Transcriptomic and Metabolomics Analysis of Different Endosperm Region under Nitrogen Treatments

Abstract
1. Introduction
2. Results
2.1. Grain Characteristics
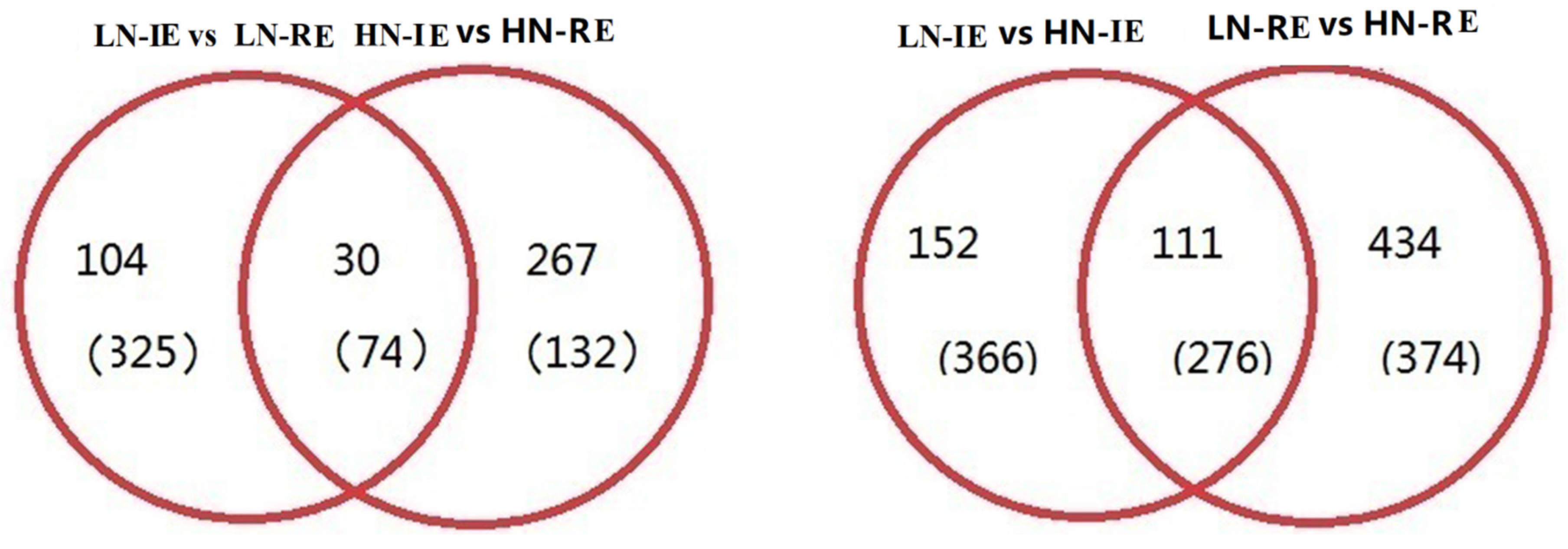
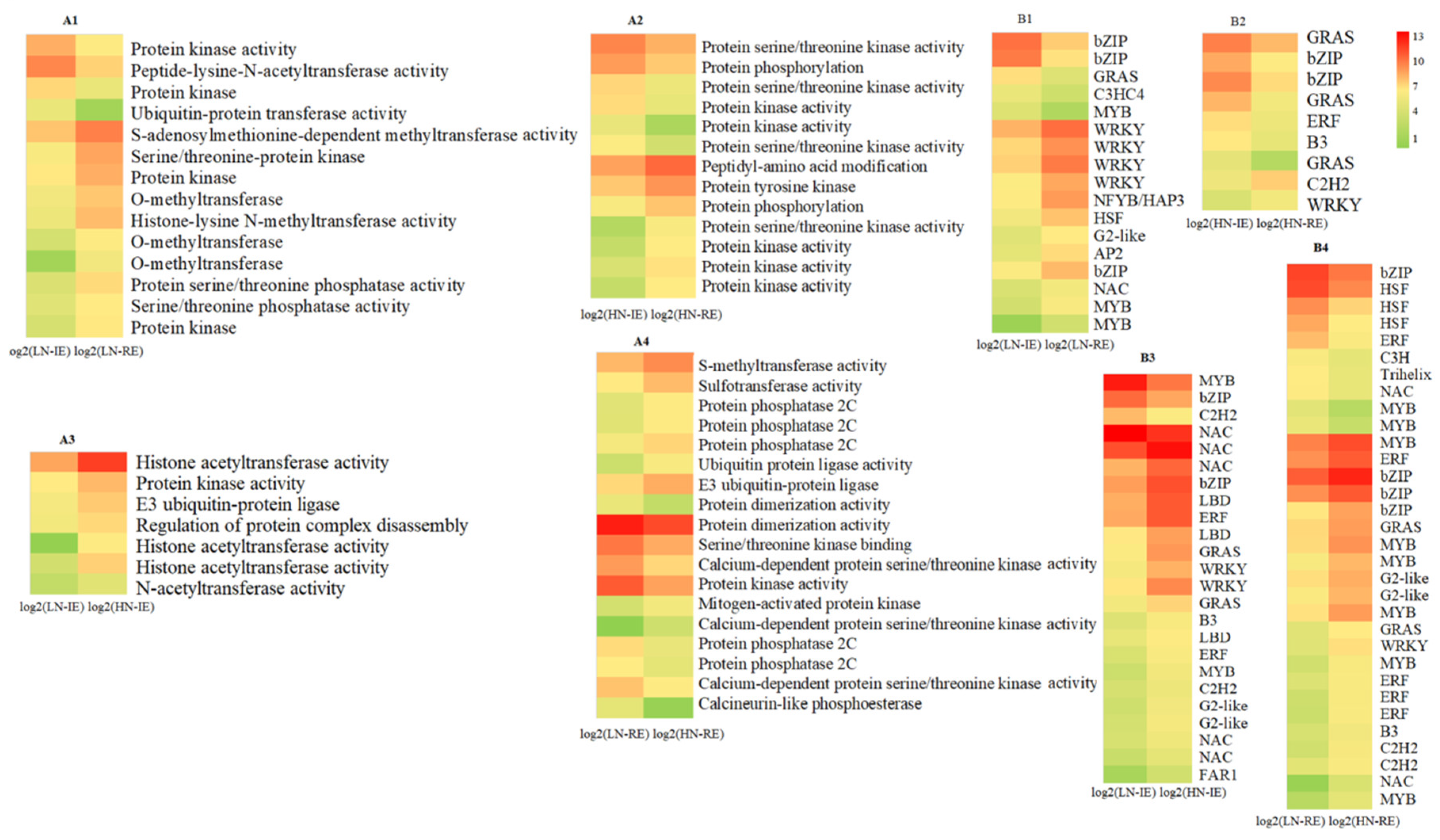
2.2. Transcriptome Analysis of the Innermost Endosperm Region and the Remianing Endosperm Region from Plants Grown at Two Nitrogen Fertilizer Levels
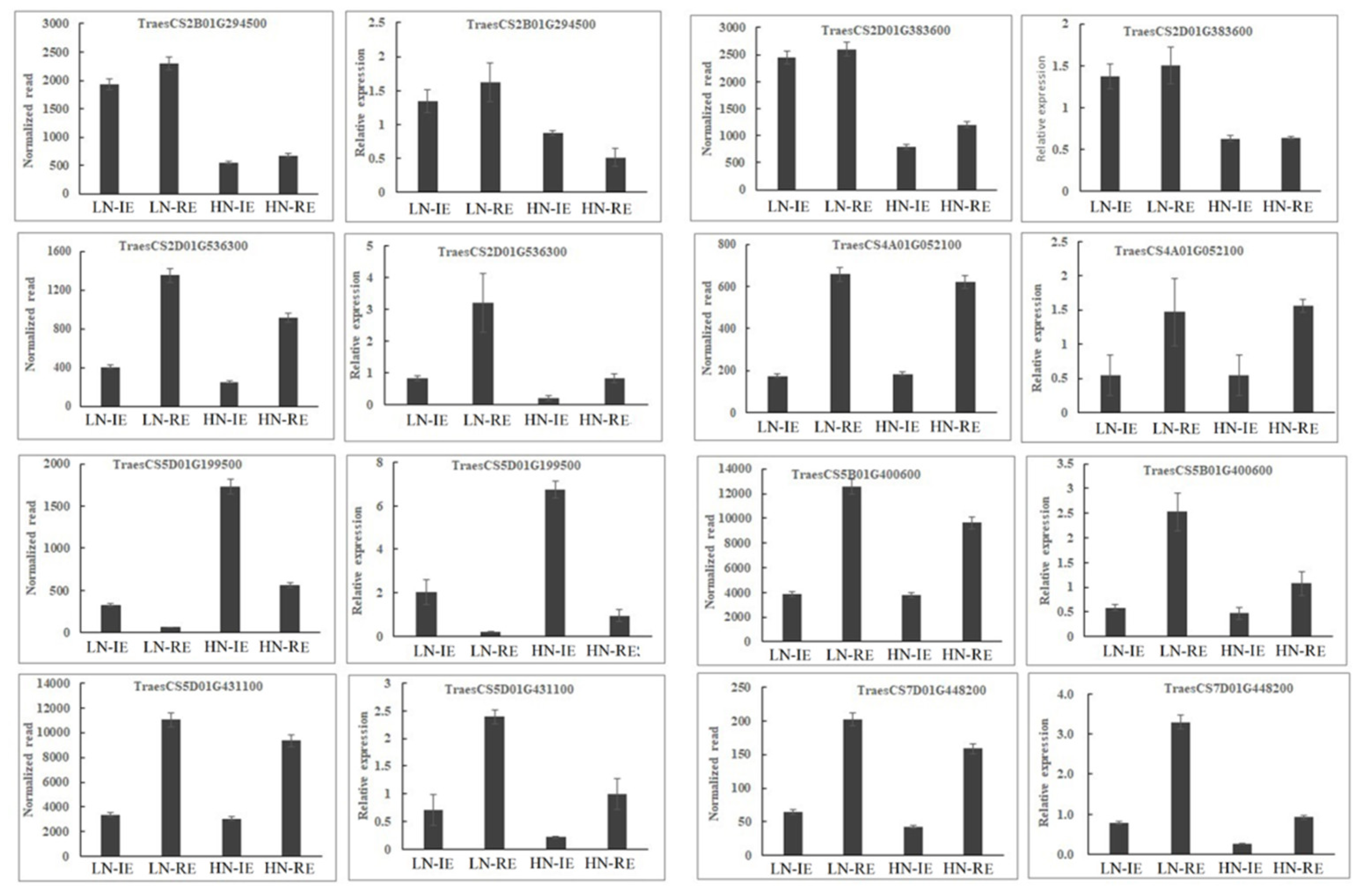
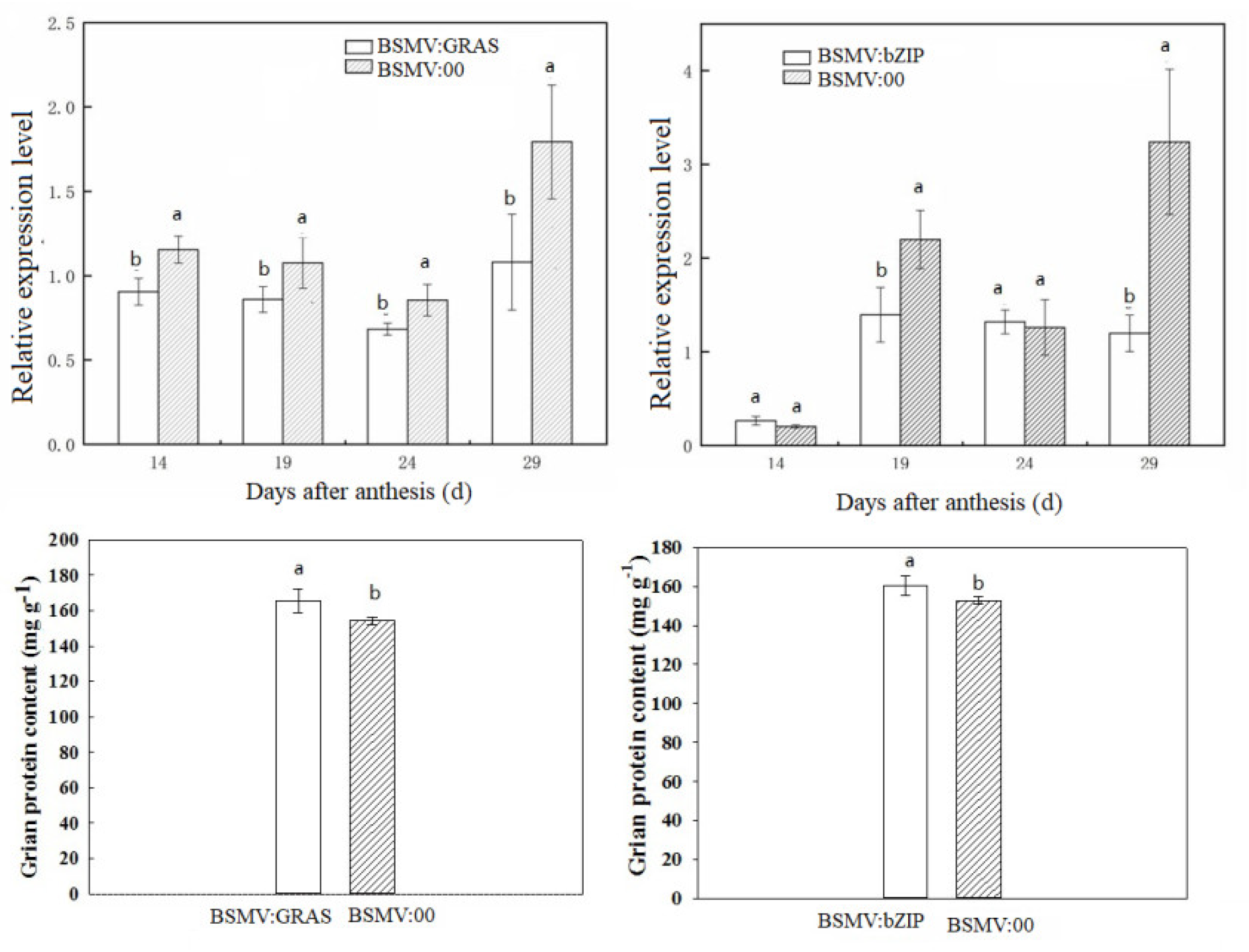
2.3. qRT-PCR and BSMV-VIGS Validation of Identified Genes
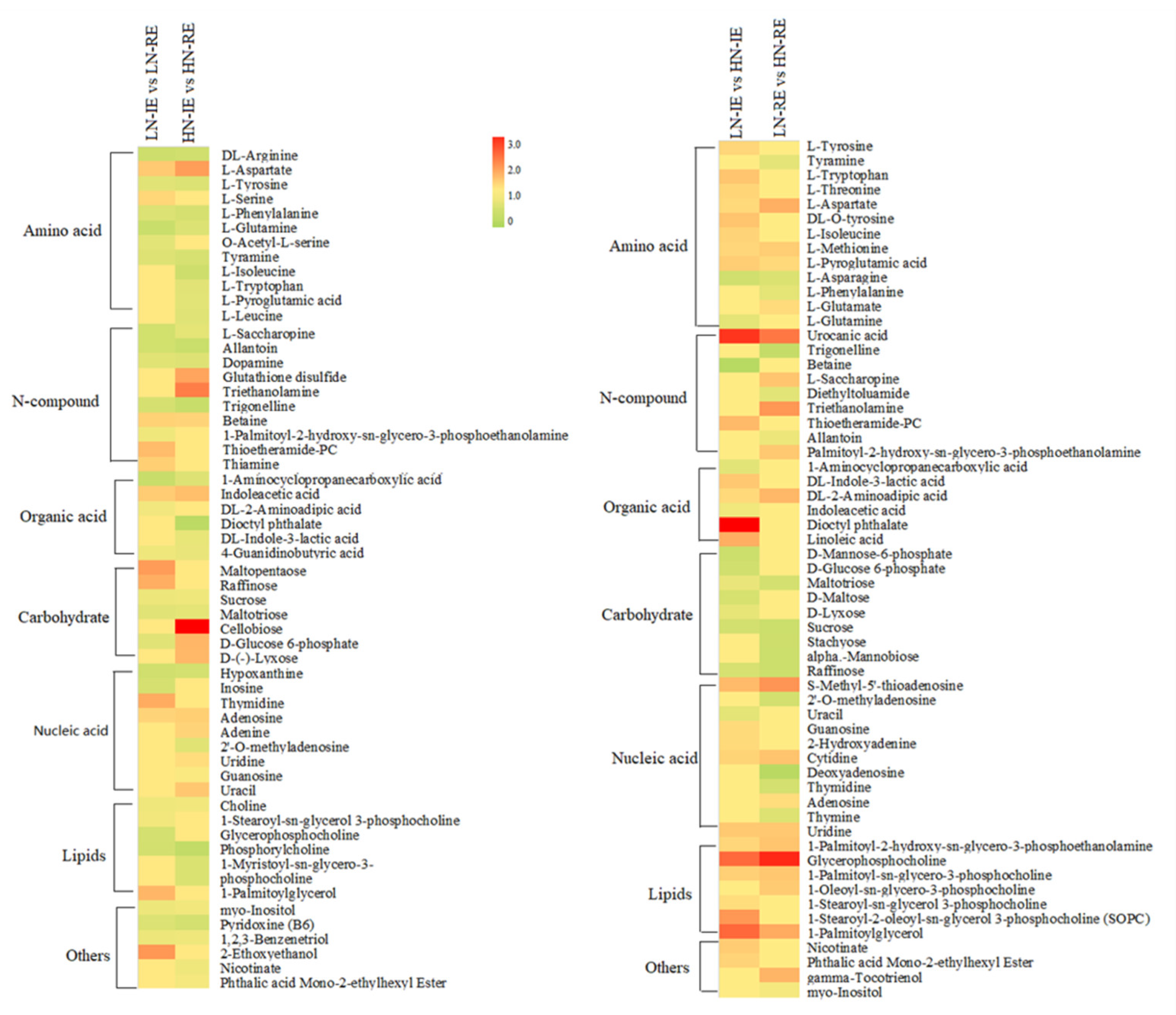
2.4. Metabolic Profiling of the Innermost Endosperm Region and the Remaining Endosperm Region under LN and HN Conditions
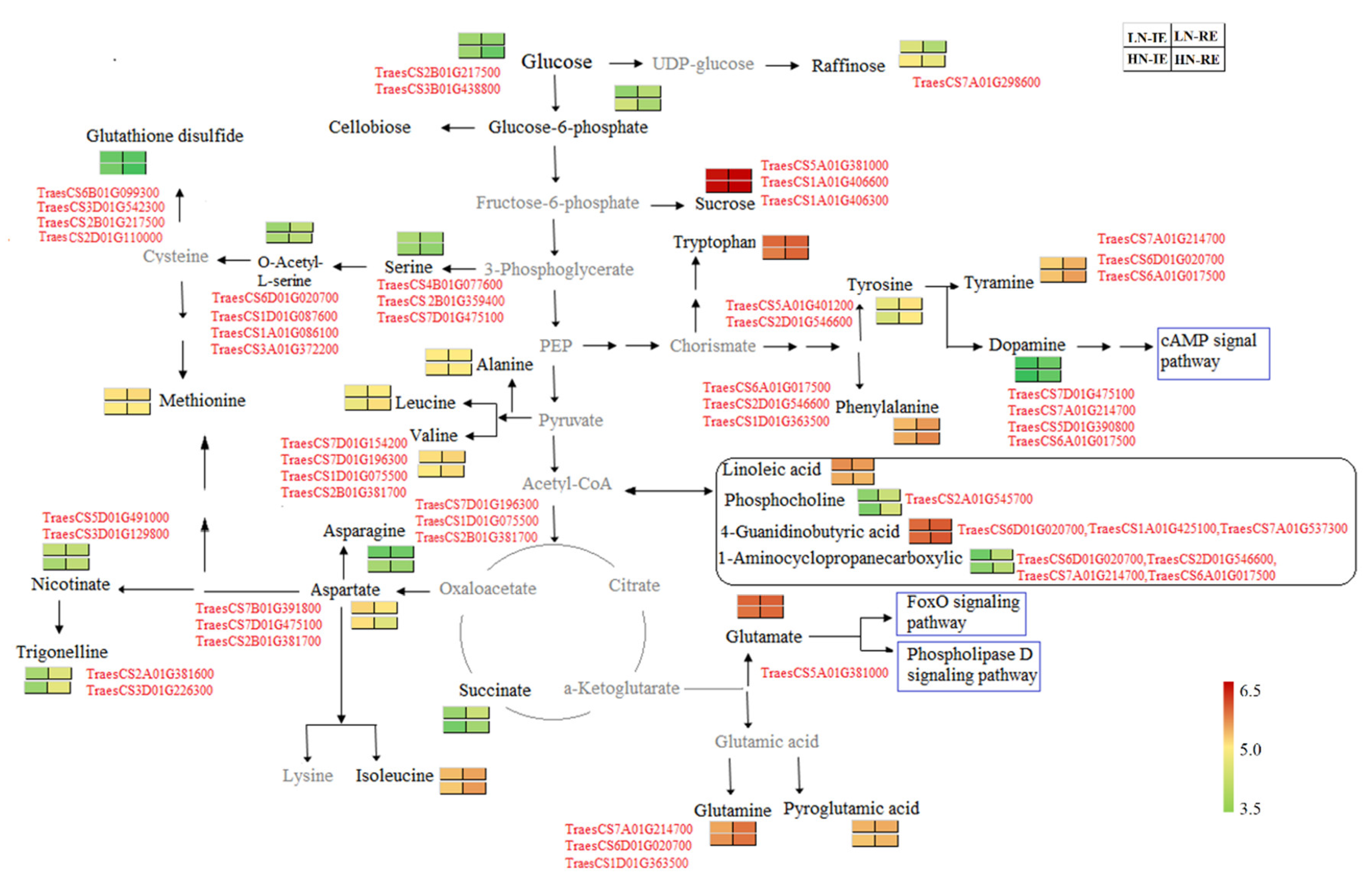
2.5. Combined Transcriptome and Metabonomics Analysis
3. Discussion
4. Materials and Methods
4.1. Experimental Design and Plant Material
4.2. Sampling
4.3. Transcript Profiling
4.4. Gene Expression Quantification Using qPCR
4.5. Grain Metabolite Extraction
4.6. Metabolomic Analysis
4.7. Functional Gene Analysis Using the Barley Stripe Mosaic Virus (BSMV)-Based Virus-Induced Gene Silencing (VIGS) System
4.8. Determination of Grain Total Protein Content and the Individual Fraction Contents
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Park, C.S.; Hong, B.H.; Baik, B. Protein quality of wheat desirable for making fresh white salted noodles and its influences on processing and texture of noodles. Cereal Chem. 2003, 80, 297–303. [Google Scholar] [CrossRef]
- Tosi, P.; Gritsch, C.S.; He, J.B.; Shewry, P.R. Distribution of gluten proteins in bread wheat (Triticum aestivum) grain. Ann. Bot. 2011, 108, 23–35. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Penson, S.; Powers, S.J.; Hawes, C.; Shewry, P.R.; Tosi, P. Spatial patterns of gluten protein and polymer distribution in wheat grain. J Agric. Food Chem. 2013, 61, 6207–6215. [Google Scholar] [CrossRef] [PubMed]
- Farrand, E.A.; Hinton, J.J.C. Study of relationships between wheat protein contents of two U.K. varieties and derived flour protein contents at varying extraction rates. II. Studies by hand-dissection of individual grains. Cereal Chem. 1974, 51, 66–73. [Google Scholar]
- Savill, G.P.; Michalski, A.; Powers, S.J.; Wan, Y.; Tosi, P.; Buchner, P.; Hawkesford, M.J. Temperature and nitrogen supply interact to determine protein distribution gradients in the wheat grain endosperm. J. Exp. Bot. 2018, 69, 3117–3126. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.Y.; Li, B.; Shao, S.S.; Wang, L.L.; Zhu, X.W.; Yang, Y.; Wang, W.J.; Yu, X.R.; Xiong, F. Accumulation characteristic of protein bodies in different regions of wheat endosperm under drought stress. J. Integr. Agr. 2016, 5, 2921–2930. [Google Scholar] [CrossRef]
- Zhou, Q.; Li, X.; Yang, J.; Zhou, L.; Cai, J.; Wang, X.; Dai, T.; Cao, W.; Jiang, D. Spatial distribution patterns of protein and starch in wheat grain affect baking quality of bread and biscuit. J. Cereal Sci. 2018, 79, 362–369. [Google Scholar] [CrossRef]
- Fustier, P.; Castaigne, F.; Turgeon, S.L.; Biliaderis, C.G. Flour constituent interactions and their influence on dough rheology and quality of semi-sweet biscuits: a mixture design approach with reconstituted blends of gluten, water-solubles and starch fractions. J Cereal Sci. 2008, 48, 144–158. [Google Scholar] [CrossRef]
- Ugalde, T.D.; Jenner, C.F. Substrate gradients and regional patterns of dry matter deposition within developing wheat endosperm. II. Amino acids and protein. Australian J. Plant Physiol. 1990, 17, 395–406. [Google Scholar] [CrossRef]
- Moore, K.L.; Tosi, P.; Palmer, R.; Hawkesford, M.J.; Grovenor, C.R.M.; Shewry, P.R. The dynamics of protein body formation in developing wheat grain. Plant Biotechnol. J. 2016, 14, 1876–1882. [Google Scholar] [CrossRef]
- Li, X.; Zhou, L.; Liu, F.; Zhou, Q.; Cai, J.; Wang, X.; Dai, T.; Cao, W.; Jiang, D. Variations in protein concentration and nitrogen sources in different position of grain in wheat. Frontiers Plant Sci. 2016, 7, 942. [Google Scholar] [CrossRef] [PubMed]
- Ayoub, M.; Guertin, S.; Fregeau-Reid, J.; Smith, D.L. Nitrogen fertilizer effect on breadmaking quality of hard red spring wheat in eastern Canada. Crop Sci. 1994, 34, 1346–1352. [Google Scholar] [CrossRef]
- Ma, D.; Guo, T.; Wang, Z.; Wang, C.; Zhu, Y.; Wang, Y. Influence of nitrogen fertilizer application rate on winter wheat (Triticuma estivum L.) flour quality and Chinese noodle quality. J Sci. Food Agric. 2009, 89, 1213–1220. [Google Scholar] [CrossRef]
- Zhang, M.W.; Ma, D.Y.; Ma, G.; Wang, C.Y.; Xie, X.D.; Kang, G.Z. Responses of glutamine synthetase activity and gene expression to nitrogen levels in winter wheat cultivars with different grain protein content. J Cereal Sci. 2017, 74, 187–193. [Google Scholar] [CrossRef]
- Wan, Y.; Gritsch, C.S.; Hawkesford, M.J.; Shewry, P.R. Effects of nitrogen nutrition on the synthesis and deposition of the ω-gliadins of wheat. Ann. Bot. 2014, 113, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Islam, S.; She, M.; Diepeveen, D.; Zhang, Y.; Tang, G.; Zhang, J.; Juhasz, A.; Yang, R.; Ma, W. Wheat grain protein accumulation and polymerization mechanisms driven by nitrogen fertilization. Plant J. 2018, 96, 1160–1177. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.; Upadyay, A.K.; Biradar, H.; Hittalmani, S. OsNAC-like transcription factor involved in regulating seed-storage protein content at different stages of grain filling in rice under aerobic conditions. J. Genet. 2019, 98, 18. [Google Scholar] [CrossRef]
- Distelfeld, A.; Korol, A.; Dubcovsky, J.; Uauy, C.; Blake, T.; Fahima, T. Colinearity between the barley grain protein content (GPC) QTL on chromosome arm 6HS and the wheat Gpc-B1 region. Mol. Breeding 2008, 22, 25–38. [Google Scholar] [CrossRef]
- Ugalde, T.D.; Jenner, C.F. Substrate gradients and regional patterns of dry matter deposition within developing wheat endosperm. I. carbohydrates. Australian J. Plant Physiol. 1990, 17, 377–394. [Google Scholar] [CrossRef]
- Tosi, P.; Parker, M.; Gritsch, C.S.; Carzaniga, R.; Martin, B.; Shewry, P.R. Trafficking of storage proteins in developing grain of wheat. J. Exp. Bot. 2009, 60, 979–991. [Google Scholar] [CrossRef]
- Okrajkova´, A.; Prieto-Linde, M.L.; Muchova´, Z.; Johansson, E. Protein concentration and composition in wheat flour mills streams. Cereal Res. Commun. 2007, 35, 119–128. [Google Scholar] [CrossRef]
- Hirai, M.Y.; Fujiwara, T.; Chino, M.; Naito, S. Effects of sulfate concentrations on the expression of a soybean seed storage protein gene and its reversibility in transgenic Arabidopsis thaliana. Plant Cell Physiol. 1995, 36, 1331–1339. [Google Scholar] [PubMed]
- Dai, Z.; Plessis, A.; Vincent, J.; Duchateau, N.; Besson, A.; Dardevet, M.; Prodhomme, D.; Gibon, Y.; Hilbert, G.; Pailloux, M. Transcriptional and metabolic alternations rebalance wheat grain storage protein accumulation under variable nitrogen and sulfur supply. Plant J. 2015, 83, 326–343. [Google Scholar] [CrossRef] [PubMed]
- Zhen, S.M.; Zhou, J.X.; Deng, X.; Zhu, G.R.; Cao, H.; Wang, Z.H.; Yan, Y.M. Metabolite profiling of the response to high-nitrogen fertilizer during grain development of bread wheat (Triticum aestivum L.). J. Cereal Sci. 2016, 69, 85–94. [Google Scholar] [CrossRef]
- Fettke, J.; Fernie, A.R. Intracellular and cell-to-apoplast compartmentation of carbohydrate metabolism. Trends Plant Sci. 2015, 20, 490–497. [Google Scholar] [CrossRef] [PubMed]
- Weichert, N.; Saalbach, I.; Weichert, H.; Kohl, S.; Erban, A.; Kopka, J.; Hause, B.; Varshney, A.; Sreenivasulu, N.; Strickert, M. Increasing sucrose uptake capacity of wheat grains stimulates storage protein synthesis. Plant Physiol. 2010, 152, 698–710. [Google Scholar] [CrossRef]
- Weber, H.; Rolletschek, H.; Heim, U.; Golombek, S.; Gubatz, S.; Wobus, U. Antisense-inhibition of ADP-glucose pyrophosphorylase in developing seeds of Vicia narbonensis moderately decreases starch but increases protein content and affects seed maturation. Plant J. 2000, 24, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Li, C.X.; Zhang, G.F.; Shi, H.E.; Shi, Y.M. Effects of nitrogen on the dynamic change of nitrate reductase activity and grain protein content of wheat. Acta Bot. Boreali-Occident. Sin. 1995, 15, 276–281. [Google Scholar]
- Ohkama-Ohtsu, N.; Kezuka, A.; Onouchi, H.; Fujiwara, T.; Naito, S. Promoter region of the beta subunit gene of beta-conglycinin responds to methionine and glutathione in transient assays using Arabidopsis protoplasts. Soil Sci. Plant Nut. 2008, 54, 128–132. [Google Scholar] [CrossRef]
- Domínguze, F.; González, M.; Cejudo, F.J. A germination-related gene encoding a serine carboxypeptidase is expressed during the differentiation of the vascular tissue in wheat grains and seedlings. Planta 2002, 215, 727–734. [Google Scholar] [CrossRef]
- Weier, D.; Mittasch, J.; Strack, D.; Milkowski, C. The genes BnSCT1 and BnSCT2 from Brassica napus encoding the final enzyme of sinapine biosynthesis: Molecular characterization and suppression. Planta 2008, 227, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Beers, E.P.; Woffenden, B.J.; Zhao, C. Plant proteolytic enzymes: possible roles during programmed cell death. Plant Mol. Biol. 2000, 44, 399–415. [Google Scholar] [CrossRef] [PubMed]
- Veliz, C.G.; Criado, M.V.; Roberts, I.N.; Echeverria, M.; Prieto, P.; Gutierrez Boem, F.H.; Caputo, C.; Prystupa, P. Phloem sugars and amino acids as potential regulators of hordein expression in field grown malting barley (Hordeum vulgare L.). J. Cereal Sci. 2014, 60, 433–439. [Google Scholar] [CrossRef]
- Sogawa, Y.; Ohkama-Ohtsu, N.; Hayashi, H.; Yoneyama, T.; Fujiwara, T. Independent roles of glutathione and O-acetyl-L-serine in regulation of sulfur-responsive gene expression in Arabidopsis thaliana. Plant Biotechnol. 2005, 22, 51–54. [Google Scholar] [CrossRef][Green Version]
- Rubio-Somoza, I.; Martinez, M.; Abraham, Z.; Diaz, I.; Carbonero, P. Ternary complex formation between HvMYBS3 and other factors involved in transcriptional control in barley seeds. Plant J. 2006, 47, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Perry, S.E. Identification of direct targets of FUSCA3, a key regulator of Arabidopsis seed development. Plant Physiol. 2013, 161, 1251–1264. [Google Scholar] [CrossRef]
- Yamamoto, M.P.; Onodera, Y.; Touno, S.M.; Takaiwa, F. Synergism between RPBF Dof and RISBZ1 bZIP activators in the regulation of rice seed expression genes. Plant Physiol. 2006, 141, 1694–1707. [Google Scholar] [CrossRef]
- Kawakatsu, T.; Yamamoto, M.P.; Touno, S.M.; Yasuda, H.; Takaiwa, F. Compensation and interaction between RISBZ1 and RPBF during grain filling in rice. Plant J. 2009, 59, 908–920. [Google Scholar] [CrossRef]
- Yang, J.; Wang, M.; Li, W.; He, X.; Teng, W.; Ma, W.; Zhao, X.; Hu, M.; Li, H.; Zhang, Y. Reducing expression of a nitrate-responsive bZIP transcription factor increases grain yield and N use in wheat. Plant Biotechnol. J. 2019. [Google Scholar] [CrossRef]
- Battaglia, M.; Rípodas, C.; Clúa, J.; Baudin, M.; Aguilar, O.M.; Niebel, A.; Zanetti, M.E.; Blanco, F.A. A nuclear factor Y interacting protein of the GRAS family is required for nodule organogenesis, infection thread progression, and lateral root growth. Plant Physiol. 2014, 164, 1430–1442. [Google Scholar] [CrossRef]
- Xu, K.; Chen, S.; Li, T.; Ma, X.; Liang, X.; Ding, X.; Liu, H.; Luo, L. OsGRAS23, a rice GRAS transcription factor gene, is involved in drought stress response through regulating expression of stress-responsive genes. BMC Plant Biol. 2015, 15, 141. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Jones, W.T.; Rikkerink, E.H.A. GRAS proteins: the versatile roles of intrinsically disordered proteins in plant signalling. Biochem. J. 2012, 442, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Szopaa, J.; Wilczyński, G.; Fiehnb, O.; Wenczela, A.; Willmitzer, L. Identification and quantification of catecholamines in potato plants (Solanum tuberosum) by GC–MS. Phytochemistry 2001, 58, 315–320. [Google Scholar] [CrossRef]
- Smith, T.A. Plant amines. Secondary Plant Products. In Encyclopedia of Plant Physiology New Series; Bell, E.A., Charlwood, B.V., Eds.; Springer: Berlin, Germany, 1980; Volume 8, pp. 433–440. [Google Scholar]
- Ciepiela, A.P.; Sempruch, C. Effect of L -3, 4-dihydroxyphenylalanine, ornithine and gamma-aminobutyric acid on winter wheat resistance to grain aphid. J. Appl. Entomol. 1999, 123, 285–288. [Google Scholar] [CrossRef]
- Sebastia, C.H.; Marsolais, F.; Saravitz, C.; Israel, D.; Dewey, R.E.; Huber, S.C. Free amino acid profiles suggest a possible role for asparagine in the control of storage-product accumulation in developing seeds of low- and high-protein soybean lines. J. Exp. Bot. 2005, 56, 1951–1963. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.; Pachter, L. Transcript assembly and abundance estimation from RNA-Seq reveals thousands of new transcripts and switching among isoforms. Nature Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]
- Ma, M.; Yan, Y.; Huang, L.; Chen, M.; Zhao, H. Virus-induced gene-silencing in wheat spikes and grains and its application in functional analysis of HMW-GS-encoding genes. BMC Plant Biol. 2012, 12, 141. [Google Scholar] [CrossRef]
- Liu, Z.H.; Cheng, F.M.; Cheng, W.D.; Zhang, G.P. Positional variations in phytic acid and protein content within a panicle of japonica rice. J. Cereal Sci. 2005, 41, 297–303. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Item. | Total Protein Content (mg g−1) | Albumin Content (mg g−1) | Globulin Content (mg g−1) | Gliadin Content (mg g−1) | Glutenin Content (mg g−1) | Grain Yield (kg ha−1) | |
|---|---|---|---|---|---|---|---|
| HN | IE | 118.5 ± 2.1 b | 32.0 ± 3.0 b | 11.5 ± 1.6 b,c | 23.1 ± 0.1 c | 46.1 ± 0.4 a,b | 7241.5 ± 84.6 a |
| RE | 137.5 ± 0.7 a | 36.9 ± 1.6 a | 19.4 ± 1.1 a | 25.6 ± 0.1 b | 49.0 ± 3.3 a | ||
| LN | IE | 101.5 ± 2.4 c | 29.6 ± 0.5 b | 8.4 ± 2.1 c | 19.9 ± 0.6 d | 43.0 ± 1.0 b | 5864.6 ± 131 b |
| RE | 116.5 ± 1.2 b | 30.8 ± 2.4 b | 12.3 ± 0.8 b | 26.5 ± 1.3 a | 44.8 ± 1.4 a,b | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, D.; Gao, H.; Du, C.; Li, L.; Sun, W.; Liu, S.; Wang, C.; Xie, Y.; Kang, G. Transcriptomic and Metabolomics Analysis of Different Endosperm Region under Nitrogen Treatments. Int. J. Mol. Sci. 2019, 20, 4212. https://doi.org/10.3390/ijms20174212
Ma D, Gao H, Du C, Li L, Sun W, Liu S, Wang C, Xie Y, Kang G. Transcriptomic and Metabolomics Analysis of Different Endosperm Region under Nitrogen Treatments. International Journal of Molecular Sciences. 2019; 20(17):4212. https://doi.org/10.3390/ijms20174212
Chicago/Turabian StyleMa, Dongyun, Honghuan Gao, Chenyang Du, Lingli Li, Wan Sun, Sujun Liu, Chenyang Wang, Yingxin Xie, and Guozhang Kang. 2019. "Transcriptomic and Metabolomics Analysis of Different Endosperm Region under Nitrogen Treatments" International Journal of Molecular Sciences 20, no. 17: 4212. https://doi.org/10.3390/ijms20174212
APA StyleMa, D., Gao, H., Du, C., Li, L., Sun, W., Liu, S., Wang, C., Xie, Y., & Kang, G. (2019). Transcriptomic and Metabolomics Analysis of Different Endosperm Region under Nitrogen Treatments. International Journal of Molecular Sciences, 20(17), 4212. https://doi.org/10.3390/ijms20174212