CNNDLP: A Method Based on Convolutional Autoencoder and Convolutional Neural Network with Adjacent Edge Attention for Predicting lncRNA–Disease Associations

Abstract
1. Introduction
2. Experimental Evaluations and Discussions
2.1. Evaluation Metrics
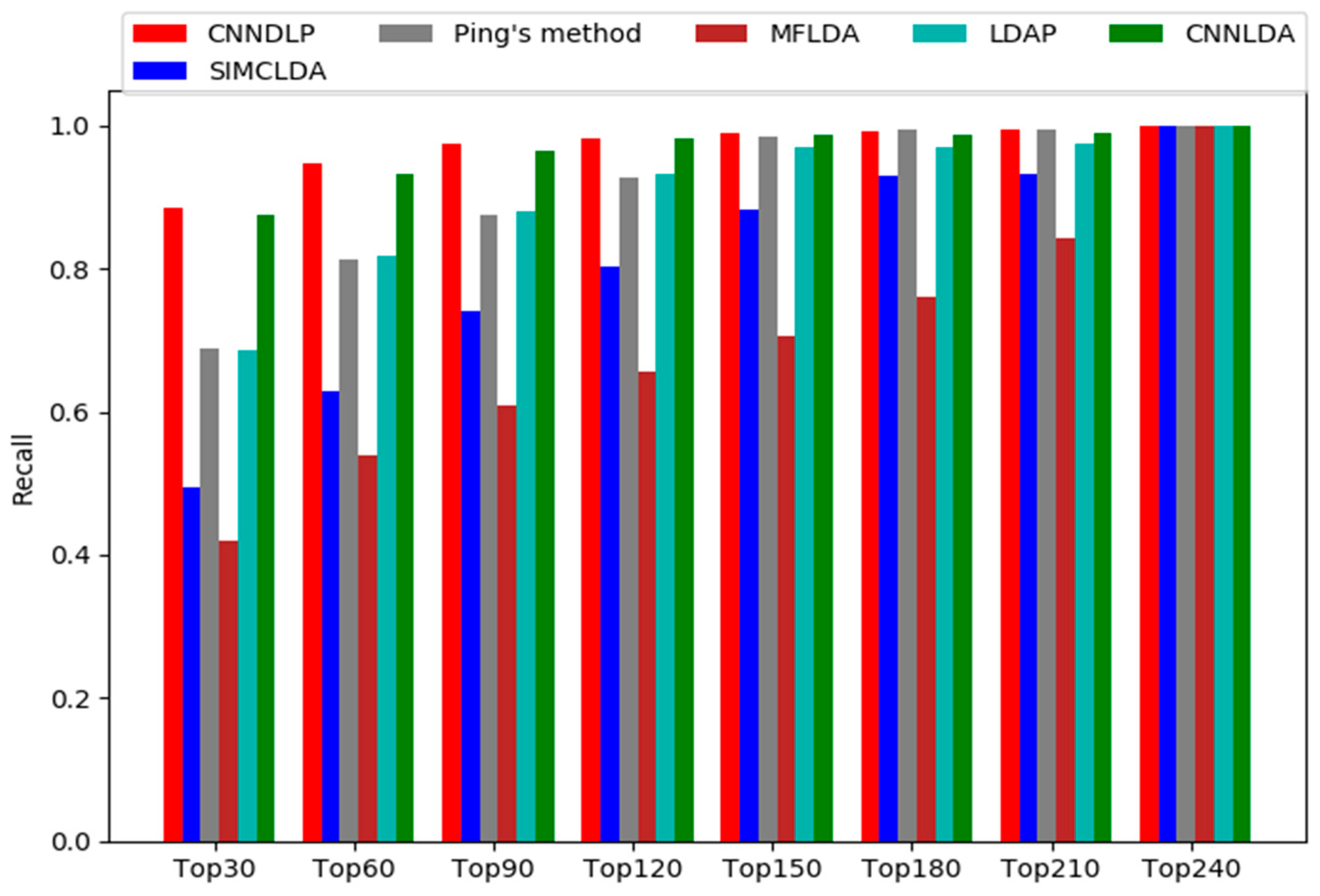
2.2. Comparison with Other Methods
2.3. Case Studies: Stomach Cancer, Breast Cancer and Prostate Cancer
2.4. Prediction of Novel Disease lncRNAs
3. Materials and Methods
3.1. Datasets for lncRNA-Disease Association Prediction
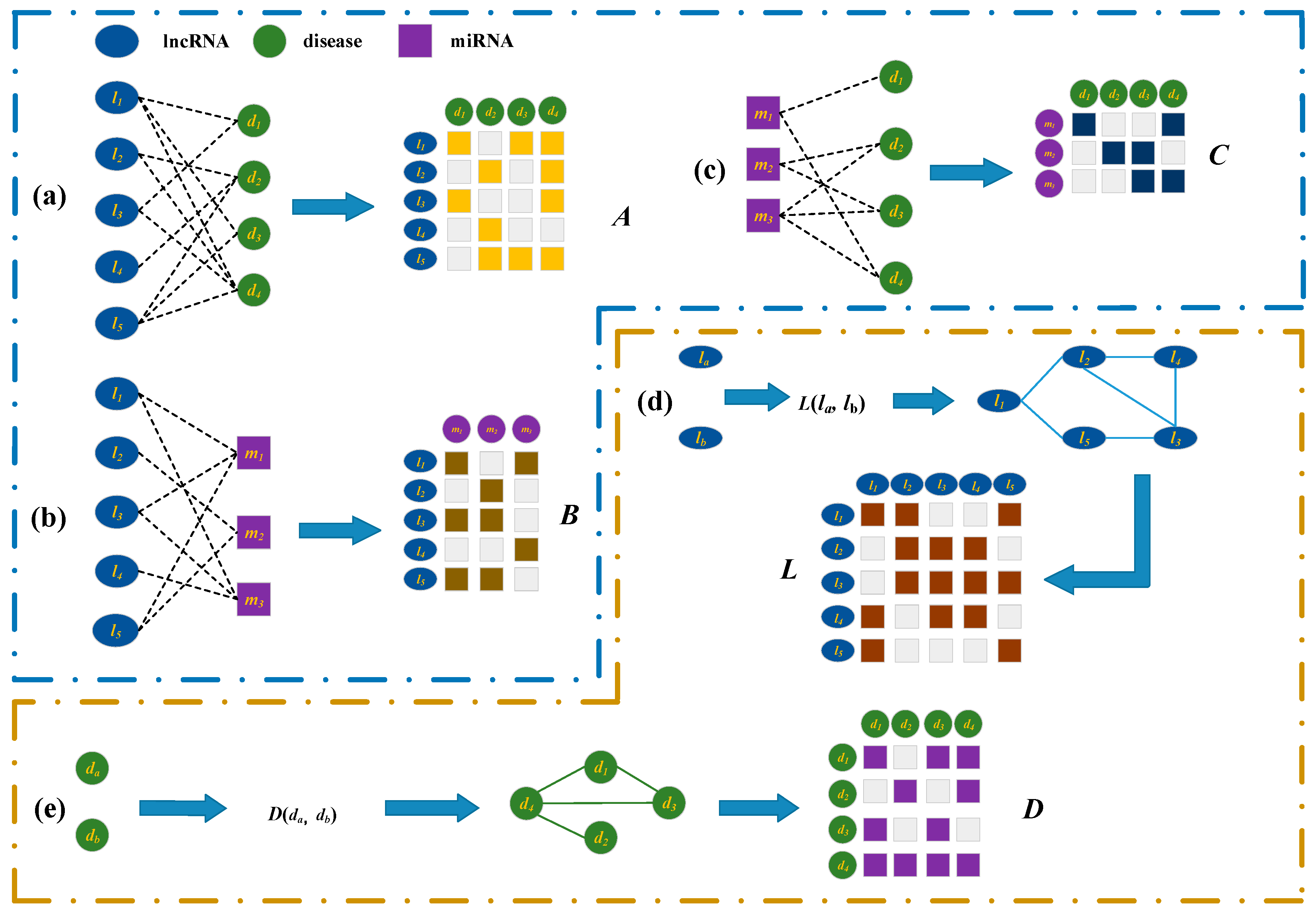
3.2. Bipartite Graphs about the lncRNAs, Diseases, miRNAs, and Representations
3.3. LncRNA-Disease Association Prediction Model Based on CNN
3.3.1. Embedding Layer on the Left
lncRNA Functional Similarity Measurement
Disease Similarity Measurement
The Left Embedding Layer for Integrating the Original Information
Attention at the Adjacent Edge Level
3.3.2. Embedding Layer on the Right
3.4. Convolutional Module on the Left
3.5. Convalutional Autoencoder Module on the Right
3.5.1. Encoding Strategy
3.5.2. Decoding Strategy
3.6. Combined Strategy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fu, X.-D. Non-coding RNA: A new frontier in regulatory biology. Natl. Sci. Rev. 2014, 1, 190–204. [Google Scholar] [CrossRef] [PubMed]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef]
- Guttman, M.; Rinn, J.L. Modular regulatory principles of large non-coding RNAs. Nature 2012, 482, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Wapinski, O.; Chang, H.Y. Long noncoding RNAs and human disease. Trends Cell Biol. 2011, 21, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yan, G.-Y. Novel human lncRNA-disease association inference based on lncRNA expression profiles. Bioinformatics 2013, 29, 2617–2624. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Xu, J.; Liu, L.; Bai, J.; Xu, C.; Xiao, Y.; Li, X.; Zhang, L. Identification of cancer-related lncRNAs through integrating genome, regulome and transcriptome features. Mol. Biosyst. 2015, 11, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Lan, W.; Huang, L.; Lai, D.; Chen, Q. Identifying Interactions Between Long Noncoding RNAs and Diseases Based on Computational Methods. Methods Mol. Biol. 2018, 1754, 205–221. [Google Scholar]
- Xuan, P.; Cao, Y.; Zhang, T.; Kong, R.; Zhang, Z. Dual Convolutional Neural Networks with Attention Mechanisms Based Method for Predicting Disease-Related lncRNA Genes. Front. Genet. 2019, 10, 416. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Fu, G.; Lu, C.; Ren, Y.; Wang, J. BRWLDA: Bi-random walks for predicting lncRNA-disease associations. Oncotarget 2017, 8, 60429–60446. [Google Scholar] [CrossRef]
- Chen, X.; You, Z.-H.; Yan, G.-Y.; Gong, D.-W. IRWRLDA: Improved random walk with restart for lncRNA-disease association prediction. Oncotarget 2016, 7, 57919–57931. [Google Scholar] [CrossRef]
- Xiao, X.; Zhu, W.; Liao, B.; Xu, J.; Gu, C.; Ji, B.; Yao, Y.; Peng, L.; Yang, J. BPLLDA: Predicting lncRNA-Disease Associations Based on Simple Paths with Limited Lengths in a Heterogeneous Network. Front. Genet. 2018, 9, 411. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Shi, H.; Wang, Z.; Zhang, C.; Liu, L.; Wang, L.; He, W.; Hao, D.; Liu, S.; Zhou, M. Inferring novel lncRNA-disease associations based on a random walk model of a lncRNA functional similarity network. Mol. Biosyst. 2014, 10, 2074–2081. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Wang, M.; Sun, D.; Li, A. TPGLDA: Novel prediction of associations between lncRNAs and diseases via lncRNA-disease-gene tripartite graph. Sci. Rep. 2018, 8, 1065. [Google Scholar] [CrossRef] [PubMed]
- Xuan, Z.; Li, J.; Yu, J.; Feng, X.; Zhao, B.; Wang, L. A Probabilistic Matrix Factorization Method for Identifying lncRNA-disease Associations. Genes 2019, 10, 126. [Google Scholar] [CrossRef] [PubMed]
- Fu, G.; Wang, J.; Domeniconi, C.; Yu, G. Matrix factorization-based data fusion for the prediction of lncRNA–disease associations. Bioinformatics 2017, 34, 1529–1537. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, Z.; Chen, Z.; Deng, L. Integrating Multiple Heterogeneous Networks for Novel LncRNA-Disease Association Inference. IEEE/ACM Trans. Comput. Biol. Bioinform. 2019, 16, 396–406. [Google Scholar] [CrossRef] [PubMed]
- Xuan, P.; Ye, Y.; Zhang, T.; Zhao, L.; Sun, C. Convolutional Neural Network and Bidirectional Long Short-Term Memory-Based Method for Predicting Drug-Disease Associations. Cells 2019, 8, 705. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Rehmsmeier, M. The Precision-Recall Plot Is More Informative than the ROC Plot When Evaluating Binary Classifiers on Imbalanced Datasets. PLoS ONE 2015, 10, e0118432. [Google Scholar] [CrossRef] [PubMed]
- Xuan, P.; Shen, T.; Wang, X.; Zhang, T.; Zhang, W. Inferring disease-associated microRNAs in heterogeneous networks with node attributes. IEEE/ACM Trans. Comput. Biol. Bioinform. 2018. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Yang, M.; Luo, F.; Wu, F.-X.; Li, M.; Pan, Y.; Li, Y.; Wang, J. Prediction of lncRNA–disease associations based on inductive matrix completion. Bioinformatics 2018, 34, 3357–3364. [Google Scholar] [CrossRef]
- Ping, P.; Wang, L.; Kuang, L.; Ye, S.; Iqbal, M.F.B.; Pei, T. A Novel Method for LncRNA-Disease Association Prediction Based on an lncRNA-Disease Association Network. IEEE/ACM Trans. Comput. Biol. Bioinform. 2019, 16, 688–693. [Google Scholar] [CrossRef] [PubMed]
- Lan, W.; Li, M.; Zhao, K.; Liu, J.; Wu, F.-X.; Pan, Y.; Wang, J. LDAP: A web server for lncRNA-disease association prediction. Bioinformatics 2016, 33, 458–460. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Shin, H.-R.; Bray, F.; Forman, D.; Mathers, C.; Maxwell Parkin, D. Estimates of worldwide burden of cancer in 2008: GLOBOCAN. Int. J. Cancer 2010, 127, 2893–2917. [Google Scholar] [CrossRef] [PubMed]
- Ning, S.; Zhang, J.; Wang, P.; Zhi, H.; Wang, J.; Liu, Y.; Gao, Y.; Guo, M.; Yue, M.; Wang, L.; et al. Lnc2Cancer: A manually curated database of experimentally supported lncRNAs associated with various human cancers. Nucleic Acids Res. 2016, 44, D980–D985. [Google Scholar] [CrossRef] [PubMed]
- Bao, Z.; Yang, Z.; Huang, Z.; Zhou, Y.; Cui, Q.; Dong, D. LncRNADisease 2.0: An updated database of long non-coding RNA-associated diseases. Nucleic Acids Res. 2019, 47, D1034–D1037. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Luo, Y.; Liang, B.; Ye, L.; Lu, G.; He, W. Potential applications of MEG3 in cancer diagnosis and prognosis. Oncotarget 2017, 8, 73282–73295. [Google Scholar] [CrossRef]
- Xu, Y.; Chen, M.; Liu, C.; Zhang, X.; Li, W.; Cheng, H.; Zhu, J.; Zhang, M.; Chen, Z.; Zhang, B. Association Study Confirmed Three Breast Cancer-Specific Molecular Subtype-Associated Susceptibility Loci in Chinese Han Women. Oncologist 2017, 22, 890–894. [Google Scholar] [CrossRef]
- Lv, X.-B.; Jiao, Y.; Qing, Y.; Hu, H.; Cui, X.; Lin, T.; Song, E.; Yu, F. miR-124 suppresses multiple steps of breast cancer metastasis by targeting a cohort of pro-metastatic genes in vitro. Chin. J. Cancer 2011, 30, 821–830. [Google Scholar] [CrossRef]
- Chen, G.; Wang, Z.; Wang, D.; Qiu, C.; Liu, M.; Chen, X.; Zhang, Q.; Yan, G.; Cui, Q. LncRNADisease: A database for long-non-coding RNA-associated diseases. Nucleic Acids Res. 2013, 41, D983–D986. [Google Scholar] [CrossRef]
- Li, J.H.; Liu, S.; Zhou, H.; Qu, L.H.; Yang, J.H. starBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014, 42, D92–D97. [Google Scholar] [CrossRef]
- Li, Y.; Qiu, C.; Tu, J.; Geng, B.; Yang, J.; Jiang, T.; Cui, Q. HMDD v2.0: A database for experimentally supported human microRNA and disease associations. Nucleic Acids Res. 2014, 42, D1070–D1074. [Google Scholar] [CrossRef] [PubMed]
- Paraskevopoulou, M.D.; Hatzigeorgiou, A.G. Analyzing MiRNA-LncRNA Interactions. Methods Mol. Biol. 2016, 1402, 271–286. [Google Scholar] [PubMed]
- Wang, D.; Wang, J.; Lu, M.; Song, F.; Cui, Q. Inferring the human microRNA functional similarity and functional network based on microRNA-associated diseases. Bioinformatics 2010, 26, 1644–1650. [Google Scholar] [CrossRef] [PubMed]
- Nair, V.; Hinton, G.E. Rectified linear units improve restricted boltzmann machines. In Proceedings of the 27th International Conference on International Conference on Machine Learning, Haifa, Israel, 21–24 June 2010; pp. 807–814. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease Name | CNNDLP | Ping’s Method | AUC LDAP | SIMCLDA | MFLDA | CNNLDA |
|---|---|---|---|---|---|---|
| Prostate cancer | 0.951 | 0.826 | 0.710 | 0.874 | 0.553 | 0.897 |
| Stomach cancer | 0.947 | 0.930 | 0.928 | 0.864 | 0.467 | 0.958 |
| Lung cancer | 0.976 | 0.911 | 0.882 | 0.790 | 0.676 | 0.940 |
| Breast cancer | 0.956 | 0.872 | 0.830 | 0.742 | 0.517 | 0.836 |
| Reproduce organ cancer | 0.943 | 0.818 | 0.742 | 0.707 | 0.740 | 0.922 |
| Ovarian cancer | 0.970 | 0.913 | 0.857 | 0.786 | 0.558 | 0.942 |
| Hematologic cancer | 0.989 | 0.908 | 0.903 | 0.828 | 0.716 | 0.934 |
| Kidney cancer | 0.984 | 0.979 | 0.977 | 0.728 | 0.677 | 0.956 |
| Liver cancer | 0.956 | 0.910 | 0.898 | 0.799 | 0.634 | 0.918 |
| Thoracic cancer | 0.921 | 0.860 | 0.792 | 0.792 | 0.649 | 0.890 |
| Average AUC of 405 diseases | 0.969 | 0.870 | 0.745 | 0.745 | 0.626 | 0.952 |
| Disease Name | CNNDLP | Ping’s Method | AUPR LDAP | SIMCLDA | MFLDA | CNNLDA |
|---|---|---|---|---|---|---|
| Prostate cancer | 0.538 | 0.333 | 0.297 | 0.176 | 0.092 | 0.390 |
| Stomach cancer | 0.373 | 0.364 | 0.094 | 0.138 | 0.008 | 0.286 |
| Lung cancer | 0.666 | 0.437 | 0.363 | 0.131 | 0.171 | 0.058 |
| Breast cancer | 0.485 | 0.403 | 0.396 | 0.047 | 0.031 | 0.964 |
| Reproduce organ cancer | 0.498 | 0.281 | 0.240 | 0.130 | 0.103 | 0.091 |
| Ovarian cancer | 0.552 | 0.483 | 0.427 | 0.027 | 0.023 | 0.526 |
| Hematologic cancer | 0.667 | 0.403 | 0.370 | 0.216 | 0.121 | 0.523 |
| Kidney cancer | 0.569 | 0.663 | 0.462 | 0.030 | 0.034 | 0.584 |
| Liver cancer | 0.630 | 0.498 | 0.511 | 0.140 | 0.110 | 0.666 |
| Thoracic cancer | 0.399 | 0.383 | 0.364 | 0.155 | 0.102 | 0.890 |
| Average AUC of 405 diseases | 0.286 | 0.152 | 0.127 | 0.059 | 0.039 | 0.251 |
| SIMCLDA | Ping’s Method | MFLDA | LDAP | CNNLDA | |
|---|---|---|---|---|---|
| p-value of ROC curve | 9.2454 × 10−6 | 0.00048 | 5.9940 × 10−7 | 0.00121 | 0.00773 |
| p-value of PR curve | 8.3473 × 10−7 | 0.04174 | 3.5037 × 10−8 | 0.00126 | 0.00024 |
| Rank | lncRNA Name | Description | Rank | lncRNA Name | Description |
|---|---|---|---|---|---|
| 1 | SPRY4-IT1 | Lnc2Cancer, LncRNADisease | 9 | CDKN2B-AS1 | LncRNADisease |
| 2 | TINCR | Lnc2Cancer, LncRNADisease | 10 | CCAT1 | Lnc2Cancer, LncRNADisease |
| 3 | H19 | Lnc2Cancer, LncRNADisease | 11 | HOTAIR | Lnc2Cancer, LncRNADisease |
| 4 | TUSC7 | Lnc2Cancer, LncRNADisease | 12 | GACAT2 | LncRNADisease |
| 5 | BANCR | Lnc2Cancer, LncRNADisease | 13 | UCA1 | Lnc2Cancer, LncRNADisease |
| 6 | MEG3 | Lnc2Cancer, LncRNADisease | 14 | PVT1 | Lnc2Cancer, LncRNADisease |
| 7 | GAS5 | Lnc2Cancer, LncRNADisease | 15 | MEG8 | literature |
| 8 | GHET1 | Lnc2Cancer, LncRNADisease |
| Rank | lncRNA Name | Description | Rank | lncRNA Name | Description |
|---|---|---|---|---|---|
| 1 | SOX2-OT | Lnc2Cancer, LncRNADisease | 9 | CCAT1 | Lnc2Cancer, LncRNADisease |
| 2 | HOTAIR | Lnc2Cancer, LncRNADisease | 10 | GAS5 | Lnc2Cancer, LncRNADisease |
| 3 | LINC00472 | Lnc2Cancer, LncRNADisease | 11 | MIR124-2HG | literature |
| 4 | BCYRN1 | LncRNADisease | 12 | XIST | Lnc2Cancer, LncRNADisease |
| 5 | LINC-PINT | literature | 13 | LINC-ROR | Lnc2Cancer, LncRNADisease |
| 6 | MALAT1 | Lnc2Cancer, LncRNADisease | 14 | PANDAR | Lnc2Cancer, LncRNADisease |
| 7 | CDKN2B-AS1 | LncRNADisease | 15 | AFAP1-AS1 | Lnc2Cancer |
| 8 | SPRY4-IT1 | Lnc2Cancer, LncRNADisease |
| Rank | lncRNA Name | Description | Rank | lncRNA Name | Description |
|---|---|---|---|---|---|
| 1 | CDKN2B-AS1 | LncRNADisease | 9 | HOTAIR | Lnc2Cancer, LncRNADisease |
| 2 | PCGEM1 | Lnc2Cancer, LncRNADisease | 10 | LINC00963 | Lnc2Cancer, LncRNADisease |
| 3 | PVT1 | Lnc2Cancer, LncRNADisease | 11 | H19 | Lnc2Cancer, LncRNADisease |
| 4 | GAS5 | Lnc2Cancer, LncRNADisease | 12 | MEG3 | Lnc2Cancer, LncRNADisease |
| 5 | HOTTIP | Lnc2Cancer, LncRNADisease | 13 | TUG1 | Lnc2Cancer, LncRNADisease |
| 6 | NEAT1 | Lnc2Cancer, LncRNADisease | 14 | PCA3 | Lnc2Cancer, LncRNADisease |
| 7 | PCAT5 | Lnc2Cancer | 15 | DANCR | Lnc2Cancer, LncRNADisease |
| 8 | PRINS | Lnc2Cancer, LncRNADisease |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xuan, P.; Sheng, N.; Zhang, T.; Liu, Y.; Guo, Y. CNNDLP: A Method Based on Convolutional Autoencoder and Convolutional Neural Network with Adjacent Edge Attention for Predicting lncRNA–Disease Associations. Int. J. Mol. Sci. 2019, 20, 4260. https://doi.org/10.3390/ijms20174260
Xuan P, Sheng N, Zhang T, Liu Y, Guo Y. CNNDLP: A Method Based on Convolutional Autoencoder and Convolutional Neural Network with Adjacent Edge Attention for Predicting lncRNA–Disease Associations. International Journal of Molecular Sciences. 2019; 20(17):4260. https://doi.org/10.3390/ijms20174260
Chicago/Turabian StyleXuan, Ping, Nan Sheng, Tiangang Zhang, Yong Liu, and Yahong Guo. 2019. "CNNDLP: A Method Based on Convolutional Autoencoder and Convolutional Neural Network with Adjacent Edge Attention for Predicting lncRNA–Disease Associations" International Journal of Molecular Sciences 20, no. 17: 4260. https://doi.org/10.3390/ijms20174260
APA StyleXuan, P., Sheng, N., Zhang, T., Liu, Y., & Guo, Y. (2019). CNNDLP: A Method Based on Convolutional Autoencoder and Convolutional Neural Network with Adjacent Edge Attention for Predicting lncRNA–Disease Associations. International Journal of Molecular Sciences, 20(17), 4260. https://doi.org/10.3390/ijms20174260