.; Karbalaie, K.; Tanhaei, S.; Esfahani, M.H.N.; Baharvand, H. Transplantation of primed or unprimed mouse embryonic stem cell-derived neural precursor cells improves cognitive function in alzheimerian rats. Differentiation 2009, 78, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Bissonnette, C.J.; Lyass, L.; Bhattacharyya, B.J.; Belmadani, A.; Miller, R.J.; Kessler, J.A. The controlled generation of functional basal forebrain cholinergic neurons from human embryonic stem cells. Stem Cells 2011, 29, 802–811. [Google Scholar] [CrossRef] [PubMed]
- Yue, W.; Li, Y.; Zhang, T.; Jiang, M.; Qian, Y.; Zhang, M.; Sheng, N.; Feng, S.; Tang, K.; Yu, X. Esc-derived basal forebrain cholinergic neurons ameliorate the cognitive symptoms associated with alzheimer’s disease in mouse models. Stem Cell Rep. 2015, 5, 776–790. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Weick, J.P.; Liu, H.; Krencik, R.; Zhang, X.; Ma, L.; Zhou, G.-m.; Ayala, M.; Zhang, S.-C. Medial ganglionic eminence–like cells derived from human embryonic stem cells correct learning and memory deficits. Nat. Biotechnol. 2013, 31, 440. [Google Scholar] [CrossRef] [PubMed]
- Shimada, I.S.; LeComte, M.D.; Granger, J.C.; Quinlan, N.J.; Spees, J.L. Self-renewal and differentiation of reactive astrocyte-derived neural stem/progenitor cells isolated from the cortical peri-infarct area after stroke. J. Neurosci. 2012, 32, 7926–7940. [Google Scholar] [CrossRef]
- Liang, Y.; Ågren, L.; Lyczek, A.; Walczak, P.; Bulte, J.W. Neural progenitor cell survival in mouse brain can be improved by co-transplantation of helper cells expressing bfgf under doxycycline control. Exp. Neurol. 2013, 247, 73–79. [Google Scholar] [CrossRef]
- Nakaji-Hirabayashi, T.; Kato, K.; Iwata, H. In vivo study on the survival of neural stem cells transplanted into the rat brain with a collagen hydrogel that incorporates laminin-derived polypeptides. Bioconjugate Chem. 2013, 24, 1798–1804. [Google Scholar] [CrossRef]
- Lee, I.-S.; Jung, K.; Kim, I.-S.; Lee, H.; Kim, M.; Yun, S.; Hwang, K.; Shin, J.E.; Park, K.I. Human neural stem cells alleviate alzheimer-like pathology in a mouse model. Mol. Neurodegener. 2015, 10, 38. [Google Scholar] [CrossRef] [PubMed]
- Ager, R.R.; Davis, J.L.; Agazaryan, A.; Benavente, F.; Poon, W.W.; LaFerla, F.M.; Blurton-Jones, M. Human neural stem cells improve cognition and promote synaptic growth in two complementary transgenic models of alzheimer’s disease and neuronal loss. Hippocampus 2015, 25, 813–826. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhu, H.; Sun, X.; Zuo, F.; Lei, J.; Wang, Z.; Bao, X.; Wang, R. Human neural stem cell transplantation rescues cognitive defects in app/ps1 model of alzheimer’s disease by enhancing neuronal connectivity and metabolic activity. Front. Aging Neurosci. 2016, 8, 282. [Google Scholar] [CrossRef] [PubMed]
- Xuan, A.; Luo, M.; Ji, W.; Long, D. Effects of engrafted neural stem cells in alzheimer’s disease rats. Neurosci. Lett. 2009, 450, 167–171. [Google Scholar] [CrossRef] [PubMed]
- Xuan, A.; Long, D.; Gu, H.; Yang, D.; Hong, L.; Leng, S. Bdnf improves the effects of neural stem cells on the rat model of alzheimer’s disease with unilateral lesion of fimbria-fornix. Neurosci. Lett. 2008, 440, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Park, D.; Yang, G.; Bae, D.K.; Lee, S.H.; Yang, Y.H.; Kyung, J.; Kim, D.; Choi, E.K.; Choi, K.C.; Kim, S.U. Human adipose tissue-derived mesenchymal stem cells improve cognitive function and physical activity in ageing mice. J. Neurosci. Res. 2013, 91, 660–670. [Google Scholar] [CrossRef] [PubMed]
- Lilja, A.M.; Malmsten, L.; Röjdner, J.; Voytenko, L.; Verkhratsky, A.; Ögren, S.O.; Nordberg, A.; Marutle, A. Neural stem cell transplant-induced effect on neurogenesis and cognition in alzheimer tg2576 mice is inhibited by concomitant treatment with amyloid-lowering or cholinergic 7 nicotinic receptor drugs. Neural Plast. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Rockenstein, E.; Desplats, P.; Ubhi, K.; Mante, M.; Florio, J.; Adame, A.; Winter, S.; Brandstaetter, H.; Meier, D.; Masliah, E. Neuro-peptide treatment with cerebrolysin improves the survival of neural stem cell grafts in an app transgenic model of alzheimer disease. Stem Cell Res. 2015, 15, 54–67. [Google Scholar] [CrossRef]
- Zhang, Q.; Wu, H.h.; Wang, Y.; Gu, G.j.; Zhang, W.; Xia, R. Neural stem cell transplantation decreases neuroinflammation in a transgenic mouse model of alzheimer’s disease. J. Neurochem. 2016, 136, 815–825. [Google Scholar] [CrossRef]
- Ross, C.A.; Akimov, S.S. Human-induced pluripotent stem cells: Potential for neurodegenerative diseases. Hum. Mol. Genet. 2014, 23, R17–R26. [Google Scholar] [CrossRef]
- Hossini, A.M.; Megges, M.; Prigione, A.; Lichtner, B.; Toliat, M.R.; Wruck, W.; Schröter, F.; Nuernberg, P.; Kroll, H.; Makrantonaki, E. Induced pluripotent stem cell-derived neuronal cells from a sporadic alzheimer’s disease donor as a model for investigating ad-associated gene regulatory networks. BMC Genom. 2015, 16, 84. [Google Scholar]
- Muratore, C.R.; Rice, H.C.; Srikanth, P.; Callahan, D.G.; Shin, T.; Benjamin, L.N.; Walsh, D.M.; Selkoe, D.J.; Young-Pearse, T.L. The familial alzheimer’s disease appv717i mutation alters app processing and tau expression in ipsc-derived neurons. Hum. Mol. Genet. 2014, 23, 3523–3536. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Najm, R.; Xu, Q.; Jeong, D.-e.; Walker, D.; Balestra, M.E.; Yoon, S.Y.; Yuan, H.; Li, G.; Miller, Z.A. Gain of toxic apolipoprotein e4 effects in human ipsc-derived neurons is ameliorated by a small-molecule structure corrector. Nat. Med. 2018, 24, 647. [Google Scholar] [CrossRef] [PubMed]
- Cha, M.Y.; Kwon, Y.W.; Ahn, H.S.; Jeong, H.; Lee, Y.Y.; Moon, M.; Baik, S.H.; Kim, D.K.; Song, H.; Yi, E.C. Protein-induced pluripotent stem cells ameliorate cognitive dysfunction and reduce aβ deposition in a mouse model of alzheimer’s disease. Stem Cells Transl. Med. 2017, 6, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, N.; Shimizu, J.; Takai, K.; Arimitsu, N.; Saito, A.; Kono, T.; Umehara, T.; Ueda, Y.; Wakisaka, S.; Suzuki, T. Restoration of spatial memory dysfunction of human app transgenic mice by transplantation of neuronal precursors derived from human ips cells. Neurosci. Lett. 2013, 557, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Hallett, P.J.; Deleidi, M.; Astradsson, A.; Smith, G.A.; Cooper, O.; Osborn, T.M.; Sundberg, M.; Moore, M.A.; Perez-Torres, E.; Brownell, A.-L. Successful function of autologous ipsc-derived dopamine neurons following transplantation in a non-human primate model of parkinson’s disease. Cell Stem Cell 2015, 16, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Rhee, Y.-H.; Ko, J.-Y.; Chang, M.-Y.; Yi, S.-H.; Kim, D.; Kim, C.-H.; Shim, J.-W.; Jo, A.-Y.; Kim, B.-W.; Lee, H. Protein-based human ips cells efficiently generate functional dopamine neurons and can treat a rat model of parkinson disease. J. Clin. Investig. 2011, 121, 2326–2335. [Google Scholar] [CrossRef]
- Kikuchi, T.; Morizane, A.; Onoe, H.; Hayashi, T.; Kawasaki, T.; Saiki, H.; Miyamoto, S.; Takahashi, J. Survival of human induced pluripotent stem cell–derived midbrain dopaminergic neurons in the brain of a primate model of parkinson’s disease. J. Parkinson’s Dis. 2011, 1, 395–412. [Google Scholar]
- Eckert, A.; Huang, L.; Gonzalez, R.; Kim, H.-S.; Hamblin, M.H.; Lee, J.-P. Bystander effect fuels human induced pluripotent stem cell-derived neural stem cells to quickly attenuate early stage neurological deficits after stroke. Stem Cells Transl. Med. 2015, 4, 841–851. [Google Scholar] [CrossRef]
- Bianco, P.; Robey, P.G.; Simmons, P.J. Mesenchymal stem cells: Revisiting history, concepts, and assays. Cell Stem Cell 2008, 2, 313–319. [Google Scholar] [CrossRef]
- Lee, J.; Kuroda, S.; Shichinohe, H.; Ikeda, J.; Seki, T.; Hida, K.; Tada, M.; Sawada, K.i.; Iwasaki, Y. Migration and differentiation of nuclear fluorescence-labeled bone marrow stromal cells after transplantation into cerebral infarct and spinal cord injury in mice. Neuropathology 2003, 23, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Ra, J.C.; Shin, I.S.; Kim, S.H.; Kang, S.K.; Kang, B.C.; Lee, H.Y.; Kim, Y.J.; Jo, J.Y.; Yoon, E.J.; Choi, H.J. Safety of intravenous infusion of human adipose tissue-derived mesenchymal stem cells in animals and humans. Stem Cells Dev. 2011, 20, 1297–1308. [Google Scholar] [CrossRef] [PubMed]
- Lykhmus, O.; Koval, L.; Voytenko, L.; Uspenska, K.; Komisarenko, S.; Deryabina, O.; Shuvalova, N.; Kordium, V.; Ustymenko, A.; Kyryk, V. Intravenously injected mesenchymal stem cells penetrate the brain and treat inflammation-induced brain damage and memory impairment in mice. Front. Pharmacol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Xie, Z.H.; Wei, L.F.; Yang, H.N.; Yang, S.N.; Zhu, Z.Y.; Wang, P.; Zhao, C.P.; Bi, J.Z. Human umbilical cord mesenchymal stem cell-derived neuron-like cells rescue memory deficits and reduce amyloid-beta deposition in an aβpp/ps1 transgenic mouse model. Stem Cell Res. Ther. 2013, 4, 76. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-S.; Kim, H.S.; Park, J.-M.; Kim, H.W.; Park, M.-k.; Lee, H.-S.; Lim, D.S.; Lee, T.H.; Chopp, M.; Moon, J. Long-term immunomodulatory effect of amniotic stem cells in an alzheimer’s disease model. Neurobiol. Aging 2013, 34, 2408–2420. [Google Scholar] [CrossRef]
- Matchynski-Franks, J.J.; Pappas, C.; Rossignol, J.; Reinke, T.; Fink, K.; Crane, A.; Twite, A.; Lowrance, S.A.; Song, C.; Dunbar, G.L. Mesenchymal stem cells as treatment for behavioral deficits and neuropathology in the 5xfad mouse model of alzheimer’s disease. Cell Transplant. 2016, 25, 687–703. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.H.; Kim, H.N.; Park, H.-J.; Shin, J.Y.; Lee, P.H. Mesenchymal stem cells increase hippocampal neurogenesis and neuronal differentiation by enhancing the wnt signaling pathway in an Alzheimer’s disease model. Cell Transplant. 2015, 24, 1097–1109. [Google Scholar] [CrossRef]
- Teixeira, F.G.; Carvalho, M.M.; Neves-Carvalho, A.; Panchalingam, K.M.; Behie, L.A.; Pinto, L.; Sousa, N.; Salgado, A.J. Secretome of mesenchymal progenitors from the umbilical cord acts as modulator of neural/glial proliferation and differentiation. Stem Cell Rev. Rep. 2015, 11, 288–297. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J. Delivery of sirna to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341. [Google Scholar] [CrossRef]
- Katsuda, T.; Tsuchiya, R.; Kosaka, N.; Yoshioka, Y.; Takagaki, K.; Oki, K.; Takeshita, F.; Sakai, Y.; Kuroda, M.; Ochiya, T. Human adipose tissue-derived mesenchymal stem cells secrete functional neprilysin-bound exosomes. Sci. Rep. 2013, 3, 1197. [Google Scholar] [CrossRef]
- Garcia, K.d.O.; Ornellas, F.L.; Matsumoto, P.; Patti, C.d.L.; Mello, L.E.; Frussa-Filho, R.; Han, S.W.; Longo, B.M. Therapeutic effects of the transplantation of vegf overexpressing bone marrow mesenchymal stem cells in the hippocampus of murine model of alzheimer’s disease. Front. Aging Neurosci. 2014, 6, 30. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Seo, S.W.; Chang, J.W.; Lee, J.I.; Kim, C.H.; Chin, J.; Choi, S.J.; Kwon, H.; Yun, H.J.; Lee, J.M. Stereotactic brain injection of human umbilical cord blood mesenchymal stem cells in patients with alzheimer’s disease dementia: A phase 1 clinical trial. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2015, 1, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Tuszynski, M.H.; Thal, L.; Pay, M.; Salmon, D.P.; Bakay, R.; Patel, P.; Blesch, A.; Vahlsing, H.L.; Ho, G.; Tong, G. A phase 1 clinical trial of nerve growth factor gene therapy for alzheimer disease. Nat. Med. 2005, 11, 551. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Stem cell treatment development in Alzheimer’s disease. AD patients or healthy donors, or a blastocyst in case of ESCs, can be the source of different stem cell types which can be used for treatment development. Stem cells can be further involved in various processes, such as differentiation, genetic manipulation, used for drug screening or tested on mice and other organisms. Finally, stem cells reach the AD patients and are applied in therapeutic purposes.
Figure 1.
Stem cell treatment development in Alzheimer’s disease. AD patients or healthy donors, or a blastocyst in case of ESCs, can be the source of different stem cell types which can be used for treatment development. Stem cells can be further involved in various processes, such as differentiation, genetic manipulation, used for drug screening or tested on mice and other organisms. Finally, stem cells reach the AD patients and are applied in therapeutic purposes.


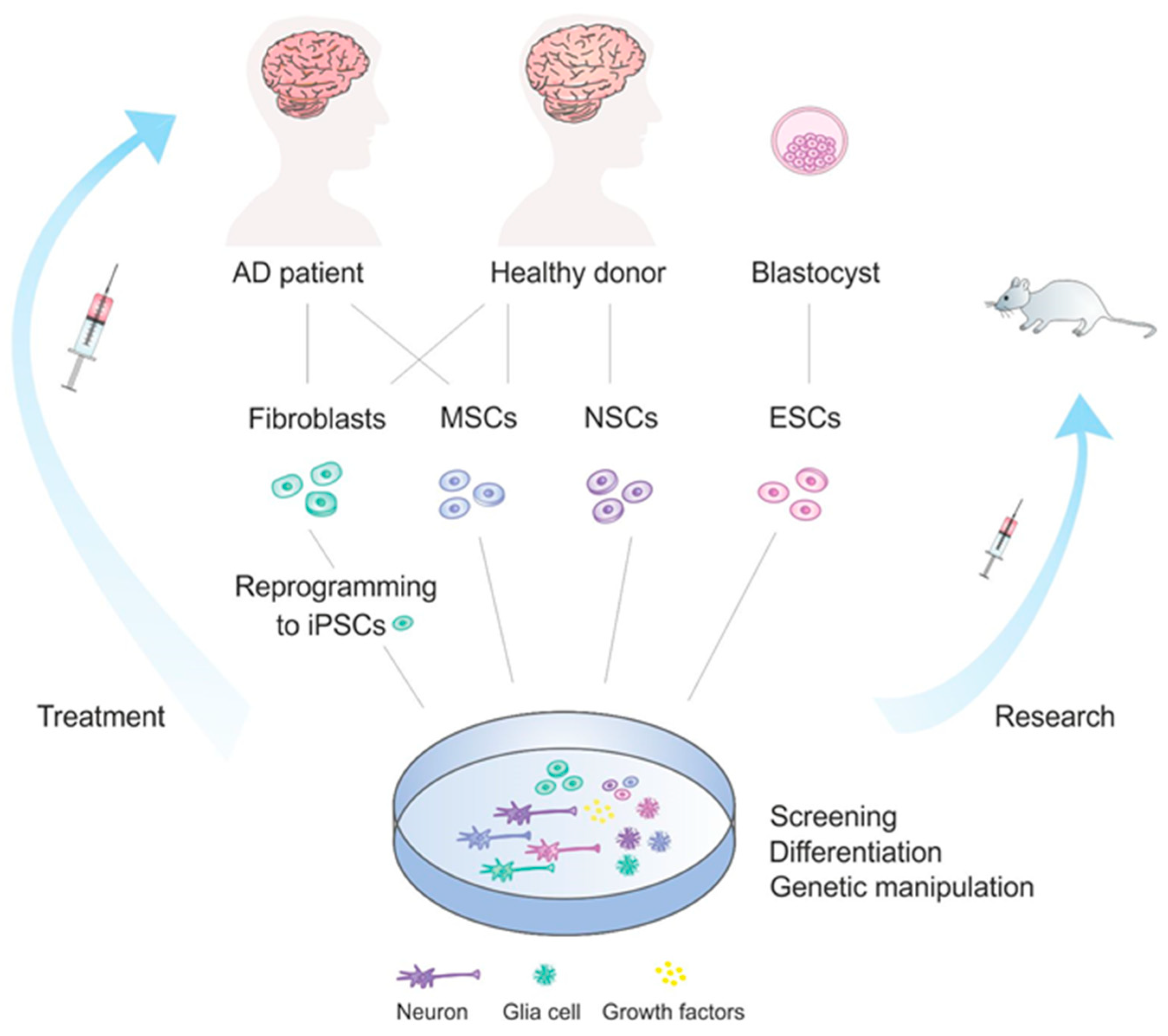{kind=link}
Table 1.
Involvement of specific cell types and the role of organelles/cell processes in the development of AD.
Table 1.
Involvement of specific cell types and the role of organelles/cell processes in the development of AD.
| Cellular System | Physiological Function | Involvement in AD |
|---|---|---|
| Microglia | Component in the determination of cognitive function, preservation of a healthy brain, attack and removal of pathogens and detritus, secretion of tissue rebuilding factors, synaptic remodeling [15,16,17,18,19,20,21,22,24,25,26,27] | Involved in the generation of neuro-inflammation, imbalance of Aβ peptide homeostasis, decrease of phagocytic activity, release of pro-inflammatory neurotoxins and cytokines/chemokines [29,30,31,33,34,35,36,37,38,39,40,41,42,43,44] |
| Astrocytes | Interactions with neurons by releasing and recycling glio-transmitter, control ion homeostasis, energy metabolism, synaptic remodeling and the modulation of oxidative stress leading to control of neurotransmission, synaptic plasticity and the modulation of cognitive functions involved in the degradation of Aβ peptides [8] | Changes in intra- and extracellular degradation of Aβ peptides, release of cytokines and chemokines, expression of ApoE, formation of hyperphosphorylated tau protein [45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63] |
| Oligodendrocytes | Form together with myelin lipid layers the envelope of the neuronal axons [8] | Specific morphological changes during AD progression, deterioration in myelin integrity and axonal destruction, killed by Aβ peptides [64,65,66,67,68,69,73] |
| NG2-glia | Oligodendrocyte precursor cells | Aβ peptides-induced inhibition of Wnt signaling pathway results in an inhibition of the differentiation of NG2-glia [74,76] |
| Neurons | Expression of a large number of molecules for protection against inflammatory attacks and induction of neurological disorders [5] | Formation of intracellular neurofibrillary tangles by hyperphosphorylated tau protein, impaired axonal transport of mitochondria resulting in energy dysfunction, generation of reactive oxygen and nitrogen species [77,78,79,80,81,82,83] |
| Mitochondria | ATP synthesis, reaction to different energy demands by balanced fission and fusion processes and directed transport along axons, protection against ROS damage by elimination of defective constituents | Aggregation of hyperphosphorylated tau in neurofibrillary tangles, perinuclear mis-localization resulting in ATP depletion, synaptic dysfunction, oxidative stress [84,85,86,87,88,89,90,91] |
| Autophagy (Mitophagy) | Degradation of organelles, proteins and lipids mediated by membranes, vesicles and lysosomes, essential for organelle turn-over, synaptic plasticity, anti-inflammatory function in glial cells, oligodendrocyte development, and the myelination process | Dysregulation leading to changes in the expression of several autophagy genes resulting in reduced energy levels, increased ROS production and impaired neuroplasticity [92,93,94,95,96,97,98,99,100,101,102,103,104,105,106] |
| Endocytic processes | Internalization of materials from the cell surface by clathrin-dependent and clathrin-independent pathways, using flotillins or caveolins as the main proteins, as well as the protection against the processing of APP and Aβ toxicity [36] | Only a few data available, volume of total endosomes increases, enhanced levels of several endocytic enzymes [107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125] |
Table 2.
Summary of advantages and disadvantages of distinct stem cell types.
| Stem Cells | Advantages | Disadvantages |
|---|---|---|
| ESCs | Unlimited self-renewal; Pluripotent [161] | Ethical issues; Uncontrolled differentiation [162,163] |
| NSCs | Multipotent [168] | Poor survival [169,170] |
| iPSCs | Autologous transplantation; Pluripotent [180] | Possible pathological phenotype [181,182] |
| MSCs | Easy handling; Multipotent; Intravenous application [190,192,193] | Low rate of neuronal differentiation [191] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Vasic, V.; Barth, K.; Schmidt, M.H.H. Neurodegeneration and Neuro-Regeneration—Alzheimer’s Disease and Stem Cell Therapy. Int. J. Mol. Sci. 2019, 20, 4272. https://doi.org/10.3390/ijms20174272
AMA Style
Vasic V, Barth K, Schmidt MHH. Neurodegeneration and Neuro-Regeneration—Alzheimer’s Disease and Stem Cell Therapy. International Journal of Molecular Sciences. 2019; 20(17):4272. https://doi.org/10.3390/ijms20174272
Chicago/Turabian StyleVasic, Verica, Kathrin Barth, and Mirko H.H. Schmidt. 2019. "Neurodegeneration and Neuro-Regeneration—Alzheimer’s Disease and Stem Cell Therapy" International Journal of Molecular Sciences 20, no. 17: 4272. https://doi.org/10.3390/ijms20174272
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.