Inhibition of Phosphoinositide 3-Kinase/Protein Kinase B Signaling Hampers the Vasopressin-dependent Stimulation of Myogenic Differentiation

 , , ,
, , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Background
2. Results
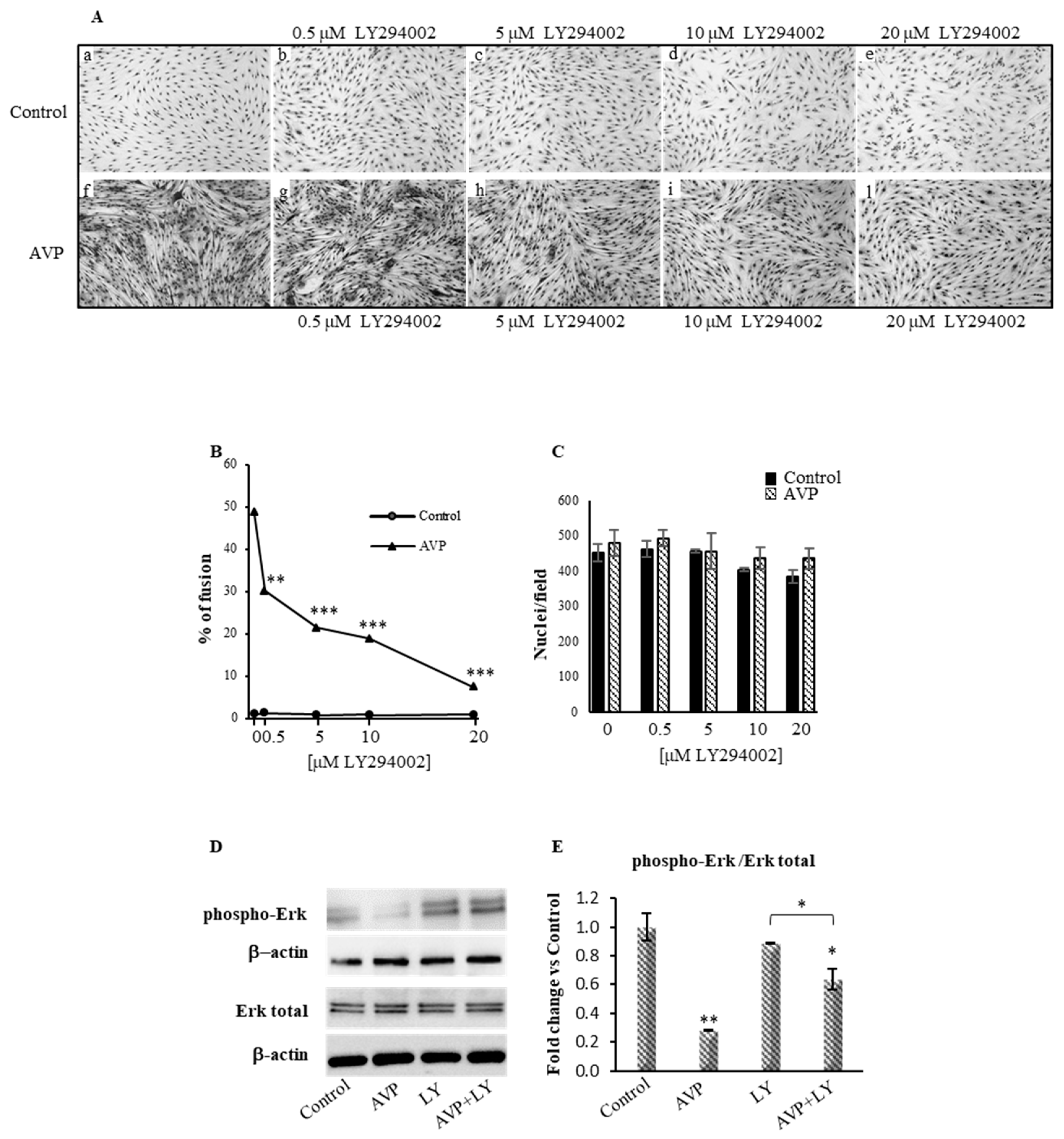
2.1. LY294002 Inhibits the AVP-Dependent Myogenic Differentiation of L6 Cells
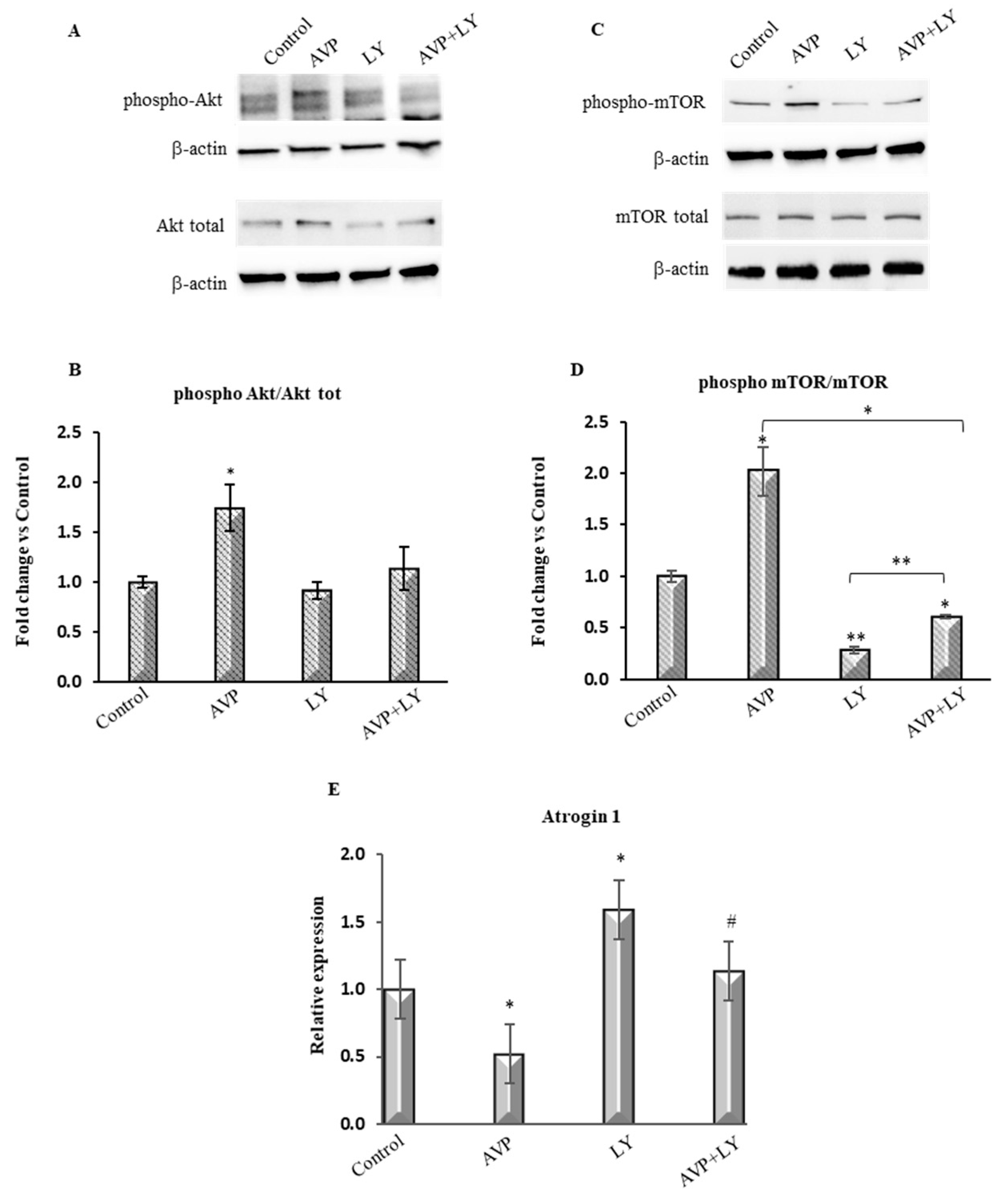
2.2. LY294002 Counteracts the AVP-Dependent Stimulation of PI3K/Akt/mTOR Signaling
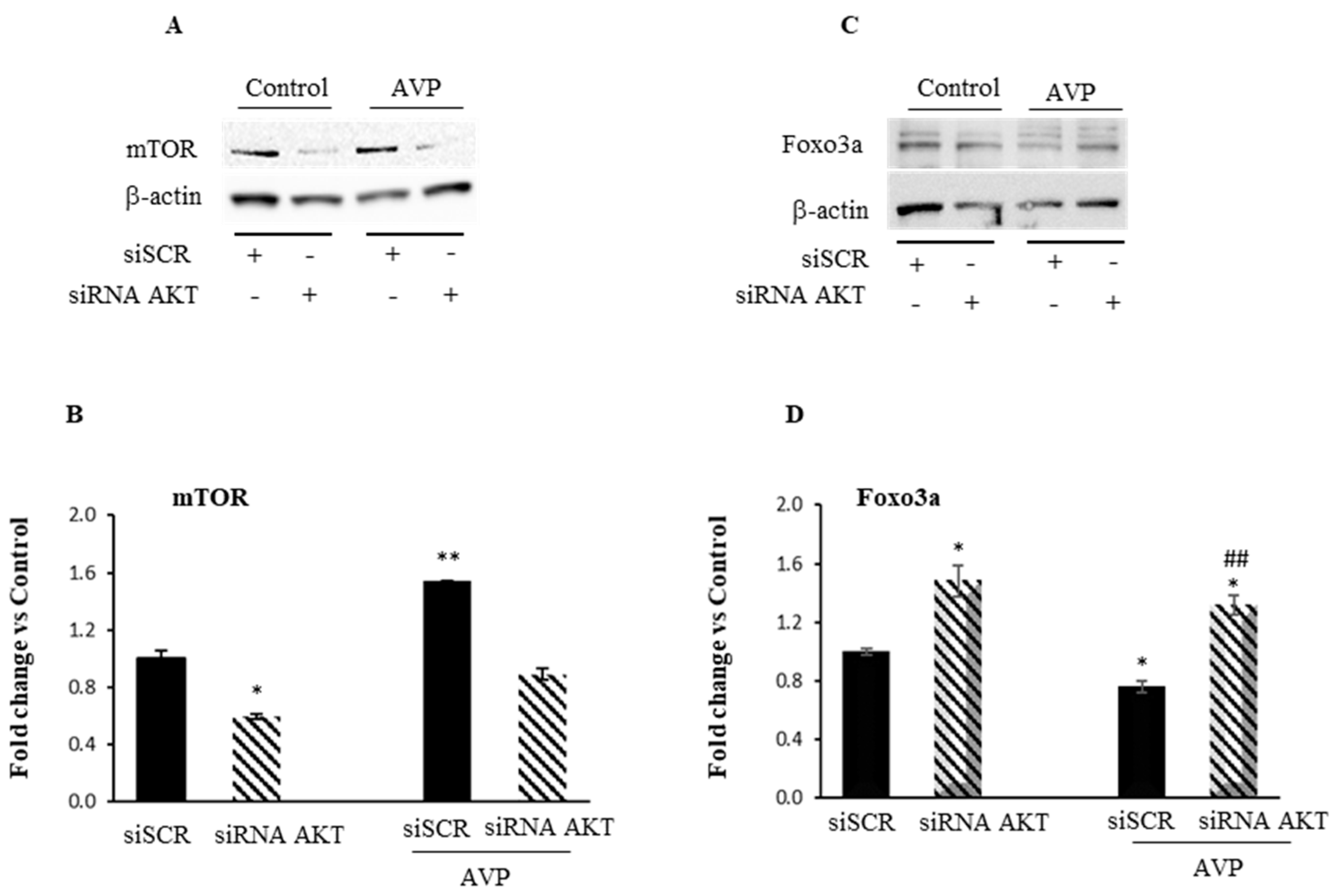
2.3. Akt Knockdown Hampers the Myogenic Effect of AVP in L6 Cells
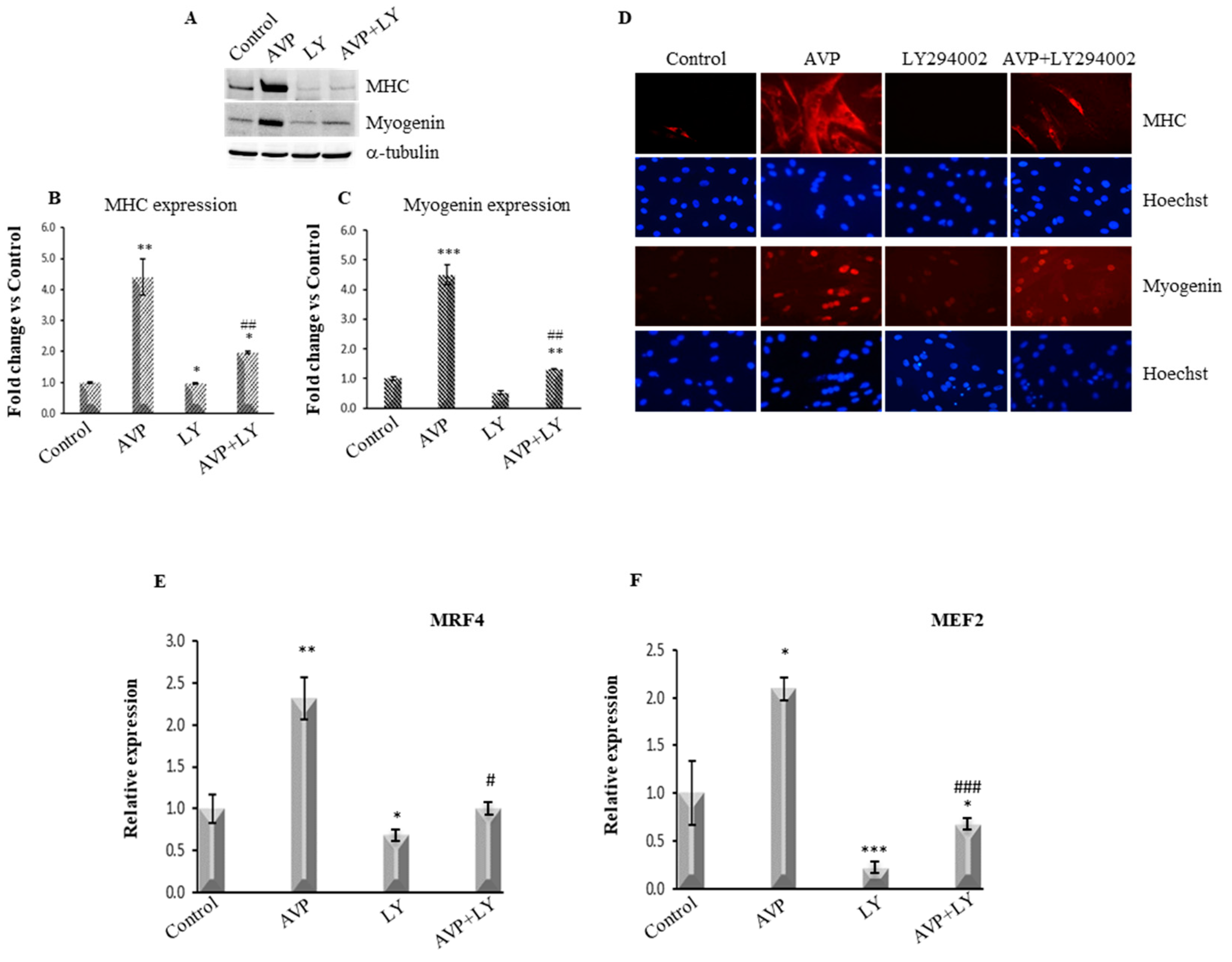
2.4. LY294002-Dependent Inhibition of PI3K/Akt Signaling Hinders the AVP-Dependent Induction of Myogenin and MHC Expression
2.5. LY294002 Treatment Inhibits the AVP-Dependent Stimulation of MRF4 and MEF2 mRNA Expression
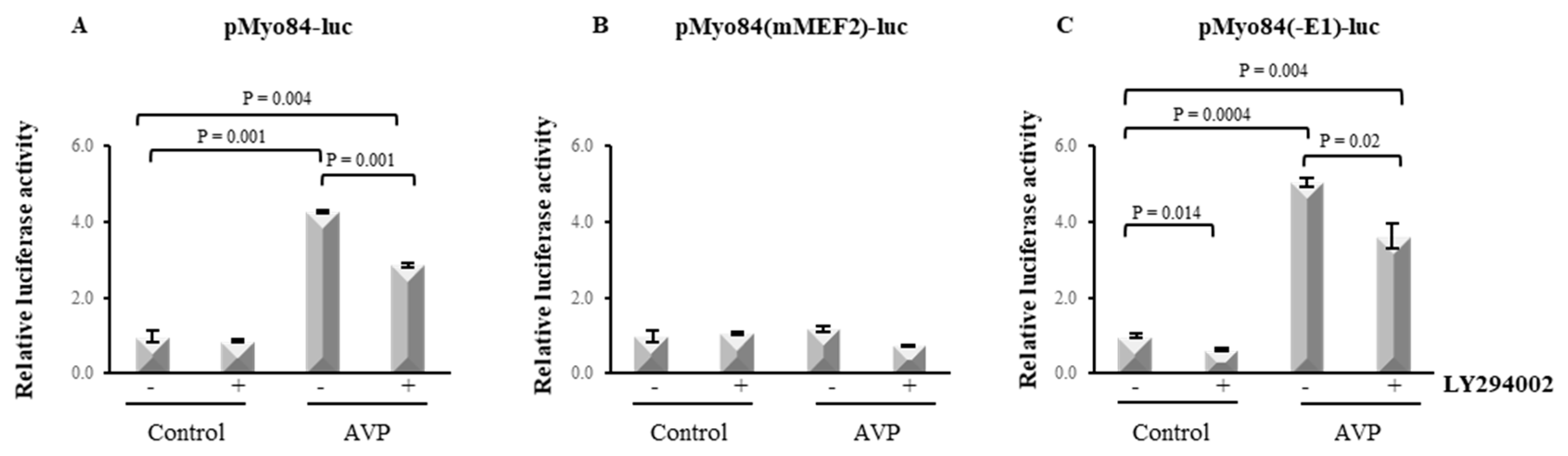
2.6. LY294002 Hampers the AVP-Dependent Activation of the 84-bp Myogenin Promoter
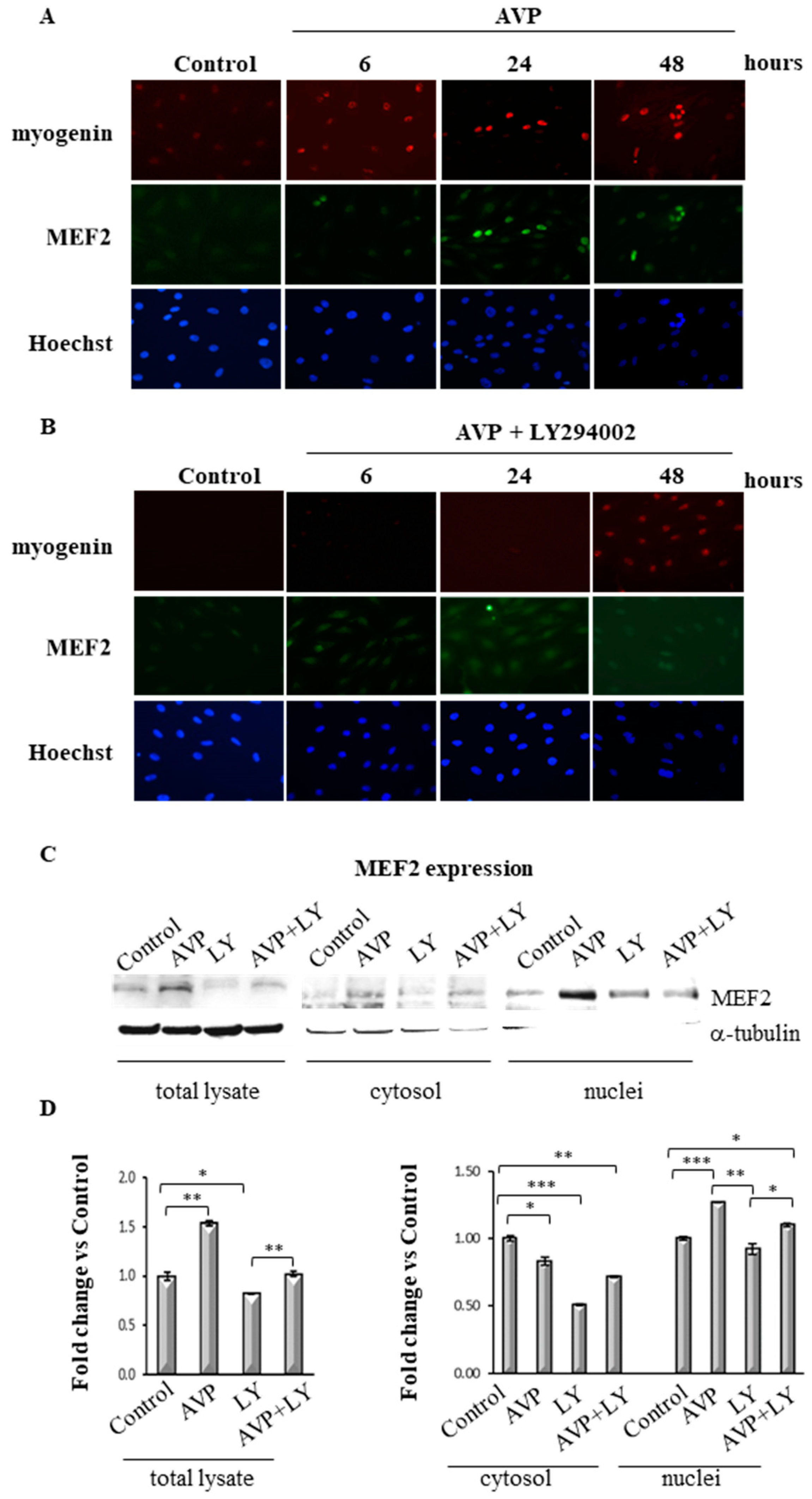
2.7. LY294002 Regulates MEF2 Expression and Cellular Localization in AVP-Treated L6 Cells
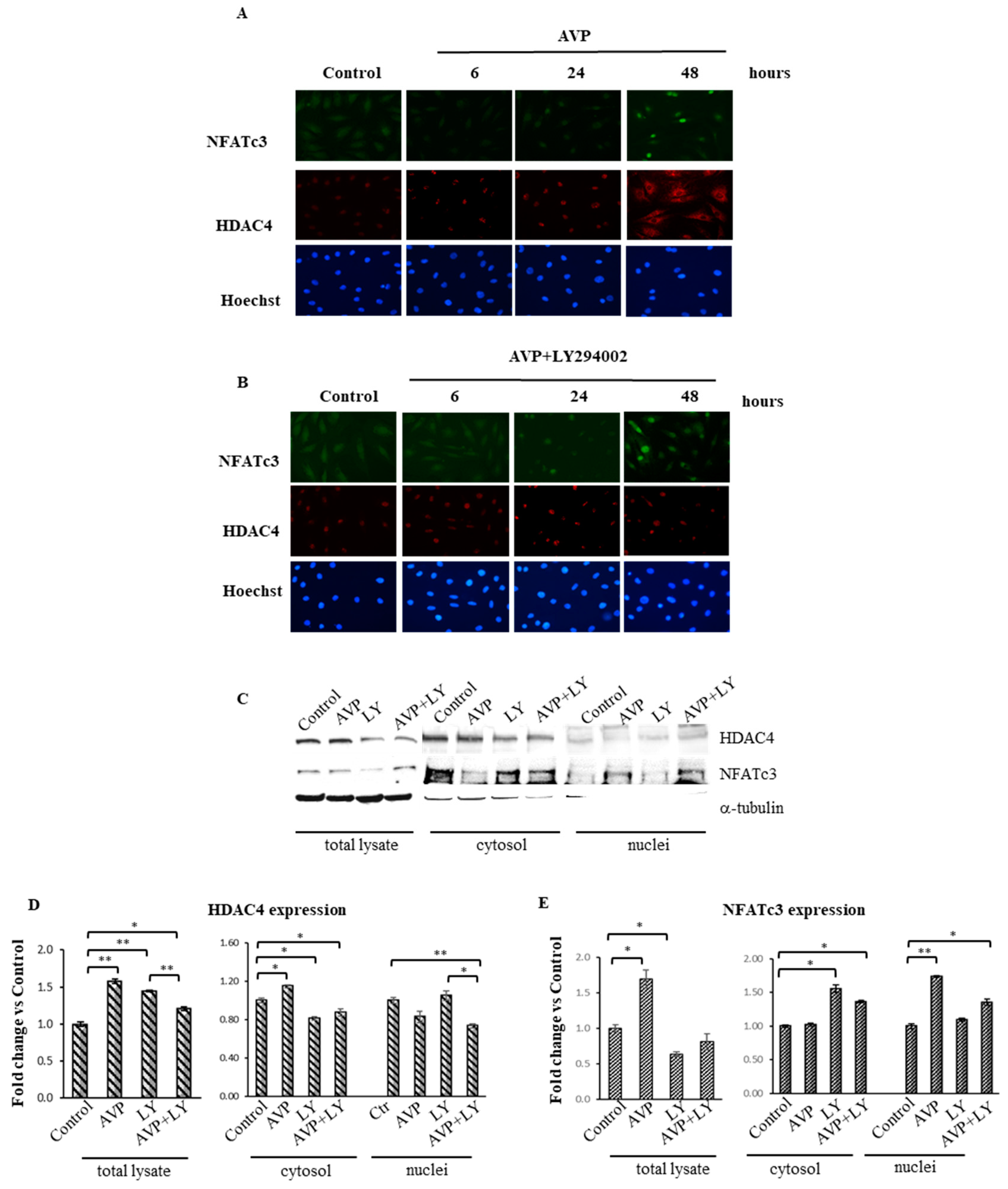
2.8. LY294002 Interferes with the AVP-Dependent Cellular Localization of HDAC4 and NFATc3 in L6 Cells
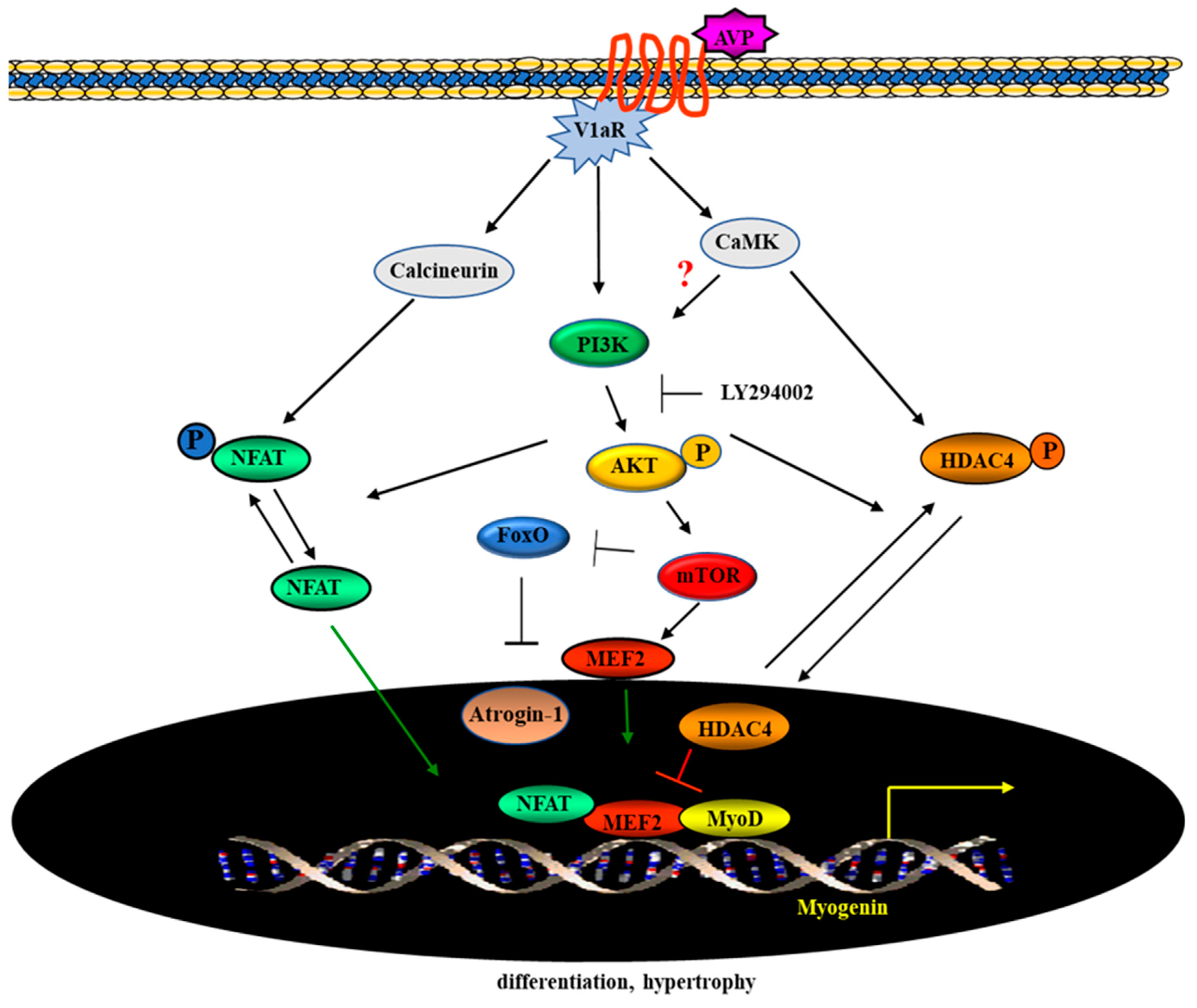
3. Discussion
4. Methods
4.1. Cell Cultures
4.2. Morphological Analysis and Measurement of Myoblast Fusion and Growth
4.3. Real-Time PCR Analysis
4.4. Western Blot Analysis
4.5. Immunofluorescence ASSAY
4.6. Transient Transfection of L6 CELLS
4.7. siRNA Transfection
4.8. Statistical Analysis
Supplementary Materials
Availability of Data Material
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Akt | Protein kinase B |
| AVP | Arg8-vasopressin |
| CaMK | Ca2+-Calmodulin-dependent protein kinase |
| CnA | Calcineurin A |
| FoxO | Forkhead box O transcription factor |
| HATs | Histone acetyltransferases |
| HDACs | Histone deacetylases |
| b-HLH-MRFs | Basic-Helix-Loop-Helix Myogenic Regulatory Factors |
| InsP3 | inositol 1,4,5-trisphosphate |
| LY294002 | 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one |
| MEF2 | Myocyte Enhancer Factor 2 |
| MHC | Myosin heavy chain |
| OT | oxytocin |
| PI3K | Phosphoinositide 3-Kinase |
| mTOR | Mammalian Target of Rapamycin |
| V1aR | V1a vasopressin Receptor |
References
- Edmondson, D.G.; Olson, E.N. A gene with homology to the myc similarity region of MyoD1 is expressed during myogenesis and is sufficient to activate the muscle differentiation program. Genes Dev. 1989, 3, 628–640. [Google Scholar] [CrossRef]
- Funk, W.D.; Ouellette, M.; Wright, W.E. Molecular biology of myogenic regulatory factors. Mol. Biol. Med. 1991, 8, 185–195. [Google Scholar]
- Nervi, C.; Benedetti, L.; Minasi, A.; Molinaro, M.; Adamo, S. Arginine-vasopressin induces differentiation of skeletal myogenic cells and up-regulation of myogenin and Myf-5. Cell Growth Differ. 1995, 6, 81–89. [Google Scholar]
- Lee, K.S.; Smith, K.; Amieux, P.S.; Wang, E.H. MBNL3/CHCR prevents myogenic differentiation by inhibiting MyoD-dependent gene transcription. Differentiation 2008, 76, 299–309. [Google Scholar] [CrossRef]
- Venuti, J.M.; Morris, J.H.; Vivian, J.L.; Olson, E.N.; Klein, W.H. Myogenin is required for late but not early aspects of myogenesis during mouse development. J. Cell Biol. 1995, 128, 563–576. [Google Scholar] [CrossRef]
- Hernández-Hernández, J.M.; García-González, E.G.; Brun, C.E.; Rudnicki, M.A. The myogenic regulatory factors, determinants of muscle development, cell identity and regeneration. Semin. Cell Dev. Biol. 2017, 72, 10–18. [Google Scholar] [CrossRef]
- Schwarz, J.J.; Chakraborty, T.; Martin, J.; Zhou, J.M.; Olson, E.N. The basic region of myogenin cooperates with two transcription activation domains to induce muscle-specific transcription. Mol. Cell. Biol. 1992, 12, 266–275. [Google Scholar] [CrossRef]
- Molkentin, J.D.; Black, B.L.; Martin, J.F.; Olson, E.N. Cooperative activation of muscle gene expression by MEF2 and myogenic bHLH proteins. Cell 1995, 83, 1125–1136. [Google Scholar] [CrossRef] [Green Version]
- Scicchitano, B.M.; Spath, L.; Musarò, A.; Molinaro, M.; Adamo, S.; Nervi, C. AVP induces myogenesis through the transcriptional activation of the myocyte enhancer factor 2. Mol. Endocrinol. 2002, 16. [Google Scholar] [CrossRef]
- Taylor, M.V.; Hughes, S.M. Mef2 and the skeletal muscle differentiation program. Semin. Cell Dev. Biol. 2017, 72, 33–44. [Google Scholar] [CrossRef]
- Sousa-Victor, P.; Muñoz-Cánoves, P.; Perdiguero, E. Regulation of skeletal muscle stem cells through epigenetic mechanisms. Toxicol. Mech. Methods 2011, 21, 334–342. [Google Scholar] [CrossRef]
- Yahi, H.; Philipot, O.; Guasconi, V.; Fritsch, L.; Ait-Si-Ali, S. Chromatin modification and muscle differentiation. Expert Opin. Ther. Targets 2006, 10, 923–934. [Google Scholar] [CrossRef]
- Bharathy, N.; Ling, B.M.T.; Taneja, R. Epigenetic Regulation of Skeletal Muscle Development and Differentiation. Sub-Cell. Biochem. 2013, 61, 139–150. [Google Scholar]
- Dressel, U.; Bailey, P.J.; Wang, S.-C.M.; Downes, M.; Evans, R.M.; Muscat, G.E.O. A Dynamic Role for HDAC7 in MEF2-mediated Muscle Differentiation. J. Biol. Chem. 2001, 276, 17007–17013. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; McKinsey, T.A.; Nicol, R.L.; Olson, E.N. Signal-dependent activation of the MEF2 transcription factor by dissociation from histone deacetylases. Proc. Natl. Acad. Sci. USA 2000, 97, 4070–4075. [Google Scholar] [CrossRef] [Green Version]
- McKinsey, T.A.; Zhang, C.L.; Olson, E.N. Activation of the myocyte enhancer factor-2 transcription factor by calcium/calmodulin-dependent protein kinase-stimulated binding of 14-3-3 to histone deacetylase 5. Proc. Natl. Acad. Sci. USA 2000, 97, 14400–14405. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Sternsdorf, T.; Bolger, T.A.; Evans, R.M.; Yao, T.-P. Regulation of MEF2 by histone deacetylase 4- and SIRT1 deacetylase-mediated lysine modifications. Mol. Cell. Biol. 2005, 25, 8456–8464. [Google Scholar] [CrossRef]
- Scicchitano, B.M.; Spath, L.; Musarò, A.; Molinaro, M.; Rosenthal, N.; Nervi, C.; Adamo, S. Vasopressin-dependent myogenic cell differentiation is mediated by both Ca2+/calmodulin-dependent kinase and calcineurin pathways. Mol. Biol. Cell 2005, 16. [Google Scholar] [CrossRef]
- Minotti, S.; Scicchitano, B.M.; Nervi, C.; Scarpa, S.; Lucarelli, M.; Molinaro, M.; Adamo, S. Vasopressin and insulin-like growth factors synergistically induce myogenesis in serum-free medium. Cell Growth Differ. 1998, 9, 155–163. [Google Scholar]
- Alvisi, M.; De Arcangelis, V.; Ciccone, L.; Palombi, V.; Alessandrini, M.; Nemoz, G.; Molinaro, M.; Adamo, S.; Naro, F. V1a vasopressin receptor expression is modulated during myogenic differentiation. Differentiation 2008, 76, 371–380. [Google Scholar] [CrossRef]
- Toschi, A.; Severi, A.; Coletti, D.; Catizone, A.; Musarò, A.; Molinaro, M.; Nervi, C.; Adamo, S.; Scicchitano, B.M. Skeletal muscle regeneration in mice is stimulated by local overexpression of V1a-vasopressin receptor. Mol. Endocrinol. 2011, 25, 1661–1673. [Google Scholar] [CrossRef]
- Costa, A.; Toschi, A.; Murfuni, I.; Pelosi, L.; Sica, G.; Adamo, S.; Scicchitano, B.M. Local overexpression of V1a-vasopressin receptor enhances regeneration in tumor necrosis factor-induced muscle atrophy. Biomed. Res. Int. 2014, 2014, 235426. [Google Scholar] [CrossRef]
- Costa, A.; Rossi, E.; Scicchitano, B.M.; Coletti, D.; Moresi, V.; Adamo, S. Neurohypophyseal Hormones: Novel Actors of Striated Muscle Development and Homeostasis. Eur. J. Transl. Myol. 2014, 24, 3790. [Google Scholar] [CrossRef]
- Breton, C.; Haenggeli, C.; Barberis, C.; Heitz, F.; Bader, C.R.; Bernheim, L.; Tribollet, E. Presence of functional oxytocin receptors in cultured human myoblasts. J. Clin. Endocrinol. Metab. 2002, 87, 1415–1418. [Google Scholar] [CrossRef]
- Thibonnier, M.; Graves, M.K.; Wagner, M.S.; Auzan, C.; Clauser, E.; Willard, H.F. Structure, sequence, expression, and chromosomal localization of the human V1a vasopressin receptor gene. Genomics 1996, 31, 327–334. [Google Scholar] [CrossRef]
- Leevers, S.J.; Vanhaesebroeck, B.; Waterfield, M.D. Signalling through phosphoinositide 3-kinases: The lipids take centre stage. Curr. Opin. Cell Biol. 1999, 11, 219–225. [Google Scholar] [CrossRef]
- Chini, B.; Fanelli, F. Molecular basis of ligand binding and receptor activity in the oxytocin and vasoressin receptor family. Exp. Physiol. 2000, 85, 59S–66S. [Google Scholar] [CrossRef]
- Teti, A.; Naro, F.; Molinaro, M.; Adamo, S. Transduction of arginine vasopressin signal in skeletal myogenic cells. Am. J. Physiol. Physiol. 1993, 265, C113–C121. [Google Scholar] [CrossRef]
- Naro, F.; Donchenko, V.; Minotti, S.; Zolla, L.; Molinaro, M.; Adamo, S. Role of phospholipase C and D signalling pathways in vasopressin-dependent myogenic differentiation. J. Cell. Physiol. 1997, 171, 34–42. [Google Scholar] [CrossRef]
- Wasilewski, M.A.; Myers, V.D.; Recchia, F.A.; Feldman, A.M.; Tilley, D.G. Arginine vasopressin receptor signaling and functional outcomes in heart failure. Cell. Signal. 2016, 28, 224–233. [Google Scholar] [CrossRef]
- Falasca, M.; Logan, S.K.; Lehto, V.P.; Baccante, G.; Lemmon, M.A.; Schlessinger, J. Activation of phospholipase C gamma by PI 3-kinase-induced PH domain-mediated membrane targeting. EMBO J. 1998, 17, 414–422. [Google Scholar] [CrossRef]
- Song, X.; Xu, W.; Zhang, A.; Huang, G.; Liang, X.; Virbasius, J.V.; Czech, M.P.; Zhou, G.W. Phox homology domains specifically bind phosphatidylinositol phosphates. Biochemistry 2001, 40, 8940–8944. [Google Scholar] [CrossRef]
- Calleja, V.; Alcor, D.; Laguerre, M.; Park, J.; Vojnovic, B.; Hemmings, B.A.; Downward, J.; Parker, P.J.; Larijani, B. Intramolecular and intermolecular interactions of protein kinase B define its activation in vivo. PLoS Biol. 2007, 5, e95. [Google Scholar] [CrossRef]
- Downward, J. PI 3-kinase, Akt and cell survival. Semin. Cell Dev. Biol. 2004, 15, 177–182. [Google Scholar] [CrossRef]
- Bonaldo, P.; Sandri, M. Cellular and molecular mechanisms of muscle atrophy. Dis. Model. Mech. 2013, 6, 25–39. [Google Scholar] [CrossRef] [Green Version]
- Glass, D.J. PI3 Kinase Regulation of Skeletal Muscle Hypertrophy and Atrophy. Curr. Top. Microbiol. Immunol. 2010, 346, 267–278. [Google Scholar]
- Schiaffino, S.; Dyar, K.A.; Ciciliot, S.; Blaauw, B.; Sandri, M. Mechanisms regulating skeletal muscle growth and atrophy. FEBS J. 2013, 280, 4294–4314. [Google Scholar] [CrossRef]
- Schiaffino, S.; Mammucari, C. Regulation of skeletal muscle growth by the IGF1-Akt/PKB pathway: Insights from genetic models. Skelet. Muscle 2011, 1, 4. [Google Scholar] [CrossRef]
- Scicchitano, B.M.; Sica, G. The Beneficial Effects of Taurine to Counteract Sarcopenia. Curr. Protein Pept. Sci. 2018, 19, 673–680. [Google Scholar] [CrossRef]
- Scicchitano, B.M.; Dobrowolny, G.; Sica, G.; Musaro, A. Molecular Insights into Muscle Homeostasis, Atrophy and Wasting. Curr. Genomics 2018, 19, 356–369. [Google Scholar] [CrossRef]
- Moresi, V.; Garcia-Alvarez, G.; Pristerà, A.; Rizzuto, E.; Albertini, M.C.; Rocchi, M.; Marazzi, G.; Sassoon, D.; Adamo, S.; Coletti, D. Modulation of caspase activity regulates skeletal muscle regeneration and function in response to vasopressin and tumor necrosis factor. PLoS ONE 2009, 4, e5570. [Google Scholar] [CrossRef]
- Dong, X.; Qin, J.; Ma, J.; Zeng, Q.; Zhang, H.; Zhang, R.; Liu, C.; Xu, C.; Zhang, S.; Huang, S.; et al. BAFF inhibits autophagy promoting cell proliferation and survival by activating Ca2+-CaMKII-dependent Akt/mTOR signaling pathway in normal and neoplastic B-lymphoid cells. Cell. Signal. 2019, 53, 68–79. [Google Scholar] [CrossRef]
- Ke, Z.; Liang, D.; Zeng, Q.; Ren, Q.; Ma, H.; Gui, L.; Chen, S.; Guo, M.; Xu, Y.; Gao, W.; et al. hsBAFF promotes proliferation and survival in cultured B lymphocytes via calcium signaling activation of mTOR pathway. Cytokine 2013, 62, 310–321. [Google Scholar] [CrossRef]
- González de Aguilar, J.L.; Gordon, J.W.; René, F.; Lutz-Bucher, B.; Kienlen-Campard, P.; Loeffler, J.P. A mouse model of familial amyotrophic lateral sclerosis expressing a mutant superoxide dismutase 1 shows evidence of disordered transport in the vasopressin hypothalamo-neurohypophysial axis. Eur. J. Neurosci. 1999, 11, 4179–4187. [Google Scholar] [CrossRef]
- Michelson, D.; Stone, L.; Galliven, E.; Magiakou, M.A.; Chrousos, G.P.; Sternberg, E.M.; Gold, P.W. Multiple sclerosis is associated with alterations in hypothalamic-pituitary-adrenal axis function. J. Clin. Endocrinol. Metab. 1994, 79, 848–853. [Google Scholar]
- Musarò, A.; Dobrowolny, G.; Rosenthal, N. The neuroprotective effects of a locally acting IGF-1 isoform. Exp. Gerontol. 2007, 42, 76–80. [Google Scholar] [CrossRef]
- Szasz, G.; Gruber, W.; Bermt, E. Creatin Kinase in serum: 1. Determination of optimum reaction conditions. Clin. Chem. 1976, 22, 650–656. [Google Scholar]
- Jiang, B.-H.; Aoki, M.; Zheng, J.Z.; Li, J.; Vogt, P.K. Myogenic signaling of phosphatidylinositol 3-kinase requires the serine-threonine kinase Akt/protein kinase B. Proc. Natl. Acad. Sci. 1999, 96, 2077–2081. [Google Scholar] [CrossRef] [Green Version]
- Puri, P.L.; Sartorelli, V. Regulation of muscle regulatory factors by DNA-binding, interacting proteins, and post-transcriptional modifications. J. Cell. Physiol. 2000, 185, 155–173. [Google Scholar] [CrossRef]
- Bober, E.; Lyons, G.E.; Braun, T.; Cossu, G.; Buckingham, M.; Arnold, H.H. The muscle regulatory gene, Myf-6, has a biphasic pattern of expression during early mouse development. J. Cell Biol. 1991, 113, 1255–1265. [Google Scholar] [CrossRef]
- Hinterberger, T.J.; Sassoon, D.A.; Rhodes, S.J.; Konieczny, S. Expression of the muscle regulatory factor MRF4 during somite and skeletal myofiber development. Dev. Biol. 1991, 147, 144–156. [Google Scholar] [CrossRef]
- Naidu, P.S.; Ludolph, D.C.; To, R.Q.; Hinterberger, T.J.; Konieczny, S.F. Myogenin and MEF2 function synergistically to activate the MRF4 promoter during myogenesis. Mol. Cell. Biol. 1995, 15, 2707–2718. [Google Scholar] [CrossRef] [Green Version]
- Musarò, A.; Rosenthal, N. Maturation of the myogenic program is induced by postmitotic expression of insulin-like growth factor I. Mol. Cell. Biol. 1999, 19, 3115–3124. [Google Scholar] [CrossRef]
- Miska, E.A.; Karlsson, C.; Langley, E.; Nielsen, S.J.; Pines, J.; Kouzarides, T. HDAC4 deacetylase associates with and represses the MEF2 transcription factor. EMBO J. 1999, 18, 5099–5107. [Google Scholar] [CrossRef]
- Coolican, S.A.; Samuel, D.S.; Ewton, D.Z.; McWade, F.J.; Florini, J.R. The Mitogenic and Myogenic Actions of Insulin-like Growth Factors Utilize Distinct Signaling Pathways. J. Biol. Chem. 1997, 272, 6653–6662. [Google Scholar] [CrossRef] [Green Version]
- Kaliman, P.; Viñals, F.; Testar, X.; Palacín, M.; Zorzano, A. Phosphatidylinositol 3-kinase inhibitors block differentiation of skeletal muscle cells. J. Biol. Chem. 1996, 271, 19146–19151. [Google Scholar] [CrossRef]
- Jiang, B.H.; Zheng, J.Z.; Vogt, P.K. An essential role of phosphatidylinositol 3-kinase in myogenic differentiation. Proc. Natl. Acad. Sci. USA 1998, 95, 14179–14183. [Google Scholar] [CrossRef] [Green Version]
- Forcina, L.; Miano, C.; Scicchitano, B.; Musarò, A. Signals from the Niche: Insights into the Role of IGF-1 and IL-6 in Modulating Skeletal Muscle Fibrosis. Cells 2019, 8, 232. [Google Scholar] [CrossRef]
- Ascenzi, F.; Barberi, L.; Dobrowolny, G.; Villa Nova Bacurau, A.; Nicoletti, C.; Rizzuto, E.; Rosenthal, N.; Scicchitano, B.M.; Musarò, A. Effects of IGF-1 isoforms on muscle growth and sarcopenia. Aging Cell 2019, 18, e12954. [Google Scholar] [CrossRef]
- Bain, J.; Plater, L.; Elliot, M.; Shpiro, N.; Hastie, C.J.; McLauchlan, H.; Klevernic, I.; Arthur, J.S.; Alessi, D.R.; Cohen, P. The selectivity of protein kinaseinhibitors: A further update. Biochem. J. 2007, 408, 297–315. [Google Scholar] [CrossRef]
- Hiroyama, M.; Wang, S.; Aoyagi, T.; Oikawa, R.; Sanbe, A.; Takeo, S.; Tanoue, A. Vasopressin promotes cardiomyocyte hypertrophy via the vasopressin V1A receptor in neonatal mice. Eur. J. Pharmacol. 2007, 559, 89–97. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sorrentino, S.; Barbiera, A.; Proietti, G.; Sica, G.; Adamo, S.; Scicchitano, B.M. Inhibition of Phosphoinositide 3-Kinase/Protein Kinase B Signaling Hampers the Vasopressin-dependent Stimulation of Myogenic Differentiation. Int. J. Mol. Sci. 2019, 20, 4188. https://doi.org/10.3390/ijms20174188
Sorrentino S, Barbiera A, Proietti G, Sica G, Adamo S, Scicchitano BM. Inhibition of Phosphoinositide 3-Kinase/Protein Kinase B Signaling Hampers the Vasopressin-dependent Stimulation of Myogenic Differentiation. International Journal of Molecular Sciences. 2019; 20(17):4188. https://doi.org/10.3390/ijms20174188
Chicago/Turabian StyleSorrentino, Silvia, Alessandra Barbiera, Gabriella Proietti, Gigliola Sica, Sergio Adamo, and Bianca Maria Scicchitano. 2019. "Inhibition of Phosphoinositide 3-Kinase/Protein Kinase B Signaling Hampers the Vasopressin-dependent Stimulation of Myogenic Differentiation" International Journal of Molecular Sciences 20, no. 17: 4188. https://doi.org/10.3390/ijms20174188
APA StyleSorrentino, S., Barbiera, A., Proietti, G., Sica, G., Adamo, S., & Scicchitano, B. M. (2019). Inhibition of Phosphoinositide 3-Kinase/Protein Kinase B Signaling Hampers the Vasopressin-dependent Stimulation of Myogenic Differentiation. International Journal of Molecular Sciences, 20(17), 4188. https://doi.org/10.3390/ijms20174188