Molecular Subtyping and Prognostic Assessment Based on Tumor Mutation Burden in Patients with Lung Adenocarcinomas

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
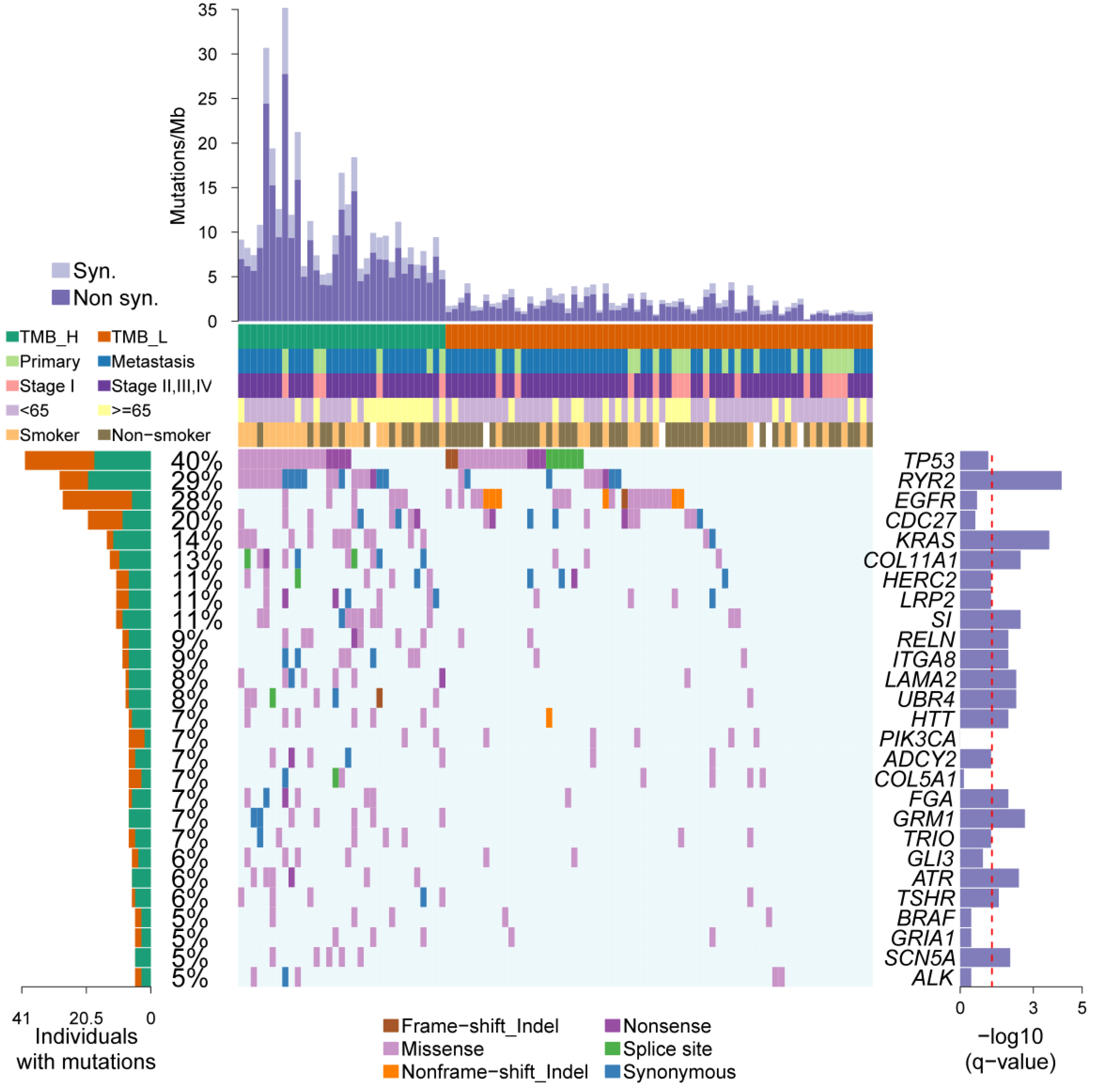
2.1. Molecular Subtyping Based on Mutation Burden
2.2. Somatic Driver Genes from Personal Mutation Background
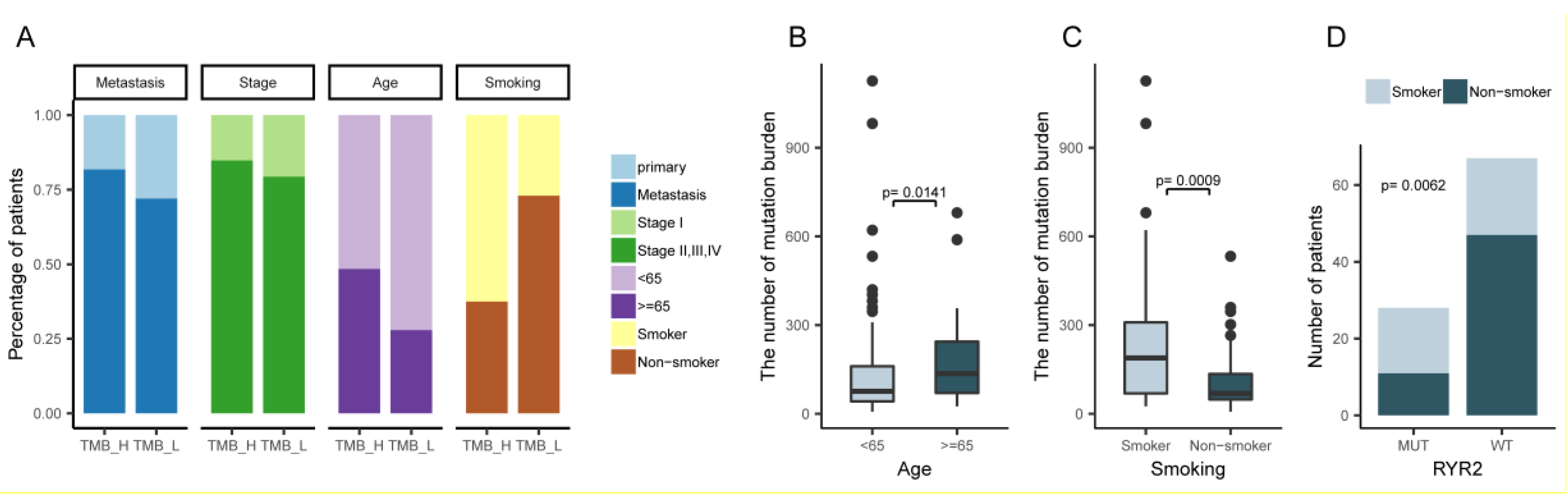
2.3. An Overview of the Clinical Implications Associated with TMB
2.4. Mutation Signature Analysis of Lung Adenocarcinoma in TMB Subtypes
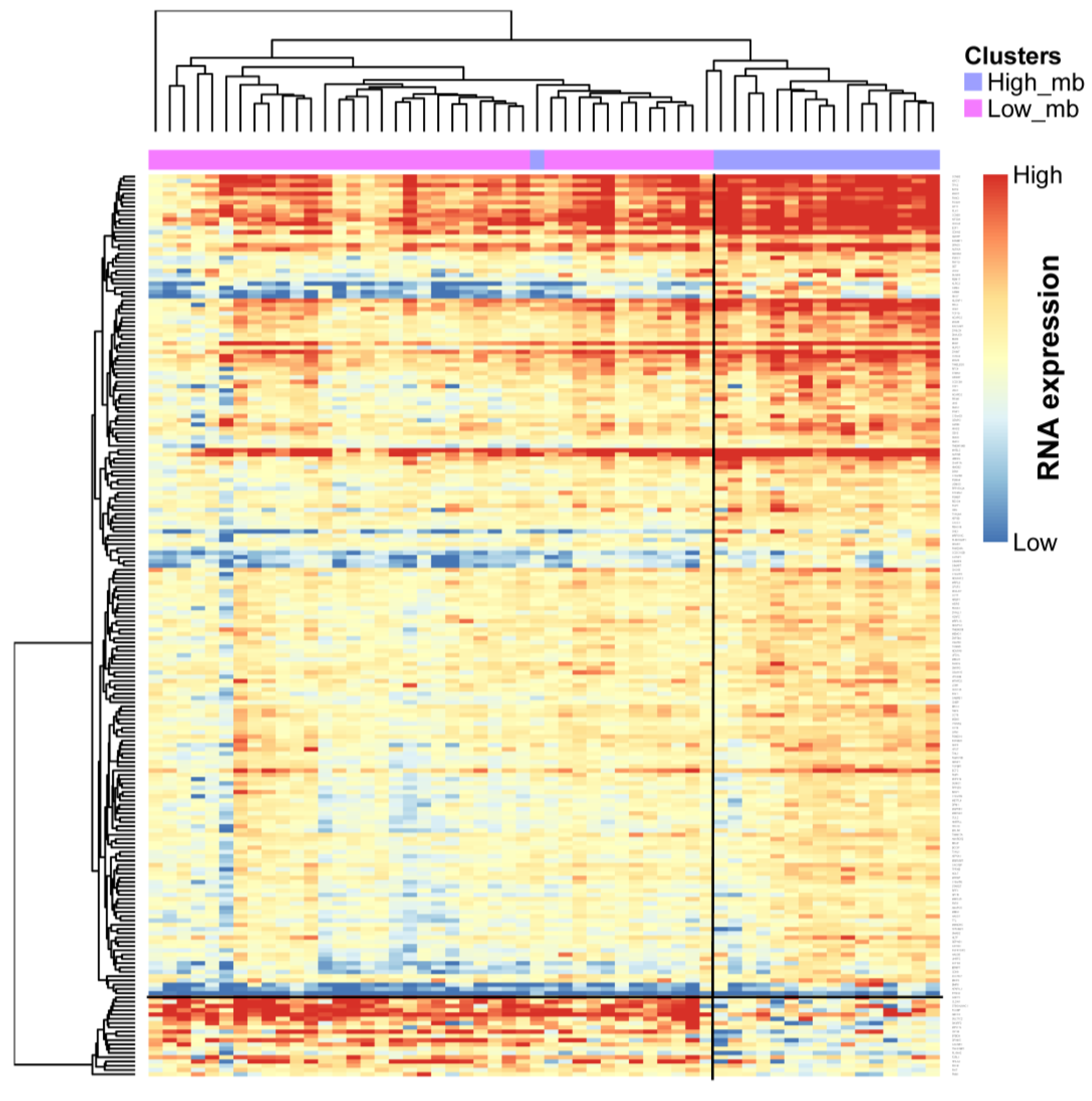
2.5. Profile of Gene Expression Level in TMB Subtypes
3. Discussion
4. Materials and Methods
4.1. Lung Adenocarcinoma Genome Data
4.2. Mutation Burden Cluster
4.3. Identification of Somatic Driver Genes
4.4. Mutation Signature Analysis
4.5. Permutation Test of Signature Enrichment Analysis
4.6. Gene Expression Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| TMB | Tumor mutation burden |
| TMB-H | High tumor mutation burden |
| TMB-L | Low tumor mutation burden |
References
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-tieulent, J.; Jemal, A. Global Cancer Statistics, 2012. CA a cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zheng, R.; Baade, P.D.; Zhang, S.; Zeng, H.; Bray, F.; Jemal, A.; Yu, X.Q.; He, J. Cancer statistics in China, 2015. CA Cancer J. Clin. 2016, 66, 115–132. [Google Scholar] [CrossRef] [PubMed]
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The cancer genome. Nature 2009, 458, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic Basis for Clinical Response to CTLA-4 Blockade in Melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability — an evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Futreal, P.A.; Coin, L.; Marshall, M.; Down, T.; Hubbard, T.; Wooster, R.; Rahman, N.; Stratton, M.R. A census of human cancer genes. Nat. Rev. Cancer 2004, 4, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Perez, A.; Perez-Llamas, C.; Deu-Pons, J.; Tamborero, D.; Schroeder, M.P.; Jene-Sanz, A.; Santos, A.; Lopez-Bigas, N. IntOGen-mutations identifies cancer drivers across tumor types. Nat. Methods 2013, 10, 1081–1082. [Google Scholar] [CrossRef] [PubMed]
- Singal, G.; Miller, P.G.; Agarwala, V.; Li, G.; Kaushik, G.; Backenroth, D.; Gossai, A.; Frampton, G.M.; Torres, A.Z.; Lehnert, E.M.; et al. Association of Patient Characteristics and Tumor Genomics With Clinical Outcomes Among Patients With Non-Small Cell Lung Cancer Using a Clinicogenomic Database. JAMA. 2019, 321, 1391–1399. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Mouw, K.W.; Polak, P.; Braunstein, L.Z.; Kamburov, A.; Tiao, G.; Kwiatkowski, D.J.; Rosenberg, J.E.; Van Allen, E.M.; D’Andrea, A.D.; et al. Somatic ERCC2 mutations are associated with a distinct genomic signature in urothelial tumors. Nat. Genet. 2016, 48, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Enk, C.D.; Jacob-Hirsch, J.; Gal, H.; Verbovetski, I.; Amariglio, N.; Mevorach, D.; Ingber, A.; Givol, D.; Rechavi, G.; Hochberg, M. The UVB-induced gene expression profile of human epidermis in vivo is different from that of cultured keratinocytes. Oncogene 2006, 25, 2601–2614. [Google Scholar] [CrossRef] [PubMed]
- Birkbak, N.J.; Kochupurakkal, B.; Izarzugaza, J.M.G.; Eklund, A.C.; Li, Y.; Liu, J.; Szallasi, Z.; Matulonis, U.A.; Richardson, A.L.; Iglehart, J.D.; et al. Tumor mutation burden forecasts outcome in ovarian cancer with BRCA1 or BRCA2 mutations. PLoS One 2013, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Danaei, G.; Vander Hoorn, S.; Lopez, A.D.; Murray, C.J.; Ezzati, M. Causes of cancer in the world: Comparative risk assessment of nine behavioural and environmental risk factors. Lancet 2005, 366, 1784–1793. [Google Scholar] [CrossRef]
- Sardari Nia, P.; Weyler, J.; Colpaert, C.; Vermeulen, P.; Marck, E. Van; Schil, P. Van Prognostic value of smoking status in operated non-small cell lung cancer. Lung Cancer 2005, 47, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Parsons, A.; Daley, A.; Begh, R.; Aveyard, P. Influence of smoking cessation after diagnosis of early stage lung cancer on prognosis: Systematic review of observational studies with meta-analysis. BMJ. 2010, 340, 251. [Google Scholar] [CrossRef]
- O’Keeffe, L.M.; Taylor, G.; Huxley, R.R.; Mitchell, P.; Woodward, M.; Peters, S.A.E. Smoking as a risk factor for lung cancer in women and men: A systematic review and meta-analysis. BMJ Open 2018, 8, e021611. [Google Scholar] [CrossRef]
- Didkowska, J.; Wojciechowska, U.; Mańczuk, M.; Łobaszewski, J. Lung cancer epidemiology: Contemporary and future challenges worldwide. Ann. Transl. Med. 2016, 4, 150. [Google Scholar] [CrossRef] [PubMed]
- Hoadley, K.A.; Yau, C.; Wolf, D.M.; Cherniack, A.D.; Tamborero, D.; Ng, S.; Leiserson, M.D.M.; Niu, B.; McLellan, M.D.; Uzunangelov, V.; et al. Multiplatform Analysis of 12 Cancer Types Reveals Molecular Classification within and across Tissues of Origin. Cell 2014, 158, 929–944. [Google Scholar] [CrossRef] [PubMed]
- Gross, A.M.; Orosco, R.K.; Shen, J.P.; Egloff, A.M.; Carter, H.; Hofree, M.; Choueiri, M.; Coffey, C.S.; Lippman, S.M.; Hayes, D.N.; et al. Multi-tiered genomic analysis of head and neck cancer ties TP53 mutation to 3p loss. Nat. Genet. 2014, 46, 939–943. [Google Scholar] [CrossRef] [PubMed]
- Akbani, R.; Akdemir, K.C.; Aksoy, B.A.; Albert, M.; Ally, A.; Amin, S.B.; Arachchi, H.; Arora, A.; Auman, J.T.; Ayala, B.; et al. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [PubMed]
- Devarakonda, S.; Rotolo, F.; Tsao, M.-S.; Lanc, I.; Brambilla, E.; Masood, A.; Olaussen, K.A.; Fulton, R.; Sakashita, S.; McLeer-Florin, A.; et al. Tumor Mutation Burden as a Biomarker in Resected Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2018, 36, 2995–3006. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Ju, Y.S.; Haase, K.; Van Loo, P.; Martincorena, I.; Nik-Zainal, S.; Totoki, Y.; Fujimoto, A.; Nakagawa, H.; Shibata, T.; et al. Mutational signatures associated with tobacco smoking in human cancer. Science 2016, 354, 618–622. [Google Scholar] [CrossRef] [PubMed]
- Kucab, J.E.; Zou, X.; Morganella, S.; Joel, M.; Nanda, A.S.; Nagy, E.; Gomez, C.; Degasperi, A.; Harris, R.; Jackson, S.P.; et al. A Compendium of Mutational Signatures of Environmental Agents. Cell 2019, 177, 821–836.e16. [Google Scholar] [CrossRef] [PubMed]
- Mouw, K.W.; Goldberg, M.S.; Konstantinopoulos, P.A.; D’Andrea, A.D. DNA Damage and Repair Biomarkers of Immunotherapy Response. Cancer Discov. 2017, 7, 675–693. [Google Scholar] [CrossRef]
- Tiso, N. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2). Hum. Mol. Genet. 2001, 10, 189–194. [Google Scholar] [CrossRef]
- McFarland, C.D.; Korolev, K.S.; Kryukov, G.V.; Sunyaev, S.R.; Mirny, L.A. Impact of deleterious passenger mutations on cancer progression. Proc. Natl. Acad. Sci. 2013, 110, 2910–2915. [Google Scholar] [CrossRef]
- Ziviani, E.; Lippi, G.; Bano, D.; Munarriz, E.; Guiducci, S.; Zoli, M.; Young, K.W.; Nicotera, P. Ryanodine receptor-2 upregulation and nicotine-mediated plasticity. EMBO J. 2011, 30, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Napolitano, C.; Tiso, N.; Memmi, M.; Vignati, G.; Bloise, R.; Sorrentino, V.; Danieli, G.A. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation 2001, 103, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Kohli, U.; Aziz, Z.; Beaser, A.D.; Nayak, H.M. A large deletion in RYR2 exon 3 is associated with nadolol and flecainide refractory catecholaminergic polymorphic ventricular tachycardia. Pacing Clin. Electrophysiol. 2019, 42, 1146–1154. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Napolitano, C.; Memmi, M.; Colombi, B.; Drago, F.; Gasparini, M.; DeSimone, L.; Coltorti, F.; Bloise, R.; Keegan, R.; et al. Clinical and Molecular Characterization of Patients With Catecholaminergic Polymorphic Ventricular Tachycardia. Circulation 2002, 106, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Croisier, H.; Tan, X.; Chen, J.; Sneyd, J.; Sanderson, M.J.; Brook, B.S. Ryanodine Receptor Sensitization Results in Abnormal Calcium Signaling in Airway Smooth Muscle Cells. Am. J. Respir. Cell Mol. Biol. 2015, 53, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; McMahon, T.J.; Zhang, Z.-S.; Stiber, J. a; Meissner, G.; Eu, J.P. Excitation-Contraction Coupling in Airway Smooth Muscle. J. Biol. Chem. 2006, 281, 30143–30151. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Stiber, J.A.; Rosenberg, P.B.; Meissner, G.; Eu, J.P. Ryanodine receptors in muscarinic receptor-mediated bronchoconstriction. J. Biol. Chem. 2005, 280, 26287–26294. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Abebe, T.; Beyene, J.; Wilke, R. a; Goldberg, A.; Woo, J.G.; Martin, L.J.; Rothenberg, M.E.; Rao, M.; Hershey, G.K.; et al. Rank-based genome-wide analysis reveals the association of Ryanodine receptor-2 gene variants with childhood asthma among human populations. Hum. Genomics 2013, 7, 16. [Google Scholar] [CrossRef] [PubMed]
- Donovan, C.; Seow, H.J.; Royce, S.G.; Bourke, J.E.; Vlahos, R. Alteration of airway reactivity and reduction of ryanodine receptor expression by cigarette smoke in mice. Am. J. Respir. Cell Mol. Biol. 2015, 53, 471–478. [Google Scholar] [CrossRef] [PubMed]
- García Sanz, M.T.; González Barcala, F.J.; Álvarez Dobaño, J.M.; Valdés Cuadrado, L. Asthma and risk of lung cancer. Clin. Transl. Oncol. 2011, 13, 728–730. [Google Scholar] [CrossRef] [PubMed]
- Rosenberger, A.; Bickeböller, H.; McCormack, V.; Brenner, D.R.; Duell, E.J.; Tjønneland, A.; Friis, S.; Muscat, J.E.; Yang, P.; Wichmann, H.-E.; et al. Asthma and lung cancer risk: A systematic investigation by the International Lung Cancer Consortium. Carcinogenesis 2012, 33, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Zhang, X.; Li, F.; Xiao, D.; Hou, Y.; Zhu, S.; Liu, D.; Ye, X.; Ye, M.; Yang, J.; et al. Frequent alterations in cytoskeleton remodelling genes in primary and metastatic lung adenocarcinomas. Nat. Commun. 2015, 6, 10131. [Google Scholar] [CrossRef] [PubMed]
- Früh, M.; Peters, S. Genomic Features of Response to Combination Immunotherapy in Lung Cancer. Cancer Cell 2018, 33, 791–793. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.; Ma, Y.; Yin, J.C.; Hong, S.; Zhou, H.; Wang, A.; Wang, F.; Bao, H.; Wu, X.; Yang, Y.; et al. Comprehensive Genomic Profiling Identifies Novel Genetic Predictors of Response to Anti–PD-(L)1 Therapies in Non–Small Cell Lung Cancer. Clin. Cancer Res. 2019, 25, 5015–5026. [Google Scholar] [CrossRef] [PubMed]
- Koay, C.G. A simple scheme for generating nearly uniform distribution of antipodally symmetric points on the unit sphere. J. Comput. Sci. 2011, 2, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Guo, Y.; Yang, H.; He, Z.; Liu, X.; Wang, K. iCAGES: Integrated CAncer GEnome Score for comprehensively prioritizing driver genes in personal cancer genomes. Genome Med. 2016, 8, 135. [Google Scholar] [CrossRef] [PubMed]
- Polak, P.; Kim, J.; Braunstein, L.Z.; Karlic, R.; Haradhavala, N.J.; Tiao, G.; Rosebrock, D.; Livitz, D.; Kübler, K.; Mouw, K.W.; et al. A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nat. Genet. 2017, 49, 1476–1486. [Google Scholar] [CrossRef] [PubMed]
- Strona, G.; Nappo, D.; Boccacci, F.; Fattorini, S.; San-Miguel-Ayanz, J. A fast and unbiased procedure to randomize ecological binary matrices with fixed row and column totals. Nat. Commun. 2014, 5, 4114. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, C.; Liang, H.; Lin, C.; Li, F.; Xie, G.; Qiao, S.; Shi, X.; Deng, J.; Zhao, X.; Wu, K.; et al. Molecular Subtyping and Prognostic Assessment Based on Tumor Mutation Burden in Patients with Lung Adenocarcinomas. Int. J. Mol. Sci. 2019, 20, 4251. https://doi.org/10.3390/ijms20174251
Wang C, Liang H, Lin C, Li F, Xie G, Qiao S, Shi X, Deng J, Zhao X, Wu K, et al. Molecular Subtyping and Prognostic Assessment Based on Tumor Mutation Burden in Patients with Lung Adenocarcinomas. International Journal of Molecular Sciences. 2019; 20(17):4251. https://doi.org/10.3390/ijms20174251
Chicago/Turabian StyleWang, Changzheng, Han Liang, Cong Lin, Fuqiang Li, Guoyun Xie, Sitan Qiao, Xulian Shi, Jianlian Deng, Xin Zhao, Kui Wu, and et al. 2019. "Molecular Subtyping and Prognostic Assessment Based on Tumor Mutation Burden in Patients with Lung Adenocarcinomas" International Journal of Molecular Sciences 20, no. 17: 4251. https://doi.org/10.3390/ijms20174251
APA StyleWang, C., Liang, H., Lin, C., Li, F., Xie, G., Qiao, S., Shi, X., Deng, J., Zhao, X., Wu, K., & Zhang, X. (2019). Molecular Subtyping and Prognostic Assessment Based on Tumor Mutation Burden in Patients with Lung Adenocarcinomas. International Journal of Molecular Sciences, 20(17), 4251. https://doi.org/10.3390/ijms20174251